

In the title compound, C16H9ClO4, the dihedral angle between the coumarin ring system [maximum deviation = 0.023 (1) Å] and the benzene ring is 73.95 (8)°.
Keywords: crystal structure, chromane, hydrogen bond, π–π interactions, quantum-chemical calculations, Hirshfeld surface analysis
Abstract
In the title compound, C16H9ClO4 the dihedral angle between the coumarin ring system [maximum deviation = 0.023 (1) Å] and the benzene ring is 73.95 (8)°. In the crystal, π–π interactions link the dimers into a three-dimensional framework. A quantum chemical calculation is in generally good agreement with the observed structure, although the calculated dihedral angle between the ring systems (85.7%) is somewhat larger than the observed value [73.95 (8)°]. Hirshfeld surface analysis has been used to confirm and quantify the supramolecular interactions.
Chemical context
Coumarin and its derivatives are widely recognized for their multiple biological activities, including anticancer (Lacy et al., 2004 ▸; Kostova, 2005 ▸), anti-inflammatory (Todeschini et al., 1998 ▸), antiviral (Borges et al., 2005 ▸), anti-malarial (Agarwal et al., 2005 ▸) and anticoagulant (Maurer et al., 1998 ▸) properties. As part of our studies in this area, we now describe the synthesis and crystal structure of the title compound, (I).
Structural commentary
In compound (I) (Fig. 1 ▸), the coumarin ring system is, as expected, almost planar [maximum deviation = 0.023 (1) Å] and is oriented at an angle of 73.95 (8)° with respect to the benzene ring. An inspection of the bond lengths shows that there is a slight asymmetry of the electronic distribution around the coumarin ring: the C3—C2 [1.335 (2) Å] and C2—C1 [1.456 (2) Å] bond lengths are shorter and longer, respectively, than those excepted for a Car—Car bond. This suggests that the electronic density is preferentially located in the C2—C3 bond at the pyrone ring, as seen in other coumarin derivatives (Gomes et al., 2016 ▸; Ziki et al., 2016 ▸).
Figure 1.

The molecular structure of compound (I), with displacement ellipsoids drawn at the 50% probability level.
Supramolecular features
In the crystal, weak aromatic π–π stacking interactions (Janiak, 2000 ▸) are present [Cg1⋯Cg2(1 − x, −y, 1 − z) = 3.4781 (10) Å and Cg2⋯Cg2(1 − x, 1 − y,1 − z) = 3.5644 (11) Å, where Cg1 is the centroid of the coumarin pyran ring and Cg2 is the centroid of the coumarin benzene ring], thus forming a three-dimensional supramolecular network. A weak C11=O4⋯Cg3(1 − x, − y, −z) (π-ring) interaction between O4 and a symmetry-related benzene ring (C6–C11, centroid Cg3) of is also present (Fig. 2 ▸).
Figure 2.

Partial packing diagram for (I), showing the π–π stacking and C—O⋯π interactions (dashed lines). The yellow dots are ring centroids. H atoms have been omitted for clarity.
Hirshfeld surface analysis
Crystal Explorer3.1 (Wolff et al., 2012 ▸) was used to generate the Hirshfeld surface and two-dimensional fingerprint (FP) plots (Rohl et al., 2008 ▸). The analysis of intramolecular and intermolecular interactions through the mapping of d norm is permitted by the contact distances d i and d e from the Hirshfeld surface to the nearest atom inside and outside, respectively. In compound (I), there are four O atoms and a Cl atom that can potentially act as acceptors for hydrogen bonds, but one of O atoms and the H atom of the chlorobenzoate moiety are involved in the establishment of intramolecular hydrogen bonds. The surface mapped over d norm displays four red spots that correspond to areas of close contact between the surface and the neighbouring environment and is shown in Fig. 3 ▸. The contributions from different contacts were selected by partial analysis of the FP plots (Fig. 4 ▸). C⋯C contacts correspond to intermolecular π–π interactions.
Figure 3.

A view of the Hirshfeld surface mapped over d norm. The contact points (red) are labelled to indicate the atoms participating in the intermolecular interactions.
Figure 4.

Two-dimensional fingerprint plots: (a) overall, and delineated into contributions from different contacts: (b) H⋯H, (c) H⋯O/O⋯H, (d) C⋯C, (e) H⋯C/C⋯H and (f) H⋯Cl/Cl⋯H.
The greatest contribution (26.5%) is from the H⋯O/O⋯H contacts, which appear as the highlighted red spot on the side of the surface (Figs. 3 ▸ and 4c ▸). The red spots in the middle of the surface correspond to C⋯C contacts appearing near d e = d i ≃1.7 and 1.8 Å (Fig. 4d ▸). As expected in organic compounds, the H⋯H contacts are important with a 24.7% contribution to Hirshfeld surface (Fig. 4b ▸). There are also H⋯C/C⋯H and H⋯Cl/Cl⋯H contacts, which make contributions of 14.5 and 12.7%, respectively (Figs. 4e and 4f ▸).
Quantum-chemical calculations
Quantum-chemical calculations were performed and the results compared with the experimental analysis. An ab-initio Hartree–Fock (HF) method was used with the standard 6-31G basis set using the GAUSSIAN03 software package (Frisch et al., 2004 ▸; Dennington et al., 2007 ▸) to obtain the optimized molecular structure. The computational results are in good agreement with the experimental crystallographic data (see Supplementary Tables S1 and S2). The dihedral angle between the coumarin ring and the chlorobenzoate ring for the calculated structure is 85.7°, which is larger than the value of 73.95 (8)° for the observed structure.
Synthesis and crystallization
To a solution of 4-chlorobenzoyl chloride (6.17 × 10 −3 mol ≃ 0.8 ml) in dry tetrahydrofuran (31 ml) was introduced dried triethylamine (3 molar equivalents ≃ 2.6 ml). While stirring strongly, 6.17 × 10 −3 mol (1 g) of chroman-2,3-dione was added in small portions over 30 min. The reaction mixture was then refluxed for 4 h and poured into a separating funnel containing 40 ml of chloroform. The solution was acidified with dilute hydrochlororic acid until the pH was 2–3. The organic layer was extracted, washed with water until neutral, dried over MgSO4 and the solvent removed. The resulting precipitate (crude product) was filtered off with suction, washed with petroleum ether and dissolved in a minimum of dichloromethane by heating under agitation. Hexane was added to this hot mixture until the formation of a new precipitate started, which dissolved in the resulting mixture upon heating. Upon cooling, yellow crystals of the title compound precipitated in a yield of 70%; m.p. 478–482 K.
Refinement details
Crystal data, data collection and structure refinement details are summarized in Table 1 ▸. H atoms were placed in calculated positions (C—H = 0.93 Å) and refined using a riding-model approximation with U iso(H) = 1.2U eq(C).
Table 1. Experimental details.
| Crystal data | |
| Chemical formula | C16H9ClO4 |
| M r | 300.68 |
| Crystal system, space group | Triclinic, P
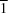
|
| Temperature (K) | 293 |
| a, b, c (Å) | 6.7866 (4), 7.1789 (3), 14.0981 (5) |
| α, β, γ (°) | 94.098 (3), 93.461 (4), 106.154 (4) |
| V (Å3) | 655.75 (5) |
| Z | 2 |
| Radiation type | Cu Kα |
| μ (mm−1) | 2.72 |
| Crystal size (mm) | 0.12 × 0.12 × 0.08 |
| Data collection | |
| Diffractometer | Agilent SuperNova Dual Source diffractometer with an Atlas detector |
| Absorption correction | Multi-scan (CrysAlis PRO; Agilent, 2014 ▸) |
| T min, T max | 0.737, 0.812 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 7634, 2409, 2109 |
| R int | 0.022 |
| (sin θ/λ)max (Å−1) | 0.606 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.039, 0.106, 1.05 |
| No. of reflections | 2409 |
| No. of parameters | 190 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.26, −0.49 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989016019538/hb7629sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989016019538/hb7629Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989016019538/hb7629Isup3.cml
CCDC reference: 1521043
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors thank the Spectropole Service of the faculty of Sciences (Aix-Marseille, France) for the use of the diffractometer.
supplementary crystallographic information
Crystal data
| C16H9ClO4 | Z = 2 |
| Mr = 300.68 | F(000) = 308 |
| Triclinic, P1 | Dx = 1.523 Mg m−3 |
| Hall symbol: -P 1 | Melting point: 478 K |
| a = 6.7866 (4) Å | Cu Kα radiation, λ = 1.54184 Å |
| b = 7.1789 (3) Å | Cell parameters from 3886 reflections |
| c = 14.0981 (5) Å | θ = 6.3–69.1° |
| α = 94.098 (3)° | µ = 2.72 mm−1 |
| β = 93.461 (4)° | T = 293 K |
| γ = 106.154 (4)° | Prism, colourless |
| V = 655.75 (5) Å3 | 0.12 × 0.12 × 0.08 mm |
Data collection
| Agilent SuperNova Dual Source diffractometer with an Atlas detector | 2409 independent reflections |
| Radiation source: fine-focus sealed tube | 2109 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.022 |
| Detector resolution: 5.3048 pixels mm-1 | θmax = 69.1°, θmin = 6.3° |
| ω scan | h = −8→7 |
| Absorption correction: multi-scan (CrysAlis PRO; Agilent, 2014) | k = −8→8 |
| Tmin = 0.737, Tmax = 0.812 | l = −17→16 |
| 7634 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.039 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.106 | H-atom parameters constrained |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0475P)2 + 0.1948P] where P = (Fo2 + 2Fc2)/3 |
| 2409 reflections | (Δ/σ)max < 0.001 |
| 190 parameters | Δρmax = 0.26 e Å−3 |
| 0 restraints | Δρmin = −0.49 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.1899 (3) | 0.0796 (2) | 0.35706 (12) | 0.0446 (4) | |
| C2 | 0.3804 (3) | 0.0653 (2) | 0.31891 (11) | 0.0422 (4) | |
| C3 | 0.5634 (3) | 0.1322 (2) | 0.36885 (11) | 0.0414 (4) | |
| H3 | 0.6815 | 0.1187 | 0.3424 | 0.050* | |
| C4 | 0.5771 (2) | 0.2250 (2) | 0.46354 (11) | 0.0384 (3) | |
| C5 | 0.7616 (3) | 0.3026 (2) | 0.52035 (13) | 0.0475 (4) | |
| H5 | 0.8848 | 0.2943 | 0.4975 | 0.057* | |
| C6 | 0.7618 (3) | 0.3918 (3) | 0.61044 (14) | 0.0549 (5) | |
| H6 | 0.8856 | 0.4451 | 0.6475 | 0.066* | |
| C7 | 0.5794 (3) | 0.4022 (3) | 0.64594 (12) | 0.0534 (5) | |
| H7 | 0.5813 | 0.4628 | 0.7067 | 0.064* | |
| C8 | 0.3938 (3) | 0.3229 (2) | 0.59161 (12) | 0.0474 (4) | |
| H8 | 0.2705 | 0.3270 | 0.6158 | 0.057* | |
| C9 | 0.3956 (2) | 0.2379 (2) | 0.50103 (11) | 0.0390 (3) | |
| C10 | 0.3047 (3) | 0.0312 (3) | 0.15135 (12) | 0.0459 (4) | |
| C11 | 0.2730 (2) | −0.1155 (3) | 0.06844 (12) | 0.0458 (4) | |
| C16 | 0.2973 (3) | −0.2994 (3) | 0.07788 (13) | 0.0525 (4) | |
| H16 | 0.3335 | −0.3326 | 0.1377 | 0.063* | |
| C15 | 0.2683 (3) | −0.4333 (3) | −0.00072 (14) | 0.0599 (5) | |
| H15 | 0.2832 | −0.5568 | 0.0058 | 0.072* | |
| C14 | 0.2170 (3) | −0.3809 (4) | −0.08894 (14) | 0.0626 (6) | |
| C13 | 0.1904 (3) | −0.1999 (4) | −0.10068 (13) | 0.0649 (6) | |
| H13 | 0.1534 | −0.1680 | −0.1607 | 0.078* | |
| C12 | 0.2200 (3) | −0.0667 (3) | −0.02135 (13) | 0.0555 (5) | |
| H12 | 0.2042 | 0.0564 | −0.0282 | 0.067* | |
| Cl1 | 0.18104 (10) | −0.55203 (13) | −0.18674 (4) | 0.0951 (3) | |
| O1 | 0.20802 (17) | 0.16486 (17) | 0.44814 (8) | 0.0444 (3) | |
| O2 | 0.0204 (2) | 0.0211 (2) | 0.31579 (10) | 0.0637 (4) | |
| O3 | 0.3603 (2) | −0.04346 (18) | 0.23221 (8) | 0.0516 (3) | |
| O4 | 0.2900 (2) | 0.1930 (2) | 0.15125 (10) | 0.0592 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0428 (9) | 0.0444 (8) | 0.0465 (9) | 0.0127 (7) | 0.0018 (7) | 0.0042 (7) |
| C2 | 0.0515 (10) | 0.0402 (8) | 0.0374 (8) | 0.0177 (7) | 0.0038 (7) | 0.0026 (6) |
| C3 | 0.0424 (9) | 0.0431 (8) | 0.0433 (8) | 0.0176 (7) | 0.0101 (7) | 0.0082 (7) |
| C4 | 0.0411 (8) | 0.0336 (7) | 0.0420 (8) | 0.0117 (6) | 0.0044 (6) | 0.0066 (6) |
| C5 | 0.0426 (9) | 0.0461 (9) | 0.0549 (10) | 0.0139 (7) | 0.0001 (7) | 0.0097 (7) |
| C6 | 0.0597 (11) | 0.0467 (9) | 0.0538 (10) | 0.0117 (8) | −0.0144 (8) | 0.0049 (8) |
| C7 | 0.0801 (13) | 0.0438 (9) | 0.0386 (8) | 0.0230 (9) | −0.0015 (8) | 0.0018 (7) |
| C8 | 0.0601 (11) | 0.0458 (9) | 0.0420 (8) | 0.0230 (8) | 0.0084 (7) | 0.0057 (7) |
| C9 | 0.0417 (8) | 0.0346 (7) | 0.0428 (8) | 0.0134 (6) | 0.0045 (6) | 0.0069 (6) |
| C10 | 0.0369 (9) | 0.0587 (10) | 0.0442 (9) | 0.0156 (7) | 0.0051 (7) | 0.0082 (7) |
| C11 | 0.0341 (8) | 0.0648 (11) | 0.0388 (8) | 0.0140 (7) | 0.0044 (6) | 0.0048 (7) |
| C16 | 0.0515 (10) | 0.0651 (11) | 0.0407 (9) | 0.0181 (8) | 0.0000 (7) | 0.0006 (8) |
| C15 | 0.0541 (11) | 0.0709 (12) | 0.0519 (10) | 0.0169 (9) | 0.0015 (8) | −0.0073 (9) |
| C14 | 0.0401 (10) | 0.0978 (16) | 0.0434 (10) | 0.0137 (10) | 0.0043 (7) | −0.0134 (10) |
| C13 | 0.0437 (10) | 0.1146 (19) | 0.0370 (9) | 0.0235 (11) | 0.0029 (7) | 0.0066 (10) |
| C12 | 0.0432 (10) | 0.0826 (13) | 0.0446 (9) | 0.0217 (9) | 0.0061 (7) | 0.0141 (9) |
| Cl1 | 0.0754 (4) | 0.1412 (6) | 0.0550 (3) | 0.0214 (4) | 0.0009 (3) | −0.0395 (4) |
| O1 | 0.0383 (6) | 0.0504 (6) | 0.0464 (6) | 0.0160 (5) | 0.0069 (5) | 0.0012 (5) |
| O2 | 0.0439 (7) | 0.0792 (9) | 0.0619 (8) | 0.0120 (6) | −0.0056 (6) | −0.0029 (7) |
| O3 | 0.0686 (8) | 0.0544 (7) | 0.0369 (6) | 0.0280 (6) | 0.0000 (5) | −0.0007 (5) |
| O4 | 0.0689 (9) | 0.0586 (8) | 0.0561 (8) | 0.0273 (7) | 0.0046 (6) | 0.0092 (6) |
Geometric parameters (Å, º)
| C1—O2 | 1.206 (2) | C8—H8 | 0.9300 |
| C1—O1 | 1.366 (2) | C9—O1 | 1.382 (2) |
| C1—C2 | 1.456 (2) | C10—O4 | 1.193 (2) |
| C2—C3 | 1.335 (2) | C10—O3 | 1.370 (2) |
| C2—O3 | 1.3809 (19) | C10—C11 | 1.479 (2) |
| C3—C4 | 1.435 (2) | C11—C16 | 1.390 (3) |
| C3—H3 | 0.9300 | C11—C12 | 1.390 (2) |
| C4—C9 | 1.393 (2) | C16—C15 | 1.381 (3) |
| C4—C5 | 1.395 (2) | C16—H16 | 0.9300 |
| C5—C6 | 1.380 (3) | C15—C14 | 1.377 (3) |
| C5—H5 | 0.9300 | C15—H15 | 0.9300 |
| C6—C7 | 1.382 (3) | C14—C13 | 1.381 (4) |
| C6—H6 | 0.9300 | C14—Cl1 | 1.738 (2) |
| C7—C8 | 1.385 (3) | C13—C12 | 1.385 (3) |
| C7—H7 | 0.9300 | C13—H13 | 0.9300 |
| C8—C9 | 1.378 (2) | C12—H12 | 0.9300 |
| O2—C1—O1 | 118.19 (16) | C8—C9—C4 | 122.08 (16) |
| O2—C1—C2 | 125.73 (17) | O1—C9—C4 | 120.91 (14) |
| O1—C1—C2 | 116.07 (14) | O4—C10—O3 | 122.83 (17) |
| C3—C2—O3 | 120.59 (15) | O4—C10—C11 | 127.29 (16) |
| C3—C2—C1 | 122.67 (15) | O3—C10—C11 | 109.86 (15) |
| O3—C2—C1 | 116.26 (15) | C16—C11—C12 | 119.31 (18) |
| C2—C3—C4 | 119.71 (15) | C16—C11—C10 | 121.73 (16) |
| C2—C3—H3 | 120.1 | C12—C11—C10 | 118.96 (18) |
| C4—C3—H3 | 120.1 | C15—C16—C11 | 120.64 (18) |
| C9—C4—C5 | 118.11 (15) | C15—C16—H16 | 119.7 |
| C9—C4—C3 | 118.07 (15) | C11—C16—H16 | 119.7 |
| C5—C4—C3 | 123.81 (15) | C14—C15—C16 | 118.9 (2) |
| C6—C5—C4 | 120.22 (17) | C14—C15—H15 | 120.5 |
| C6—C5—H5 | 119.9 | C16—C15—H15 | 120.5 |
| C4—C5—H5 | 119.9 | C15—C14—C13 | 121.89 (19) |
| C5—C6—C7 | 120.47 (17) | C15—C14—Cl1 | 118.0 (2) |
| C5—C6—H6 | 119.8 | C13—C14—Cl1 | 120.05 (16) |
| C7—C6—H6 | 119.8 | C14—C13—C12 | 118.72 (18) |
| C6—C7—C8 | 120.40 (17) | C14—C13—H13 | 120.6 |
| C6—C7—H7 | 119.8 | C12—C13—H13 | 120.6 |
| C8—C7—H7 | 119.8 | C13—C12—C11 | 120.5 (2) |
| C9—C8—C7 | 118.70 (17) | C13—C12—H12 | 119.7 |
| C9—C8—H8 | 120.6 | C11—C12—H12 | 119.7 |
| C7—C8—H8 | 120.6 | C1—O1—C9 | 122.54 (13) |
| C8—C9—O1 | 117.01 (15) | C10—O3—C2 | 118.78 (14) |
| O2—C1—C2—C3 | −179.51 (17) | O4—C10—C11—C12 | 0.0 (3) |
| O1—C1—C2—C3 | −0.5 (2) | O3—C10—C11—C12 | −178.60 (15) |
| O2—C1—C2—O3 | −7.4 (3) | C12—C11—C16—C15 | −0.3 (3) |
| O1—C1—C2—O3 | 171.59 (13) | C10—C11—C16—C15 | −179.53 (16) |
| O3—C2—C3—C4 | −172.69 (13) | C11—C16—C15—C14 | 0.7 (3) |
| C1—C2—C3—C4 | −0.9 (2) | C16—C15—C14—C13 | −1.1 (3) |
| C2—C3—C4—C9 | 1.5 (2) | C16—C15—C14—Cl1 | −179.94 (15) |
| C2—C3—C4—C5 | −178.72 (15) | C15—C14—C13—C12 | 1.1 (3) |
| C9—C4—C5—C6 | −1.0 (2) | Cl1—C14—C13—C12 | 179.94 (14) |
| C3—C4—C5—C6 | 179.23 (15) | C14—C13—C12—C11 | −0.7 (3) |
| C4—C5—C6—C7 | 1.1 (3) | C16—C11—C12—C13 | 0.3 (3) |
| C5—C6—C7—C8 | 0.2 (3) | C10—C11—C12—C13 | 179.57 (16) |
| C6—C7—C8—C9 | −1.5 (3) | O2—C1—O1—C9 | −179.48 (15) |
| C7—C8—C9—O1 | −178.39 (14) | C2—C1—O1—C9 | 1.4 (2) |
| C7—C8—C9—C4 | 1.6 (2) | C8—C9—O1—C1 | 179.09 (14) |
| C5—C4—C9—C8 | −0.4 (2) | C4—C9—O1—C1 | −0.9 (2) |
| C3—C4—C9—C8 | 179.42 (14) | O4—C10—O3—C2 | 6.6 (3) |
| C5—C4—C9—O1 | 179.60 (13) | C11—C10—O3—C2 | −174.68 (14) |
| C3—C4—C9—O1 | −0.6 (2) | C3—C2—O3—C10 | −115.15 (18) |
| O4—C10—C11—C16 | 179.25 (18) | C1—C2—O3—C10 | 72.60 (19) |
| O3—C10—C11—C16 | 0.6 (2) |
References
- Agarwal, A., Srivastava, K., Puri, S. K. & Chauhan, P. M. S. (2005). Bioorg. Med. Chem. 13, 4645–4650. [DOI] [PubMed]
- Agilent. (2014). CrysAlis PRO. Agilent Technologies Ltd, Yarnton, England.
- Borges, F., Roleira, F., Milhazes, N., Santana, L. & Uriarte, E. (2005). Curr. Med. Chem. 12, 887–916. [DOI] [PubMed]
- Dennington, R., Keith, T. & Millam, J. (2007). GAUSSVIEW4.1. Semichem Inc., Shawnee Mission, KS, USA.
- Frisch, M. J., et al. (2004). GAUSSIAN03. Gaussian Inc., Wallingford, CT, USA.
- Gomes, L. R., Low, J. N., Fonseca, A., Matos, M. J. & Borges, F. (2016). Acta Cryst. E72, 926–932. [DOI] [PMC free article] [PubMed]
- Janiak, C. (2000). J. Chem. Soc. Dalton Trans. pp. 3885–3889.
- Kostova, I. (2005). Curr. Med. Chem. Anti-Cancer Agents, 5, 29–46. [DOI] [PubMed]
- Lacy, A. & O’Kennedy, R. (2004). Curr. Pharm. Des. 10, 3797–3811. [DOI] [PubMed]
- Maurer, H. H. & Arlt, J. W. (1998). J. Chromatogr. B Biomed. Sci. Appl. 714, 181–195. [DOI] [PubMed]
- Rohl, A. L., Moret, M., Kaminsky, W., Claborn, K., McKinnon, J. J. & Kahr, B. (2008). Cryst. Growth Des. 8, 4517–4525.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Todeschini, A. R., de Miranda, A. L. P., da Silva, K. C. M., Parrini, S. C. & Barreiro, E. J. (1998). Eur. J. Med. Chem. 33, 189–199.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Wolff, S. K., Grimwood, D. J., McKinnon, J. J., Turner, M. J., Jayatilaka, D. & Spackman, M. A. (2012). Crystal Explorer. The University of Western Australia.
- Ziki, E., Yoda, J., Djandé, A., Saba, A. & Kakou-Yao, R. (2016). Acta Cryst. E72, 1562–1564. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989016019538/hb7629sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989016019538/hb7629Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989016019538/hb7629Isup3.cml
CCDC reference: 1521043
Additional supporting information: crystallographic information; 3D view; checkCIF report