

Abstract
Epilepsy is linked to mutations in KCNQ channels. KCNQ channels including KCNQ2 and KCNQ3 are enriched in neurons, regulating action potential generation and modulation. Here, we showed that properties of KCNQ2 channel in rat hippocampal cultured neurons are regulated by ubiquitous calcium sensor calmodulin. We analyzed calmodulin function on the KCNQ2 channel in both HEK293 cells and neurons. We used shRNAs to suppress expression of calmodulin protein. On the other hand, we used cDNA to over-express calmodulin in HEK293 and neuron cells. In wild type and mis-sense mutations of KCNQ2 proteins, calmodulin over-expression enhanced outward K+ current and decreased neuronal activity. Meanwhile, calmodulin knockdown reduced KCNQ2 current and increased neuronal activity, showing that hippocampal neuronal excitability is regulated by expression level of calmodulin protein. Our data suggest that calmodulin performs a major function in regulating KCNQ2 properties via direct binding to KCNQ2 protein, indicating that calmodulin could be a target of as gene therapy in epilepsy.
Keywords: Epilepsy, KCNQ2, calmodulin, gene therapy
Introduction
Neuronal KCNQ channels are voltage-dependent potassium (K+) channels composed mostly of KCNQ2 (Kv7.2) and KCNQ3 (Kv7.3) subunits, which are found in the nervous system including the hippocampus neurons [1,2]. KCNQ2 and KCNQ3 genes are associated with benign familial neonatal seizures (BFNS) and autosomal dominant epilepsy of infancy [3]. The heteromeric Kv7.2/Kv7.3 tetramer generates the wild-type (WT) muscarinic-regulated potassium currents (M-currents), which is a slowly activating, non-inactivating K+ current that regulates neuronal excitability in the sub-threshold range for action potential generation. KCNQ channels regulate resting membrane potential and contribute to action potential properties. Pharmacological inhibition of KCNQ channels results in the excessive firing of action potentials typical of an epileptic seizure [4]. On the other hand, drug-induced enhancement of the M-current, by impeding excessive neuronal activity, exerts potent anticonvulsant effects, thus revealing a novel role for this K+ current as a primary pharmacological target for epilepsy [5]. Loss-of-function mutations in the KCNQ2 or KCNQ3 genes cause benign familial neonatal seizures (BFNS) and autosomal dominant epilepsy of infancy. Wang et al. identified a large, four-generation family in which all 17 members carrying Kv7.2 G271V mutation had infantile seizures with onset at age 2-4 months, with two of them also manifesting seizures later in life accompanied with either choreoathetosis or myokymia [6]. The molecular basis responsible for the functional characteristics of G271V mutant KCNQ2 channel by means of a combined biochemical, immunocytochemical, and electrophysiological approach were reported in Wang’s work [6].
KCNQ2 C-terminal tail contains two helical domains (helices A and B) that bind to calmodulin, which is a calcium (Ca2+) sensor [7]. Helix A contains the consensus CaM binding IQ motif whereas helix B mediates Ca2+ -dependent CaM binding [8]. Since CaM interaction with KCNQ2 is required for functional expression of KCNQ channels, here we tested the function of CaM in KCNQ channels by utilizing CaM cDNA plasmid, CaM shRNA, or mutations that enhance or reduce CaM activity. The result showed that CaM is a regulator for KCNQ2 activities in heterologous cells and cultured hippocampal neurons. Furthermore, we examined whether the G271V mutation-induced functional K+ channel impairment could be affected in the presence or absence of calmodulin in transiently transfected human embryonic kidney (HEK) 293 cells and hippocampus neuron cells.
Materials and methods
DNA constructs, mutagenesis and materials
The plasmids pcDNA3 containing KCNQ2-GFP, plasmids pJPA7 containing wild-type calmodulin (CaM) were used as described previously [9]. Mutations (G271V, A343D, and R353G) of human KCNQ2 cDNAs were cloned into pcDNA3.0 (Invitrogen, Milan, Italy) using the Quik Change site-directed mutagenesis kit (Stratagene, CA, USA). KCNQ2 and its mutations were incorporated in a cDNA construct encoding for a fusion protein Enhanced Green Fluorescent Protein (EGFP). The DNA construct was sequenced to rule out the introduction of undesired mutations using an ABI PRISM 310 sequencing apparatus (Applied Biosystems, Foster City, CA). Calmodulin shRNA was synthesized according to the method described recently [10]. Retigabine and XE-991 were purchased from Sigma-Aldrich Company.
Cell culture and heterologous expression of KCNQ2 and CaM
HEK293 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Biowhittaker), supplemented with 10% fetal bovine serum (Lonza), penicillin 50 U/ml (Lonza), streptomycin 100 U/ml (Lonza) and nonessential amino acids 0.1 mM (Lonza), and were maintained in a humidified 5% CO2 incubator at 37°C. 2 g DNA constructs were introduced into HEK293 cells through transient transfection using Lipofectamine 2000 as described by the manufacturer (Invitrogen, CA, USA). When needed, a plasmid encoding for EGFP (Clontech, Palo Alto, CA, USA) was used as transfection marker. Transfection efficiency was estimated to be approximately 80% to 90% (Figure 1).
Figure 1.
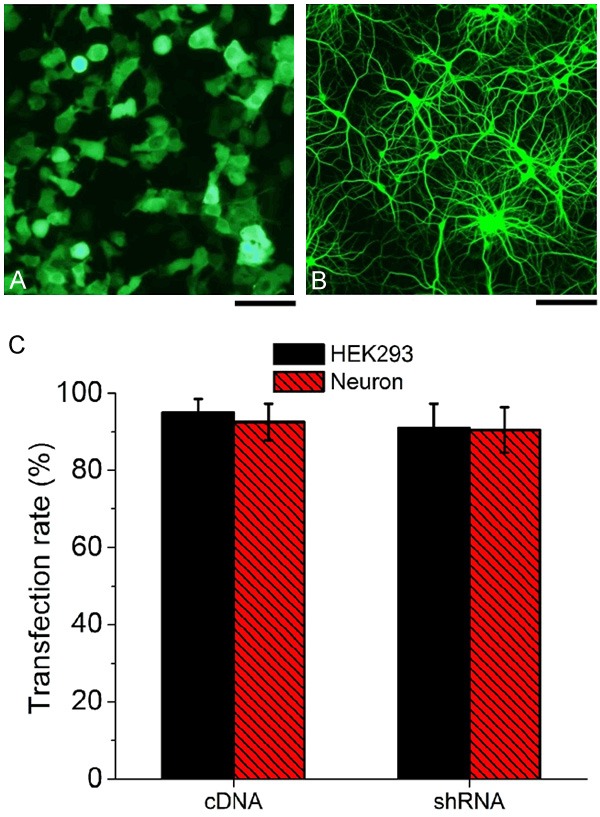
Analysis of transfection efficiency of GFP fused CaM cDNA and shRNA using confocal microscopy, showing both HEK293 (A) and neurons (B) expressed GFP in the cell bodies and neuron axons. (C) > 90% transfection rate in both cultured HEK293 cells and neurons transfected with CaM cDNA or CaM shRNA vectors (means ± S.E.M., n = 15, scale bar = 10 m).
Neuronal cell culture and transfection
Primary dissociated hippocampal cultures were prepared from hippocampi of 18 to 19 day embryonic rats as described [11]. Briefly, neurons were placed on 12 mm glass cover-slips (Warner Instruments, 105 cells per cover-slip), or 30 mm cell culture dishes (BD Biosciences, 7×105 cells per dish) coated with poly L-lysine (0.1 mg/mL). Neurons were maintained in neuro-basal medium supplemented with B27 extract, 200 mM L-glutamine, and 50 U/mL penicillin and 100 U/mL streptomycin in a cell culture incubator (37°C, 5% CO2) for 5 to 9 days in vitro. At 2 to 3 days in vitro, neurons were transfected with plasmids (~2 μg) using Lipofectamine 2000 (Invitrogen, CA, USA) according to manufacturer’s protocol.
Quantitative RT-PCR
Total RNA from 106 HEK293 cells or hippocampus neurons transfected with CaM cDNA plasmid or CaM shRNA constructs was reverse transcribed with the Vilo RT kit from Life Technologies. Obtained cDNAs were then used as input for quantitative PCR analysis with the following primers: β-actin forward, 5’-GCA CCA CAC CTT CTA CAA TG 3’ and reverse, 5’-TGC TTG CTG ATC CAC ATC TG-3’; CaM forward, 5’-CAG ATA TTG ATG GAG ACG GA-3’ and reverse, 5’-GAG CAC ACG AAG TAC AAG AG-3’.
Western blot
Western blot analysis of HEK293 cell extracts was conducted with Kv7.2 antibodies according to the protocol of Alomone Labs (Jerusalem, Israel). Briefly, proteins (2 μg/lane) from the lysate of transfected and non-transfected cells were resolved by SDS-PAGE gel before transfer onto a nitrocellulose membrane (Amersham Biosciences, Chalfont St. Giles, UK). As an internal standard for equal protein loading, β-tubulin was used. Nonfat dry milk (5%; Difco, Detroit, MI) was used as the blocking agent. Incubation was carried out with the first antibody (1:500 dilution) in blocking solution for 2 h and with the secondary antibody of horseradish peroxidase-conjugated goat antirabbit (1:10,000 dilution, Amersham Biosciences) in nonfat dry milk for 1 h. Blots were visualized by Super Signal West Pico Chemiluminescent Substrate (Pierce Biotechnology, USA). Chemiluminescent signals were collected by ChemiDoc XRP Imaging System and analyzed by Quantity One software (Bio-Rad Life Science). Each band was quantified as the total pixel value after subtraction of the background and normalized to the loading control protein GAPDH.
For hippocampal neurons, at 5 to 9 days in vitro, the cultured hippocampal neurons were washed with ice-cold ACSF and harvested in ice-cold lysis buffe (in mM). The lysis buffer contains 50 Tris, 150 NaCl, 2 EGTA, 1 EDTA, 1% Trition, 0.5% deoxycholic acid, 0.1% SDS (pH 7.4) supplemented with Halt protease inhibitors (Thermo Fisher Scientific). The resulting lysates were run on SDS-PAGE gels, transferred to a polyvinyl difluoride (PVDF) membrane (Immobilon, Millipore), and analyzed by immuno-blotting. Briefly, the membranes were blocked in 5% milk and 0.1% Tween-20 in Tris buffered saline for 1 h, and then incubated with rabbit anti-CaM (1:500 dilution) or anti-GAPDH antibody (1:1000 dilution) in wash buffer (1% milk and 0.1% Tween-20 in Tris buffered saline) overnight at 4°C. After incubating with horse radish peroxidase-conjugated secondary antibodies in wash buffer for 1 h, the blots were treated with enhanced chemifluorescence substrate (ECL, Thermo Fisher Scientific), and developed with a Konica SRX-101A film processor.
Electrophysiology in HEK 293 cells
KCNQ2 and its mutation (G271V) transfected HEK293 cells marked by GFP expression were selected using an upright fluorescence microscope (Zeiss Axioscope). The extra-cellular recording solution consisted of (in mM) 140 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1 NaH2PO4, and 5 Dextrose (pH 7.4, ~310 mOsm) titrated with NaOH. The internal solution contained (in mM) 130 K-gluconate, 1 MgCl2, 3 K2ATP, 5 K2-creatine phosphate, 10 EGTA and 5 HEPES (pH 7.3, ~285 mOsm) titrated with KOH. Borosilicate glass electrodes had tip resistances of 3 to 4 MΩ when filled with the pipette solution. Membrane currents were recorded using Axonpatch 200B amplifier (Molecular Devices). The capacitance and series resistance were compensated throughout the experiments. The currents were not leak-subtracted. Currents were sampled at 10 kHz and analyzed using pClamp 10.2 and Clampfit 10.2 software. All experiments were performed at room temperature (23 to 25°C). As a control, electrophysiological experiments were performed on un-transfected cells (GFP negative) and cells transfected with 2 μg empty vector (only GFP). To evaluate the function of Calmodulin protein in KCNQ2 channels, the same recordings in the whole-cell recording mode were performed in the presence XE-991 (3 M), or retigabine (10 μM) in bath solutions.
Electrophysiology in neurons
Cover-slips containing dissociated rat hippocampal neurons were transfected, and 24-48 h after transfection. The cover-slips were transferred to the whole-cell patch clamp recording chamber in external solution containing (in mM): 140 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1 NaH2PO4, and 5 Dextrose, bubbled with 95% O2 and 5% CO2 (pH 7.4, ~310 mOsm). The GFP-negative (un-transfected) or GFP-positive (transfected) pyramidal neurons were visually identified using an upright fluorescence microscope (Zeiss Axioscope) and the whole-cell patch clamp recordings were carried out immediately. All recordings were performed in current clamp mode at room temperature (23 to 25°C). Recording pipettes were pulled from borosilicate glass capillaries with an outer diameter of 1.5 mm on a micropipette puller (P-97; Sutter Instruments), and had a resistance of 3 to 4 MΩ when filled with internal solution containing (in mM): 135 KCl, 10 HEPES, 3 MgCl2, 1 EGTA and 3 ATP (pH 7.3, ~285 mOsm). Whole-cell recordings were made using an Axonpatch 200B amplifier (Molecular Devices). Neurons were held at -60 mV. Action potential firing rates (Hz) were measured upon delivering constant current pulses of 500 ms in the range 0 to 250 pA, and were averaged from 3 to 5 individual sweeps per current injection. Neurons were eliminated from further analysis if the access resistance changed by more than 20% over the recording period. Recordings were filtered at 5 kHz and digitized at 10 kHz. Data was acquired and analyzed with a Digidata 1440A interface (Molecular Devices) and the pClamp10.2 software (Molecular Devices). Recording analyses were performed using Clampfit10.2 software (Molecular Devices).
Statistical analyses
All fluorescence intensity and electrophysiology analyses were reported as mean ± SEM. ANOVA and post-ANOVA Tukey’s multiple comparison tests were performed to identify the statistically significant difference between groups of three or more, whereas the Student t test was performed for groups of two by Microsoft Excel with QI Macros 2013 plug-in. A priori value p < 0.05 was considered statistically significant. The number of separate transfected cells for immuno-staining and electrophysiology was reported as the sample number n.
Results
Expression of CaM in HEK293 cells
Confocal microscopy imaging showed that GFP expressed robustly in HEK 293 cells (Figure 1A) and hippocampal neurons (Figure 1B). We notice that both axon and cell body in neurons showed robust GFP fluorescence, indicating successful transfection in hippocampal neurons by using our cDNA or shRNA vectors (Figure 1B). Both CaM cDNA and shRNA showed a very high expression rate in both HEK293 cells and neurons as shown in Figure 1C, over 90% of cells were GFP positive with no obvious difference between HEK293 cells and neurons.
Next, we performed quantitative RT-PCR (q RT-PCR) analysis of CaM RNA from HEK293 cells and hippocampus neurons (Figure 2A, 2B). In both HEK 293 cells and hippocampus neurons, CaM expression increased by 1.5 fold in HEK293 cells and 1.2 fold in neurons after transfected with CaM cDNA. However, CaM expression decreased by ~70% in HEK293 cells and by ~50% in neurons when treated with CaM shRNA (Figure 2C).
Figure 2.
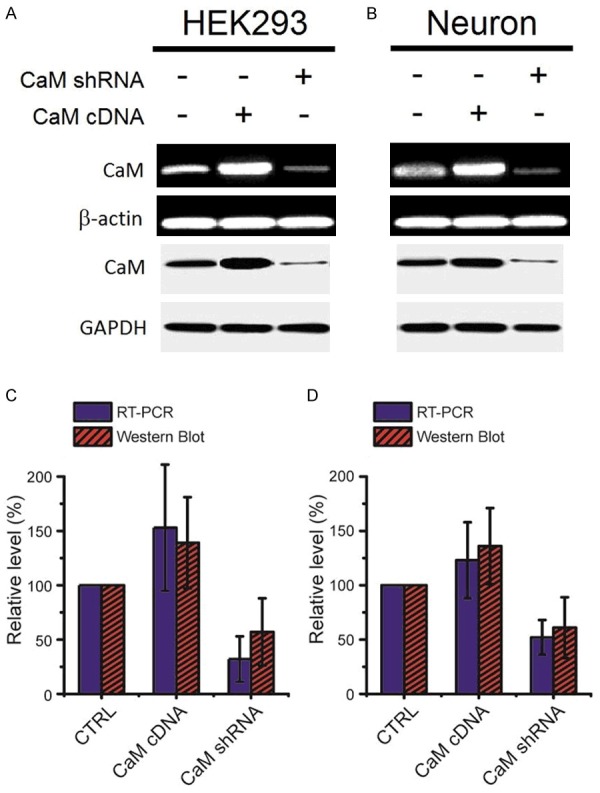
Calmodulin cDNA enhances calmodulin expression and CaM shRNA knocks down calmodulin expression in both HEK293 and neuron cells. A. qRT-PCR and Western blots of calmodulin from HEK293 cells in control and with transfection of calmodulin cDNA and shRNA. B. qRT-PCR and Western blots of calmodulin from neuron cells in control and with transfection of calmodulin cDNA and shRNA. C. Relative level of mRNA and protein after transfection with CaM cDNA and shRNA in HEK293 cells. D. Relative level of mRNA and protein after transfection with CaM cDNA and shRNA in neuron cells. β-actin and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control for RT-PCR and western-blot, respectively.
To further test the protein level, we used western blot confirm the expression change of calmodulin proteins in HEK293 cells and hippocampus neurons after transfection with CaM cDNA and shRNA (Figure 2A, 2B). The western blot results showed that HEK293 cells and hippocampus neurons transfected with CaM cDNA plasmid exhibited an enhanced band (~1.4 fold in HEK293 cells and ~1.3 fold in neurons). On the contrary, cells transfected with CaM shRNA had a reduced band (by ~50% in HEK293 cells and ~40% in neurons) (Figure 2C). The single band was approximately 96 kDa, which was consistent with the molecular weight of calmodulin. The CaM cDNA and shRNA constructs thus appeared to be functioning in both HEK293 cells and hippocampus neurons. β-actin and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control for RT-PCR and western-blot, respectively.
Effect of CaM expression on whole-cell currents from KCNQ2 and G271V mutation
To investigate the electrophysiological function of the effect of CaM on the KCNQ2 channels, KCNQ2 was expressed in HEK293 cells and functionally characterized using the Patch clamp method. Furthermore, a point mutation G271V in KCNQ2 was also recorded 48 h after transfection. Cells were held at -60 mV, and voltage steps in 10 mV increments from -120 mV to +100 mV followed with 160 ms duration followed a step at -40 mV (Figure 3).
Figure 3.
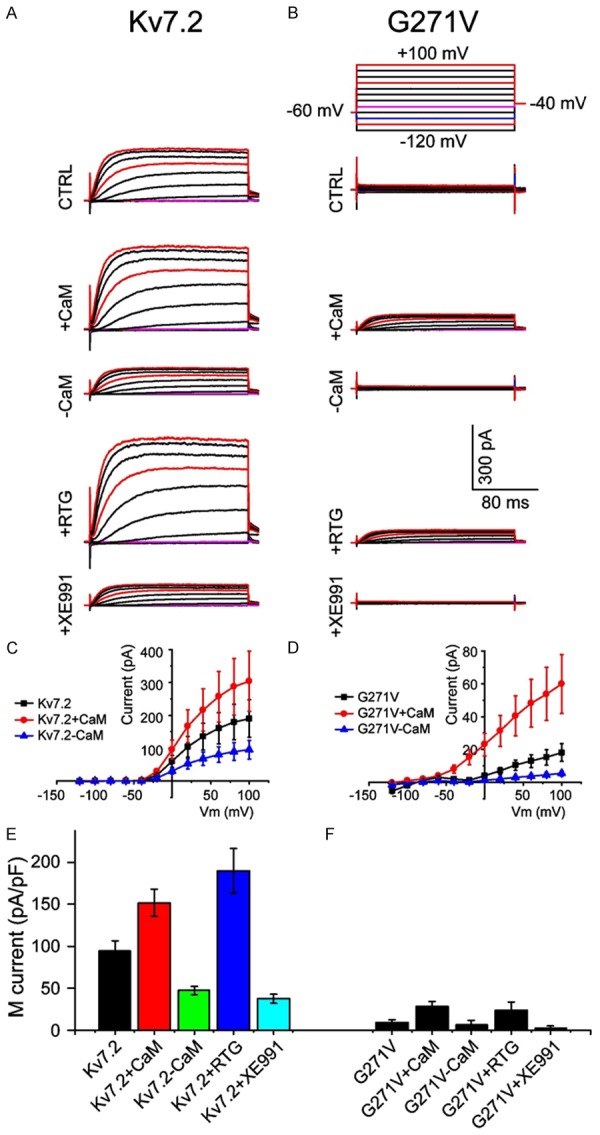
Electrophysiological properties of KCNQ2 and G271V expressed in HEK293 cells. Representative current traces for (A) KCNQ2 and (B) G271V channels recorded during voltage steps ranging from -120 mV to +100 mV at different conditions. +CaM: overexpression of CaM by cDNA; -CaM: knockdown of CaM by shRNA. Current-voltage curve expressing (C) KCNQ2 and (D) G271V mutant channels (n = 15, P < 0.05). Comparison of current densities of (E) KCNQ2 and (F) G271V (n = 15, P < 0.05).
Expression of KCNQ2 generated typical time-and voltage-dependent outward KCNQ currents (Figure 3A), whereas G271V mutation showed much reduced currents (Figure 3B), with only ~20 pA at a holding potential of +100 mV. After transfected with CaM cDNA or shRNA, the KCNQ2 current at increased CaM expression (+CaM) and reduced CaM expression (-CaM) were also measured as shown in Figure 3A and 3B. As shown in Figure 3A and 3C, the +CaM group exhibited robust increase in KCNQ2 currents, whereas the -CaM group channels had much reduced voltage-dependent outward KCNQ2 currents. Interestingly, CaM had the same effect on the KCNQ2 current in G271V mutant (Figure 3B, 3D), even thought the current was smaller in G271V mutant. Same results were shown in both KCNQ2 and G271V currents, which increased by > 50% in +CaM group but decreased by about 50% in -CaM group (Figure 3C, 3D), suggesting a pronounced functional change with changing the CaM expression. Current densities (pA/pF) of KCNQ2 currents were calculated by dividing the peak current at the end of the test pulse of +100 mV divided by the cell capacitance (Figure 3E, 3F). The current density of G271V expression (~10±0.41 pA/pF, n = 15, Figure 3F) was only ~10% of KCNQ2 current (~100 pA/pF, n = 15, p < 0.001, Figure 3E). The +CaM increased KCNQ2 current density to ~150 pA/pF and ~30 pA/pF for KCNQ2 and G271V, respectively. Surprisingly, the CaM effect was very similar to the effect of retigabine (RTG) and XE-991 (Figure 3A, 3B, 3E, 3F). RTG has demonstrated efficacy in a number of preclinical seizure and epilepsy models as a first-in-class, anti-epileptic drug for treatment resistant partial-onset seizures, while XE-991 is a KCNQ inhibitor. Increased current at RTG was comparable to the current in +CaM group; while reduced current at XE991 resembled the current change in the -CaM group.
CaM function in hippocampal neurons
The spontaneous firing and the firing rate evoked by current injection in G271V transfected neurons were higher than in KCNQ2 controls (Figure 4A) due to the inhibition of outward potassium current [12,13]. We next studied the function of CaM in the firing properties of the neurons trasfected with G271V mutation. The result of G271V group was similar to KCNQ2 group. When calmodulin protein was over-expressed in the neuron, we observed a marked reduction in evoked firings of hippocampus neurons in +CaM group, while the knockdown of calmodulin protein (-CaM) markedly increased neuronal excitability (Figure 4A, 4B). From our data, we see the same effect of CaM in both KCNQ2 and G271V groups. Taken together, these results indicate that increased firing rate and hyper-excitability in hippocampal neurons might be attributable to CaM knockdown [14].
Figure 4.
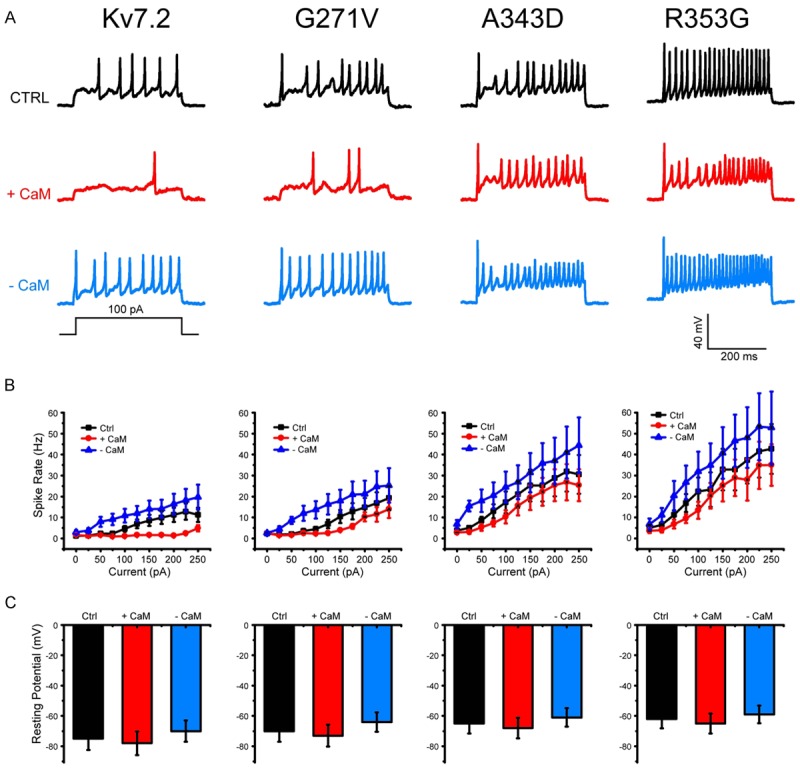
Excitability of Hippocampus Neurons. A. Representative traces of firing response in the neurons transfected with KCNQ2, G271V, A343D, and R353G plasmid, showing firing properties changes under the conditions of CaM over-expression (+CaM) and knockdown (-CaM). B. Mean value of firing frequency in the neurons at an injected current from 0 to 250 pA (n = 15 in each test), showing decreased rate in +CaM group, and increased rate in -CaM group. C. Resting membrane potentials of hippocampal inter-neurons (mean ± S.E.M., n = 15 in each test, P < 0.05).
In addition, we further demonstrated that expression of KCNQ2 A343D and R353G mutations deficient in CaM binding have big effect on action potential firing, resulting in much higher firing rate, this may be due to their inability to exit the ER and express on the axonal surface [15]. The A343D and R353G mutations KCNQ2 have been shown to disrupt KCNQ2 interaction with CaM [16]. We here demonstrated that over-expression of calmodulin (+CaM) the A343D and R353G mutation in KCNQ2 channel significantly decreased firing rate in hippocampal neurons, while knockdown of calmodulin (CaM) can increase action potential firing by regulating interaction between KCNQ2 channels and calmodulin molecules.
Figure 4C shows the effect of calmodulin on the resting potential of hippocampus neurons. In contrast to control, over-expression of CaM (+CaM) hyperpolarized the cell with a slightly negative shift in resting membrane potential, while knockdown of CaM (-CaM) made a positive shift of resting potential, resulting in the depolarization in these cells (Figure 4C). Shifts in resting membrane potential were consistent with the firing rate in the neurons, suggesting interaction between calmodulin and KCNQ channels regulates the outward potassium current of neurons. These results indicate that over-expressing calmodulin is necessary to partially reduce the action potential firing rate caused by mutations in wild-type KCNQ2.
The slight hyper-polarization and the markedly reduced excitability in +CaM group is comparable to the effect of KCNQ activator RTG as described by Grunnet et al. [17], which is also consistent with current increase at RTG in HEK293 cells (Figure 3), the effect that is completely reversed by compound wash-out (data not shown). Calmodulin knockdown (-CaM) has a slight depolarization and increased action potential frequency, which resemble the function KCNQ channel inhibitor XE991 as described by Gribkoff [18].
Discussion
The genes responsible for epilepsy and seizures were identified by positional cloning and are novel voltage-dependent potassium channel genes called KCNQ2 and KCNQ3 [19,20], and mutations in KCNQ channels have been associated to epilepsy and seizures. KCNQ2 channel opens at around -60 mV with quite slow activation kinetics (Figure 3) and display very little inactivation at prolonged de-polarizations. This makes KCNQ channels ideally adapted to control neuronal resting potentials as well as action potentials in neurons (Figure 4).
Calmodulin is one of the auxiliary proteins necessary for activation of KCNQ2 channel [21]. According to the previous study, the putative binding site of calmodulin locates at the IQ motif in helix A in KCNQ2 channel as shown in Figure 5. Here we studied the role of calmodulin in KCNQ2 function by changing calmodulin expression, including over-expressing calmodulin with cDNA, knockdown of calmodulin with shRNA. Furthermore, we genetically interrupted the interaction between KCNQ2 and calmodulin. We found that calmodulin plays an important role in epilepsy associated to KCNQ2 channel function. These calmodulin level manipulations significantly affected both KCNQ2 outward current in HEK293 cells and action potentials in cultured hippocampal neurons of mice. The activation of KCNQ2 channel can be achieved by over-expressing calmodulin protein, tending to hyperpolarize neurons and decrease excitability. Whereas KCNQ2 was inhibited by calmodulin knockdown leading to slight depolarization and increased neuron excitability (Figure 4). We propose that CaM interaction with the KCNQ2 C-terminal tail is important for regulating the KCNQ function in neurons (Figure 5), suggesting that disruption the interaction between KCNQ and CaM would lead to increasing spontaneous firing of action potentials and then enhanced seizure susceptibility (Figure 4). Our findings suggest a novel therapeutic approach to increase CaM expression resembling KCNQ agonist retigabine may result in an efficacious therapy for a variety of neurological disorders.
Figure 5.
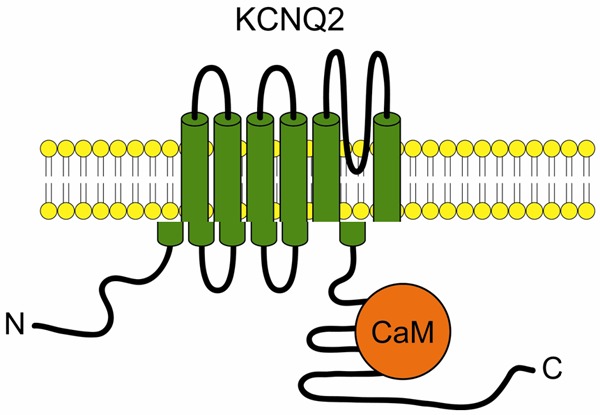
Schematic drawing of interaction between KCNQ2 and Calmodulin.
In conclusion, in this study, we elucidated the mechanisms for activation of KCNQ2 and found that calmodulin protein plays an important role in KCNQ2 functions. According to the present results, we suggest that calmodulin serve as a KCNQ regulator in neuron system. Understanding the regulation of calmodulin will allow the development of novel classes of pharmaceutical compounds in treatment of neurological diseases, including epilepsy and seizure.
Acknowledgements
This research was supported by Nanjing Medical University of Science and Technology Development Fund (Key project # 2014NJMUZD023).
Disclosure of conflict of interest
None.
References
- 1.Kalappa BI, Soh H, Duignan KM, Furuya T, Edwards S, Tzingounis AV, Tzounopoulos T. Potent KCNQ2/3-Specific Channel Activator Suppresses In Vivo Epileptic Activity and Prevents the Development of Tinnitus. J Neurosci. 2015;35:8829–8842. doi: 10.1523/JNEUROSCI.5176-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bierbower SM, Choveau FS, Lechleiter JD, Shapiro MS. Augmentation of M-type (KCNQ) potassium channels as a novel strategy to reduce stroke-induced brain injury. J Neurosci. 2015;35:2101–2111. doi: 10.1523/JNEUROSCI.3805-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, Taglialatela M. arly-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci. 2015;35:3782–3793. doi: 10.1523/JNEUROSCI.4423-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Migliore M, Cilio MR, Taglialatela M. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc Natl Acad Sci U S A. 2013;110:4386–4391. doi: 10.1073/pnas.1216867110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soh H, Niday Z, Tzingounis AV. Cortical KCNQ2/3 channels: insights from knockout mice. Channels (Austin) 2014;8:389–390. doi: 10.4161/19336950.2014.948755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Li Y, Hui Z, Cao M, Shi R, Zhang W, Geng L, Zhou X. Functional analysis of potassium channels in Kv7.2 G271V mutant causing early onset familial epilepsy. Brain Res. 2015;1616:112–122. doi: 10.1016/j.brainres.2015.04.060. [DOI] [PubMed] [Google Scholar]
- 7.Ambrosino P, Alaimo A, Bartollino S, Manocchio L, De Maria M, Mosca I, Gomis-Perez C, Alberdi A, Scambia G, Lesca G, Villarroel A, Taglialatela M, Soldovieri MV. Epilepsy-causing mutations in Kv7.2 C-terminus affect binding and functional modulation by calmodulin. Biochim Biophys Acta. 2015;1852:1856–1866. doi: 10.1016/j.bbadis.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 8.Liu W, Devaux JJ. Calmodulin orchestrates the heteromeric assembly and the trafficking of KCNQ2/3 (Kv7.2/3) channels in neurons. Mol Cell Neurosci. 2014;58:40–52. doi: 10.1016/j.mcn.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Miceli F, Striano P, Soldovieri MV, Fontana A, Nardello R, Robbiano A, Bellini G, Elia M, Zara F, Taglialatela M, Mangano S. A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia. 2015;56:e15–20. doi: 10.1111/epi.12887. [DOI] [PubMed] [Google Scholar]
- 10.Pang ZP, Xu W, Cao P, Sudhof TC. Calmodulin suppresses synaptotagmin-2 transcription in cortical neurons. J Biol Chem. 2010;285:33930–33939. doi: 10.1074/jbc.M110.150151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hani AJ, Mikati HM, Mikati MA. Genetics of Pediatric Epilepsy. Pediatr Clin North Am. 2015;62:703–722. doi: 10.1016/j.pcl.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Etxeberria A, Aivar P, Rodriguez-Alfaro JA, Alaimo A, Villacé P, Gómez-Posada JC, Areso P, Villarroel A. Calmodulin regulates the trafficking of KCNQ2 potassium channels. FASEB J. 2008;22:1135–1143. doi: 10.1096/fj.07-9712com. [DOI] [PubMed] [Google Scholar]
- 13.Yoshioka K, Namiki K, Sudo T, Kasuya Y. p38α controls self-renewal and fate decision of neurosphere-forming cells in adult hippocampus. FEBS Open Bio. 2015;5:437–444. doi: 10.1016/j.fob.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X, Song M, Chen D, Wei L, Yu SP. Potential role of KCNQ/M-channels in regulating neuronal differentiation in mouse hippocampal and embryonic stem cell-derived neuronal cultures. Exp Neurol. 2011;229:471–483. doi: 10.1016/j.expneurol.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou X, Wei J, Song M, Francis K, Yu SP. Novel role of KCNQ2/3 channels in regulating neuronal cell viability. Cell Death Differ. 2011;18:493–505. doi: 10.1038/cdd.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrovic MM, Nowacki J, Olivo V, Tsaneva-Atanasova K, Randall AD, Mellor JR. Inhibition of post-synaptic Kv7/KCNQ/M channels facilitates long-term potentiation in the hippocampus. PLoS One. 2012;7:e30402. doi: 10.1371/journal.pone.0030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavaretta JP, Sherer KR, Lee KY, Kim EH, Issema RS, Chung HJ. Polarized axonal surface expression of neuronal KCNQ potassium channels is regulated by calmodulin interaction with KCNQ2 subunit. PLoS One. 2014;9:e103655. doi: 10.1371/journal.pone.0103655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alaimo A, Gómez-Posada JC, Aivar P, Etxeberría A, Rodriguez-Alfaro JA, Areso P, Villarroel A. Calmodulin activation limits the rate of KCNQ2 K+ channel exit from the endoplasmic reticulum. J Biol Chem. 2009;284:20668–20675. doi: 10.1074/jbc.M109.019539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grunnet M, Strøbæk D, Hougaard C, Christophersen P. Kv7 channels as targets for anti-epileptic and psychiatric drug-development. Eur J Pharmacol. 2014;726:133–137. doi: 10.1016/j.ejphar.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Gribkoff VK. The therapeutic potential of neuronal KCNQ channel modulators. Expert Opin Ther Targets. 2003;7:737–748. doi: 10.1517/14728222.7.6.737. [DOI] [PubMed] [Google Scholar]
- 21.Hani AJ, Mikati HM, Mikati MA. Genetics of Pediatric Epilepsy. Pediatr Clin North Am. 2015;62:703–722. doi: 10.1016/j.pcl.2015.03.013. [DOI] [PubMed] [Google Scholar]