

Abstract
Purpose
This study utilizes FLT PET/CT imaging to characterize changes in tumor cell proliferation and vasculature during intermittent treatment with VEGR-TKI axitinib.
Methods
Patients with metastatic solid malignancies underwent 3-week treatment cycles with axitinib (7 and 5 mg BID for safety and pharmacodynamic cohorts, respectively). Cycles consisted of 2 weeks of treatment (dosing period) followed by a 1-week treatment break (washout period). Patients in the pharmacodynamic cohort had up to six FLT PET/CT scans (three scans in each cycle 1 and cycle 3) and had plasma VEGF concentrations measured at imaging timepoints. Changes in tumor SUVs and VEGF within and across drug cycles were investigated.
Results
Eight patients enrolled in the safety cohort where it was determined 7 mg axitinib was not tolerable due to severe adverse events, including three patients who experienced significant hypertension and thrombovascular effects. Sixteen patients enrolled in the pharmacodynamic cohort demonstrated significant decreases in SUVs and increases in VEGF during dosing periods. This was followed by significant increases in SUVs and decreases in VEGF during drug washout periods. No significant differences in SUVs or VEGF were found when comparing cycle 1 with cycle 3. A mixed effects model demonstrated significant negative correlation between SUV and VEGF.
Conclusions
Response to axitinib included diminished FLT uptake during dosing periods followed by increased FLT uptake during drug washout periods. These changes were not different when comparing treatment cycle 1 versus cycle 3, suggesting that the pharmacodynamic effect of intermittent axitinib is similar across multiple drug cycles.
Keywords: Axitinib, Tyrosine kinase inhibitor, VEGF, FLT PET, Pharmacodynamic, Anti-angiogenic therapym, Tumor cell proliferation
Introduction
Angiogenesis is utilized by tumors to provide nutrients necessary for continued growth. It has been hypothesized that inhibiting angiogenesis could stop tumor growth [1]. This hypothesis led to the development of anti-angiogenic treatments; some of which have been successful in treating specific cancers [2]. Many of these agents target the vascular endothelial growth factor and its receptors (VEGFR) due to their well-documented role in angiogenesis [3].
Axitinib is a tyrosine kinase inhibitor (TKI) of VEG-FRs-1, 2, and 3. It is a second generation anti-angiogenic agent that has shown improved potency over previous agents [4] and has been approved for the treatment of metastatic renal cell carcinoma. Despite the promise of VEGFR-TKIs agents such as axitinib, many patients acquire resistance to treatment. This has motivated a number of studies investigating drug pharmacokinetics and pharmacodynamics in an effort to develop improved treatment strategies. For example, pharmacokinetic modeling showed that increased exposure to axitinib in renal cell carcinoma patients tolerating therapy was associated with improved response rate [5]. It has also been hypothesized that combining VEGFR-TKI treatments with other types of treatments might improve efficacy [6]. Unfortunately, a number of studies combining VEGFR-TKI agents and cytotoxic chemotherapy have yielded no significant improvement over monotherapy [7-9]. Based on results from our previous studies [10, 11], we hypothesize that decreases in tumor cell proliferation and vasculature during VEGFR-TKI treatment antagonizes effects of concurrent treatment with cell-cycle-specific chemotherapy. Furthermore, cessation of VEGFR-TKIs initiated an acute treatment withdrawal flare characterized by increases in tumor cell proliferation and vasculature during the drug washout period [10, 11]. This withdrawal flare offers a synergistic target for sequential chemotherapy applied during the washout phase of intermittent VEGFR-TKI treatment. However, further characterization of VEGFR-TKI pharmacodynamic effects during intermittent regimens is necessary to determine the feasibility of sequential treatment strategies. This includes understanding how different doses of VEGFR-TKIs affect the withdrawal flare as well as determining whether the withdrawal flare occurs in later cycles of intermittent treatment.
Medical imaging offers a convenient noninvasive method for measuring drug pharmacodynamic effects. 3’-Deoxy-3’-[18F]fluorothymidine (FLT) positron emission tomography (PET) is an imaging modality utilized for quantifying tumor cell proliferation and vasculature characteristics making it well suited for monitoring anti-angiogenic treatments [12, 13]. Specific to the context of VEGFR-TKIs, it has shown utility in a handful of clinical trials for assessing drug pharmacodynamic effects [6, 10, 11, 14].
This study evaluates the optimal timing of novel pharmacodynamic assessment of VEGFR-TKI treatment using FLT PET/CT imaging. The primary goal is to characterize the acute treatment withdrawal flare during drug washout periods. This trial expands upon aforementioned research by: utilizing a higher dose of axitinib (7 mg BID rather than the standard FDA approved dose of 5 mg BID) and characterizing the withdrawal flare in both the first and third cycle of intermittent treatment. The ultimate clinical goal is to exploit the withdrawal flare to improve therapeutic index of cell–cycle-specific chemotherapy for applications in the neoadjuvant or metastatic setting.
Patients and methods
Patient selection
Patients with histologically or cytologically confirmed solid malignancy (excluding lymphoma) that was metastatic or unresectable and for which no standard therapy existed were enrolled on this study. Other key inclusion criteria included the following: normal organ and bone marrow function, measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST 1.1) guidelines [15], appropriate target tumors for FLT PET/CT assessment (minimum 1.5 cm, located in a region of body with low motion artifact, reasonable ability to delineate tumor boundaries on CT and PET scans, e.g., non-hepatic tumors due to high background FLT uptake in liver). Patients with prior anti-VEGF treatment were excluded. Other exclusion criteria include concomitant coumarin-derivative anticoagulation, history of brain metastases, and any concomitant use of CYP3A4 or CYP1A2 inducers. All patients signed informed consent documents approved by the Institutional Review Board at the University of Wisconsin. Additional approval by the Radioactive Drug Research Committee at the University of Wisconsin was obtained given use of an experimental tracer. This study was conducted in accordance with the Declaration of Helsinki.
Drug administration and study design
There were two cohorts in this study: (1) an initial safety cohort of patients to establish the safety and toxicity of axitinib at 7 mg BID and (2) a pharmacodynamic cohort that would undergo FLT PET/CT imaging during treatment. Since patients in the safety cohort experienced dose-limiting toxicities, the dose was reduced to the standard 5 mg BID for the pharmacodynamic cohort. All patients underwent 3-week treatment cycles with axitinib taken orally, twice daily with food on days 1–14, followed by a 1-week drug break (days 15–21).
Patients in the pharmacodynamic cohort underwent up to 6 FLT PET/CT scans at three timepoints during both cycles 1 and 3 (Fig. 1). No imaging was obtained during cycle 2. The rationale for FLT PET/CT imaging during cycle 3 was to determine whether the withdrawal flare was present in later cycles of intermittent treatment while still maintaining an evaluable number of patients on the study. The imaging timepoints for cycle 1 and cycle 3 included: (1) baseline (−3 to 0 days prior to treatment), (2) peak drug exposure (12–14 days into the dosing period), and (3) near end of drug washout period (5–7 days into the treatment break). In order to be evaluable for FLT PET/CT imaging patients needed to complete >90% of scheduled axitinib doses prior to completion of the third FLT PET/CT scan.
Fig. 1.
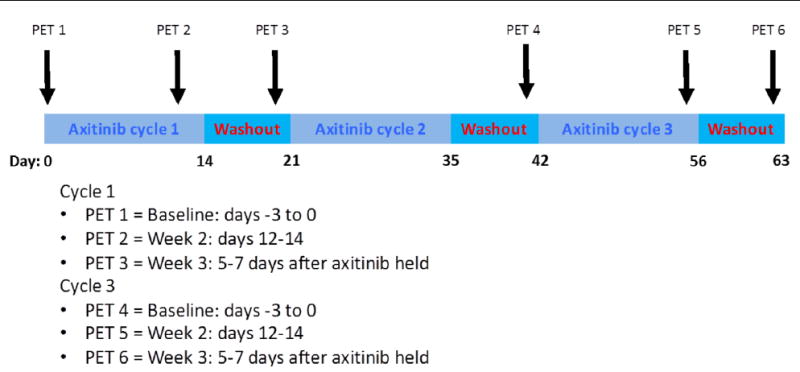
Treatment schedule and FLT PET/CT imaging timepoints
All patients were assessed for response every three cycles of therapy using RECIST 1.1 guidelines [15]. Patients were also assessed for progression by a treating physician, which was based on a number of factors available to the physician including any adverse side effects of treatment.
FLT PET/CT scans and analysis
Patients were injected with up to 300 MBq of FLT. FLT PET/CT scans were initiated 60 min post-injection (7 bed positions, 5 min per bed position) on a Discovery VCT PET/CT scanner (GE, Waukesha, WI). An ordered-subsets expectation maximization (OSEM) algorithm was used for three-dimensional image reconstruction with CT data used for attenuation correction. Parameters for the reconstruction included: 256 × 256 matrix, 35 subsets, 2 iterations, and a 3-mm Gaussian post-filter. The resulting image was a 256 × 256 × 263 matrix with voxel dimensions of 2.73, 2.73, and 3.27 mm, respectively.
Each tumor (up to five per patient) was identified by a nuclear medicine physician using both PET and CT images. Manual segmentation of tumors was performed by the same individual for all scans using Amira software (Visage Imaging Inc.). Imaging metrics analyzed included standardized uptake values (SUVs), which were corrected for injected activity and patient weight (resulting in SUVs with units of g/mL). SUVs were calculated for each voxel and summarized for each patient giving global SUVs (including SUVmean, SUVmax, and SUVtotal). SUVmean was defined as the average SUV of all tumors within a patient. SUVmax was defined as the SUV in voxel with highest SUV of all tumors within a patient. SUVtotal was defined as the product of the sum of SUVs from all tumors within a patient and the voxel volume. In addition to extracting SUVs for each time point, relative percent changes in SUVs were calculated for the treatment and washout periods within cycles 1 and 3. Equation 1 shows example calculation for the relative change in SUV during the treatment period in cycle 1.
| (1) |
VEGF and axitinib plasma concentrations
Plasma samples for analysis of axitinib and vascular endothelial growth factor ligand (VEGF) concentrations were collected on the same days as PET/CT scans: −3 to 0 days prior to dosing, after 12–14 days of dosing (week 2) and after 5–7 days of drug washout (week 3). VEGF concentrations were measured by a commercially available 96-well plate quantitative sandwich immunoassay (Quantikine® human VEGF, R&D Systems, Minneapolis, MN) according to manufacturer’s instructions. Axitinib plasma concentrations were measured by a validated LC/MS/MS as previously described [11].
Statistical methods
Relative changes in SUVs and plasma markers were summarized in terms of medians and ranges. Since the distributions of the relative changes in SUVs and plasma markers were highly skewed, the nonparametric Wilcoxon signed-rank test was utilized to evaluate changes across time points. A linear mixed effects model with subject-specific random effects was used to examine the association between SUVs and VEGF measurements. An autoregressive correlation structure was utilized to account for correlation of measurements arising from the same patient. Both the SUV and VEGF measurements were log-transformed before inclusion in the linear mixed effects model. All reported P-values are two-sided and P < 0.05 was used to define statistical significance. Data analysis was conducted using R software version 3.2.0.
Results
Patient characteristics
A total of twenty-four patients were enrolled on this study (Table 1). Eight patients were enrolled in the safety cohort. Sixteen patients were enrolled in the pharmacodynamic cohort. Two of the patients within the pharmacodynamic completed only the baseline FLT PET/CT scan and were not included in the imaging analysis. The remaining fourteen patients in the pharmacodynamic cohort completed three or more of the scheduled FLT PET/CT scans.
Table 1.
Patient demographics
| Safety N = 8 | Pharmacodynamic N = 16 | Overall N = 24 | |
|---|---|---|---|
| Age (years) | |||
| Median (range) | 57 (28–72) | 70 (38–82) | 65 (28–82) |
| Number of patients | |||
| Male | 5 (62.5%) | 11 (69%) | 16 (67%) |
| Female | 3 (37.5%) | 5 (31%) | 8 (33%) |
| Primary disease site | |||
| Prostate | 1 (12.5%) | 5 (31%) | 6 (25%) |
| Colorectal | 1 (12.5%) | 3 (19%) | 4 (17%) |
| Urothelial | 0 (0%) | 2 (13%) | 2 (8%) |
| Ovarian | 1 (12.5%) | 1 (6%) | 2 (8%) |
| Lung | 1 (12.5%) | 1 (6%) | 2 (8%) |
| Other | 4 (50%) | 4 (25%) | 8 (34%) |
| Number of organs with metastases | |||
| 1 | 2 (25%) | 5 (31%) | 7 (29%) |
| 2 | 3 (37.5%) | 5 (31%) | 8 (34%) |
| 3+ | 3 (37.5%) | 6 (38%) | 9 (37%) |
Twelve of twenty-four patients came off study due to radiographic progression as defined by RECIST 1.1 criteria. Four patients withdrew consent after beginning treatment. The remaining eight patients came off study due to either a drug-related adverse event or physician discretion. For patients in the safety cohort, the median cycles of therapy received was 3 (range 1–6 cycles). For patients in the pharmacodynamic cohorts, the median cycles of therapy received was 4 (range 2–18).
Drug-related adverse events
All eight patients (100%) in the safety cohort had an adverse event that was at least possibly related to the study drug. Three patients in the safety cohort had a serious adverse event that was at least possibly related to the study drug. These three patients developed significant hypertension and thrombovascular effects such that axitinib treatment was held for several days, and thus, these patients did receive >90% of the agent, which would have been protocol dosing requirement for adequate assessment of their disease by FLT PET/CT imaging had they been in the pharmacodynamic cohort. Therefore, prior to enrollment of the pharmacodynamic cohort, the axitinib dose was redefined as the standard 5 mg BID in order to assure that an adequate number of patients were evaluable for FLT PET/ CT analysis.
Fourteen out of sixteen patients (88%) in pharmacodynamic cohort had an adverse event that was at least possibly related to the study drug. Three patients in the pharmacodynamic cohort had a serious adverse event that was at least possibly related to the study drug.
Two patients died while enrolled on this clinical trial. One Grade 5 thromboembolic event resulting in death was seen in a patient with metastatic non-small lung cancer, who was on axitinib for 47 days. The patient was hospitalized for progressive dysphagia due to an enlarging right paraesophageal mass causing extrinsic compression of the esophagus. The patient was also noted to have a pulmonary emboli and anticoagulated with enoxaparin. They did not receive any additional axitinib after the event. One patient died of unknown causes while on axitinib therapy. This patient had metastatic squamous cell carcinoma of unknown primary origin that had a good response to therapy with a decrease of 40–50% reduction in size overall; however, the partial response was not confirmed due to death. The patient was found dead at home while still lying in bed and it was unclear the cause of death.
FLT PET/CT imaging
Table 2 summarizes median percent changes in SUVmean, SUVmax, and SUVtotal during treatment and washout periods for pharmacodynamic patients. SUVmean decreased significantly during dosing period in both cycle 1 (median −15%, P = 0.02) and cycle 3 (median −21%, P = 0.008). No statistical significant differences were found when comparing the percent change in SUVmean during dosing period in cycle 1 versus cycle 3 (P = 0.8). SUVmean significantly increased during washout period in both cycle 1 (median +20%, P < 0.001) and cycle 3 (median +27%, P = 0.03). No significant difference was found when comparing the percent change in SUVmean during washout period in cycle 1 versus cycle 3 (P = 0.7). Similar trends occurred for SUVmax and SUVtotal.
Table 2.
Median percent changes in tumor SUV during dosing and washout periods (range shown in parenthesis)
| Median % change during drug dosing period
|
Median % change during drug washout period
|
|||
|---|---|---|---|---|
| Cycle 1 (n = 14) | Cycle 3 (n = 8) | Cycle 1 (n = 14) | Cycle 3 (n = 6) | |
| SUVmean | −15 (−48 to 16) | −21 (−42 to −6) | 20 (1 to 58) | 27 (16 to 33) |
| SUVmax | −23 (−68 to 23) | −26 (−43 to −7) | 28 (−29 to 159) | 33 (14 to 69) |
| SUVtotal | −49 (−91 to 23) | −23 (−91 to −4) | 50 (−15 to 1492) | 62 (−14 to 1100) |
Figure 2 highlights a tumor that had decreases in SUVs during dosing periods and increases in SUVs during washout periods in both cycle 1 and cycle 3. Although the tumor in Fig. 2 shows increased SUV in cycle 3 relative to cycle 1, as a whole, the population of patients did not exhibit significantly different SUVs when comparing corresponding time points in cycle 1 versus cycle 3 (e.g., for SUVmean: baseline cycle 1 vs. baseline cycle 3, P = 0.9; peak drug cycle 1 vs. peak drug cycle 3, P = 1.0; washout cycle 1 vs. washout cycle 3, P = 0.5).
Fig. 2.
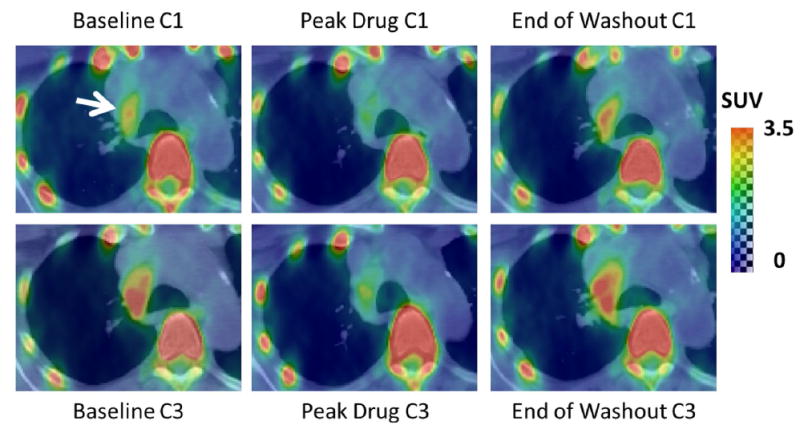
Axial PET/CT slice with increased SUV in right paratracheal lymph node (white arrow). Note reduced SUV at peak drug in both cycle 1 and cycle 3 which subsides by the end of washout. C1 cycle 1, C3 cycle 3
Plasma VEGF concentration
The last row of Table 3 summarizes the relative percent changes in VEGF for patients in the pharmacodynamic and safety cohorts. For the pharmacodynamic cohort, VEGF concentrations increased during dosing period in both cycle 1 (median +140%, P = 0.002) and cycle 3 (median +193%, P = 0.03). There was no difference between the percent changes in VEGF during dosing period for cycle 1 versus cycle 3 (P = 0.1). VEGF concentrations decreased during washout in cycle 1 (median −63%, P < 0.001) and cycle 3 (median −67%, P = 0.25). There was no significant difference between the percent changes in VEGF during washout for cycle 1 versus cycle 3 (P = 1.0). Percent changes in VEGF for the safety cohort were not significantly different from the pharmacodynamic cohort for the dosing or washout periods in cycle 1.
Table 3.
Median percent changes in plasma VEGF during dosing and washout periods (range shown in parenthesis)
| Median % change during drug dosing period
|
Median % change during drug washout period
|
|||
|---|---|---|---|---|
| Cycle 1 | Cycle 3 | Cycle 1 | Cycle 3 | |
| PD cohort (5 mg BID) | 140 (−40 to 3787) | 193 (15–1363) | −63 (−95 to 0) | −67 (−82 to −39) |
| Safety cohort (7 mg BID) | 99 (27 to 442) | – | −27 (−52 to −4) | – |
Comparison between cycle 1 and cycle 3 could not be completed for the safety cohort due to patients coming off study prior to completing cycle 3
PD pharmacodynamic
A linear mixed effects model with VEGF as a predictor variable, demonstrated significant negative correlation (P < 0.001) between log-transformed VEGF and SUVtotal (slope = −0.42, 95% CI = −0.57 to −0.27). The fitted model and measured data are shown in Fig. 3. The model indicates that a doubling in patient’s VEGF concentration would correspond to approximately 25% decrease in SUVtotal.
Fig. 3.
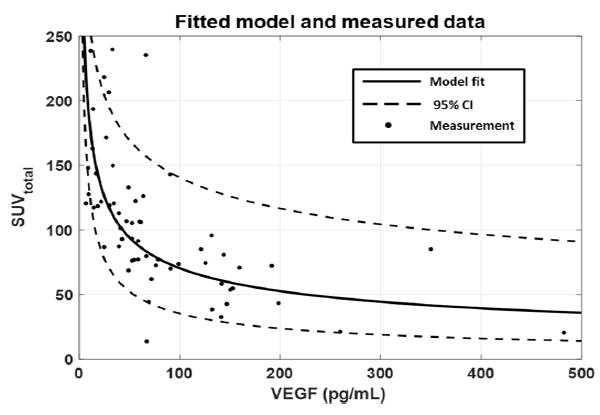
Tumor SUVtotal as a function of plasma VEGF concentration is shown for both the measured data as well as the fitted linear mixed effects model. In general, higher VEGF measurements indicated a lower SUVtotal. CI confidence interval
Axitinib plasma concentration
The median plasma concentration of axitinib in cycle 1 week 2 (after 12–14 days of dosing) was 8 ng/mL (range 1–33 ng/mL). The median concentration in cycle 3 week 2 was 11 ng/mL (range 1–46 ng/mL). No significant difference in axitinib concentration during week 2 was found in cycle 1 versus cycle 3 (P = 0.6). Axitinib concentrations measured prior to dosing and after 5–7 days of washout were below the limit of quantification. No significant correlations were found between axitinib plasma concentrations and either plasma VEGF concentrations or SUV metrics.
Discussion
The safety cohort of this study demonstrated that higher dose of axitinib (7 mg BID) given on an intermittent regimen was not tolerable for patients, as demonstrated by dose-limiting toxicities. Patients were unable to tolerate this dose consistently without experiencing significant cardiovascular side effects and the axitinib dose was subsequently reduced to the standard 5 mg BID for the pharmacodynamic cohort. Interestingly, the percent changes in plasma VEGF concentrations were not significantly different for treatment or washout periods when comparing safety versus pharmacodynamic cohort. This provides evidence that the pharmacodynamic response as measured by plasma VEGF is not markedly different for 5 mg and 7 mg BID intermittent axitinib dosing in this cohort of patients. Given little change in the pharmacodynamic response and increased dose-limiting toxicities seen for 7 mg BID, we recommend future treatments with axitinib in similar cohorts of patients continue with the standard 5 mg BID dosing.
Significant increases in tumor FLT uptake during the washout periods indicating acute treatment withdrawal flare were observed in both cycle 1 and cycle 3. Furthermore, tumor FLT uptake during the third cycle of treatment was not significantly different than uptake in first cycle of treatment. These results suggest that the pharmacodynamic effect of axitinib is similar and consistent across multiple drug cycles. This effect is characterized by decreases in tumor cell proliferation and vasculature during dosing periods followed by subsequent increases during drug washout periods.
These results suggest cell-cycle-specific chemotherapy given concurrently with VEGFR-TKI might be antagonized by the diminished tumor cell proliferation that was observed during dosing periods. On the other hand, the rebound in tumor cell proliferation during axitinib washout periods offers a potential target for cell-cycle-specific chemotherapy given sequentially. An intermittent VEGFR-TKI treatment regimen with cell-cycle-specific chemotherapy applied during the VEGFR-TKI treatment breaks would be an ideal treatment strategy based on the results of this study.
Significant increases in plasma VEGF levels during dosing period were followed by decreases in VEGF levels during the washout periods. This is in agreement with our previous study in a similar cohort of patients being treated with axitinib [5]. Additionally this study shows these trends are present in both the first and third cycles of treatment with no significant differences between cycles. It has been hypothesized that acquired resistance to VEGR-TKI treatment might be attributed to increasing tumor secretion of the VEGF ligand; however, the results of this study showed no difference between VEGF plasma concentration in early and later cycles. This indicates some patients acquire resistance by means other than increased VEGF secretion, such as recruitment of additional angiogenic pathways to circumvent the VEGF pathway [16].
A significant negative correlation was found between FLT uptake metric SUVtotal and plasma VEGF concentration. An increase in a patient’s plasma VEGF concentration indicated a corresponding decrease in their SUVtotal. This negative correlation could be explained by the fact that a greater degree of tumor VEGFR inhibition (leading to increased VEGF secretion) is accompanied by a greater drop in tumor cell proliferation. This confirms an on-target effect of axitinib and lends support to targeting the VEGF pathway to inhibit tumor cell proliferation and growth.
Based on the results of this study, diminished tumor cell proliferation and vasculature during anti-angiogenic treatment likely contribute to negative results of previous studies combining anti-angiogenic agents and chemotherapy. Increases in tumor cell proliferation and vasculature during drug washout periods offer potential for sequential combination with chemotherapy. Based in part on the results presented here, we have an ongoing clinical trial evaluating a novel VEGFR-TKI in sequential combination with a cytotoxic agent in order to capitalize on the tumor withdrawal flare that occurs during washout periods. FLT PET/CT imaging will again be used to shed insight into drug pharmacodynamic effects and generate novel therapeutic strategies.
Acknowledgments
The authors would like to thank the nurses and research specialists of the UWCCC Phase I Program for their efforts in managing this trial, the WIMR PET imaging personnel for acquiring images, and the patients for their participation in this study.
Funding This clinical trial was funded by a Pfizer Investigator-Initiated Research Grant (PI: Liu) (WS1610530) as well as a Prostate Cancer Foundation Young Investigator Award (MSN133051). This trial was conducted within the Cancer Therapy Discovery and Development (CTD2) research program with support from the Translational Imaging Research Working Group (TIRWG) at the UW Carbone Cancer Center.
Footnotes
Conflict of interest The authors have no conflicts of interest to disclose.
Compliance with ethical standards
Human and animal rights All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent Informed consent was obtained from all individual participants included in the study.
References
- 1.Folkman J. Angiogenesis inhibitors: a new class of drugs. Cancer Biol Ther. 2003;2:S127–S133. [PubMed] [Google Scholar]
- 2.Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014;17:471–494. doi: 10.1007/s10456-014-9420-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bautch V. VEGF-directed blood vessel patterning: from cells to organisms. In: Klagsburn MD, Amore P, editors. Angiogensis biology and pathology. Cold Spring Harbor; New York: 2012. pp. 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sonpavde G, Hutson T, Rini B. Axitinib for renal cell carcinoma. Expert Opin Investig Drugs. 2008;17(5):741–748. doi: 10.1517/13543784.17.5.741. [DOI] [PubMed] [Google Scholar]
- 5.Rini BI, Garrett M, Poland B, Dutcher JP, Rixe O, Wilding G, et al. Axitinib in metastatic renal cell carcinoma: results of a pharmacokinetic and pharmacodynamic analysis. J Clin Phara-mcol. 2013;53(5):491–504. doi: 10.1002/jcph.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoh CK, Burris HA, Bendell JC, Tarazi J, Rosbrook B, Kim S, Infante JR, Reid TR. Intermittent dosing of axitinib combined with chemotherapy is supported by 18FLT-PET in gastrointestinal tumours. Br J Cancer. 2014;110(4):875–881. doi: 10.1038/bjc.2013.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scagliotti G, Novello S, von Pawel J, Reck M, Pereira JR, Thomas M, Abrão Miziara JE, Balint B, De Marinis F, Keller A, Arén O, Csollak M, Albert I, Barrios CH, Grossi F, Krzakowski M, Cupit L, Cihon F, Dimatteo S, Hanna N. Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:1835–1842. doi: 10.1200/JCO.2009.26.1321. [DOI] [PubMed] [Google Scholar]
- 8.Rugo H, Stopeck AT, Joy AA, Chan S, Verma S, Lluch A, Liau KF, Kim S, Bycott P, Rosbrook B, Bair AH, Soulieres D. Randomized, placebo-controlled, double-blind, phase ii study of axitinib plus docetaxel versus docetaxel plus placebo in patients with metastatic breast cancer. J Clin Oncol. 2011;29(18):2459–2465. doi: 10.1200/JCO.2010.31.2975. [DOI] [PubMed] [Google Scholar]
- 9.Kindler H, Ioka T, Richel DJ, Bennouna J, Létourneau R, Okusaka T, Funakoshi A, Furuse J, Park YS, Ohkawa S, Springett GM, Wasan HS, Trask PC, Bycott P, Ricart AD, Kim S, Van Cutsem E. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol. 2011;12(3):256–262. doi: 10.1016/S1470-2045(11)70004-3. [DOI] [PubMed] [Google Scholar]
- 10.Liu G, Jeraj R, Vanderhoek M, Perlman S, Kolesar J, Harrison M, Simoncic U, Eickhoff J, Carmichael L, Chao B, Marnocha R, Ivy P, Wilding G. Pharmacodynamic study using FLT PET/CT in patients with renal cell cancer and other solid malignancies treated with sunitinib malate. Clin Cancer Res. 2011;17:7634–7644. doi: 10.1158/1078-0432.CCR-11-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruce J, Scully PC, Carmichael LL, Eickhoff JC, Perlman SB, Kolesar JM, Heideman JL, Jeraj R, Liu G. Pharmacodynamic study of axitnib in patients with advanced malignancies assessed with 18F-3’deoxy-3’fluoro-l-thymidine positron emission tomorgraphy/computed tomography. Cancer Chemother Pharmacol. 2015;76(1):187–195. doi: 10.1007/s00280-015-2779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shields A, Grierson J, Dohmen M, Machulla H, Stayanoff J, Lawhorn-Crews J, Obradovich J, Muzik O, Mangner T. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998;4:1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- 13.Yang W, Zhang Y, Fu Z, Sun X, Mu D, Yu J. Imaging proliferation of 18F-FLT PET/CT correlated with the expression of microvessel density of tumour tissue in non-small-cell lung cancer. Eur J Nucl Med Mol Imaging. 2012;39:1289–1296. doi: 10.1007/s00259-012-2126-8. [DOI] [PubMed] [Google Scholar]
- 14.Horn K, Yap J, Agarwal N, Morton KA, Kadrmas DJ, Beardmore B, Butterfield RI, Boucher K, Hoffman JM. FDG and FLT-PET for early measurment of response to 37.5 mg daily sunitinib therapy in metastatic renal cell carcinoma. Cancer Imaging. 2015;15:15. doi: 10.1186/s40644-015-0049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]