

Abstract
Our greater understanding of the importance of N-linked glycosylation in biological systems has spawned the field of glycomics and development of analytical tools to address the many challenges regarding our ability to characterize and quantify this complex and important modification as it relates to biological function. One of the unmet needs of the field remains a systematic method for characterization of glycans in new biological systems. This study presents a novel workflow for identification of glycans using Individuality Normalization when Labeling with Isotopic Glycan Hydrazide Tags (INLIGHT™) strategy developed in our lab. This consists of monoisotopic mass extraction followed by peak pair identification of tagged glycans from a theoretical library using an in-house program. Identification and relative quantification could then be performed using the freely available bioinformatics tool Skyline. These studies were performed in the biological context of studying the N-linked glycome of differentiating xylem of the poplar tree, a widely studied model woody plant, particularly with respect to understanding lignin biosynthesis during wood formation. Through our workflow we were able to identify 502 glycosylated proteins including 12 monolignol enzymes and 1 peroxidase (PO) through deamidation glycosite analysis. Finally, our novel semi-automated workflow allowed for rapid identification of 27 glycans by intact mass and by NAT/SIL peak pairing from a library containing 1573 potential glycans, eliminating the need for extensive manual analysis. Implementing Skyline for relative glycan quantification allowed for improved accuracy and precision of quantitative measurements over current processing tools which we attribute superior algorithms correction for baseline variation and MS1 peak filtering.
Graphical Abstract
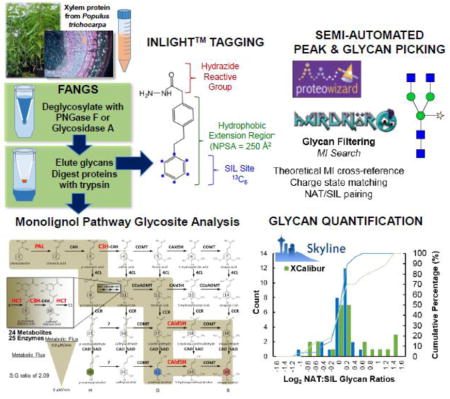
Introduction
More than 50% of all proteins in eukaryotes are known to be glycosylated [1], and it is well established that the N-linked glycosylation patterns of proteins significantly influence their biological activity. Similar to other eukaryotes, N-linked glycosylation in plants begins in the endoplasmic reticulum (ER) with the co-translational or post-translational transfer of a dolichol lipid-linked oligosaccharide precursor Glc3Man9GlcNAc2 to an asparagine residue located within an N-linked glycosylation consensus sequence (Asn–X–Ser/Thr where X is any amino acid except Pro), though more recent studies have suggested that other non-consensus sequences also exist, including Asn-X-Cys in plants [2]. Once attached, the N-linked glycan undergoes a series of steps involving the removal of glucose and/or mannose residues to generate high mannose type glycans or the addition of new sugar residues in the ER and golgi apparatus to produce complex type glycans. While high mannose type glycans are structurally similar in plant and mammalian glycoproteins, complex type glycans in plants are structurally different from their mammalian counterparts. For example, in plants the bisecting mannose contains β1,2-xylose, while sialic acids often modify glycans in mammals [3]. Moreover, in addition to the α1–6-fucose attached to N-acetylglucosamine core in mammals, plants commonly contain an additional α1–3-fucose.
Previous work has shown that glycosylation is a necessary requirement for cellulose biosynthesis and that the absence of complex N-glycans severely affects overall plant growth and development [4]. While the molecular mechanisms and the functional importance of N-linked glycosylation has been demonstrated, much is unknown regarding the composition of the glycoproteome and glycome present in the differentiating xylem of plants as well as their potential regulatory role in secondary wall biosynthesis. Our previous work in Populus trichocarpa has focused on quantifying monolignol proteins to model the flux of the monolignol biosynthetic pathway [5–9]; however, we have also shown functional significance of post-translational modifications such as phosphorylation on regulating the flux of the pathway [10]. While glycosylation of peroxidases associated with lignification has been previously observed [11]; no study within this system has ascertained glycan structural and glycosylation site-specific information on many of the enzymes involved in lignin biosynthesis.
Recent developments in preparation methods and mass spectrometry (MS) instrumentation have made it possible to more deeply and accurately characterize the location of glycosylation sites and the structure of glycans [12]. Deamidation is induced through enzymatic release of glycans, most commonly by PNGase F [13]. Glycosidase A is another glycosidase that may be used to aid in hydrolysis of glycans containing a core α1–3-fucose, whose structures known to have low cleavage efficiency by PNGase F [14]. This results in the enzymatic conversion of an asparagine/glutamine residue to its corresponding acidic form aspartic/glutamic acid via loss of NH3 and addition of H2O. This modification is a commonly encountered chemical modification in the field of proteomics. It can occur spontaneously, particularly at high pH during sample preparation [15, 16], or in vivo as a modification on glutamine and asparagine residues of proteins [17]. It has been shown that a mass accuracy of less than 5ppm is necessary to accurately identify deamidated (+0.984Da) peptides [18]. With mass accuracy greater than 5ppm, the M+1 (13C1) peak of the amidated peptide may be misidentified as a deamidated form. High resolving power mass spectrometry has made it possible to resolve the very small mass difference.
The use of 18O water has been demonstrated to be effective in accounting for background chemical deamidation during sample preparation and native biological deamidation [19]; however, it is costly and cannot account for non-specific deamidation which occurs during deglyosylation. Alternatively, filter-aided N-glycan separation (FANGS) is a filter-aided sample preparation (FASP) method which was recently designed to minimize background deamidation to proteins to less than 5% in plasma [12], allowing deamidation to be a more specific marker for glycosylation, and is subsequently employed in the following study to assess this method in a more complex biological matrix. The primary advantage of this approach is that it is cost effective and proteomic and glycomic workflows can be combined into a single workflow.
The chemical nature of released N-glycans also presents analytical challenges. Glycans hydrophilic properties lower their ionization efficiency and their heterogeneous, non-linear structures require extensive MS/MS and MSn data interpretation to obtain structural information. Sensitivity challenges may be overcome with off-line derivatization strategies conferring hydrophobicity [20], such as the Individuality Normalization when Labeling with Isotopic Glycan Hydrazide Tags (INLIGHT™) workflow [21, 22]. The hydrophobic tag permits separation by C18 chromatography, and this may be completed in sequence with proteomic analysis. Furthermore, incorporation of an isotopic (13C6) labeled tag allows accurate relative quantification of glycans by differential labeling of samples.
One of the major challenges in characterizing glycans in a new system is identifying compositions and structures from the thousands of potential structures contained within a theoretical database. When mixing replicate samples tagged with equimolar amounts of light and heavy labeled hydrazide reagent, glycans must be validated manually by extracting chromatograms for all potential glycan structures contained within a given library (often >1,000 glycans), verifying the presence of the monoisotopic masses of both the light and heavy species at approximately 1-to-1 abundance, and ensuring the correct isotopic distributions. For high-throughput screening of glycans, an automated method for reducing the theoretical library of glycans to a manageable list is necessitated. While many programs claim to be capable of profiling N-glycans, they often fail to correctly isolate the monoisotopic masses of glycans in the presence of contaminants or co-eluting glycans (both isomeric and unique). Other programs, such as SimGlycan [23], rely on consistent and high-quality MS/MS generation, which is often not feasible in discovery-based, complex biological LC-MS/MS experiments. The freely available software Skyline, developed by MacCoss and co-workers, presents an alternative to those in the field of glycomics for a more rapid and robust method of data processing. The recently developed targeted workflow has been applied to analysis of lipids [24] and other small molecules [25]. However, this platform has not yet been evaluated for glycans or compared to currently used and accepted tools.
In this current study we present deamidated glycosite profiling of differentiating xylem in P. trichocarpa as well as a workflow for identification of unknown glycans. Through this we identified potential glycosylation sites for over 500 proteins, including 12 enzymes and 1 PO involved in lignin biosynthesis as well as identification of 27 glycans. Further, we demonstrate Skyline’s improved accuracy and precision of relative quantitation for glycans over our existing platform in XCalibur. We attribute this improvement to the algorithms correction for baseline variation and MS1 peak filtering. This presents an excellent foundation of which to build an automated workflow identification and relative quantification of glycans.
Experimental
Materials
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). All solvents were HPLC-grade from Honeywell Burdick & Jackson (Muskegon, MI). Unless otherwise stated all solutions were made in 100mM ammonium bicarbonate pH 7.5 (PNGase F Digest buffer).
FANGS-INLIGHT
Crude xylem protein extraction was performed as described previously [26] and processed according to the FANGS-INLIGHT protocol [11]. 1000 μg of crude xylem protein was split into 4 × 250 μg per FASP filters. Sample was loaded onto a 0.5 ml, Amicon Ultra 10kDa MW-cutoff filter (EMD Millipore Billerica, MA) and 2 μl of 1 M dithiothreitol solution (DTT) was added. The sample was diluted with 200 μl PNGase digest buffer and incubated at 56°C for 30 min. To give a final concentration of 200 mM, iodoacetamide (1 M) was added and then the sample was incubated at 37 °C for 60 min. The denatured glycoprotein was concentrated onto the filter by centrifuging samples at 14,000 x g for 15 min, discarding the flow through. The sample was then washed twice with 100 μl PNGase digest buffer, concentrating the glycoprotein on the filter at 14,000 x g for 15 min, discarding the flow through. After transfer to a clean vial, 2 μl of glycerol-free PNGase F (New England Biolabs, Ipswich, MA) 75,000 x units/ml, 25μL (0.5 mU) of N-Glycosidase A, or no enzyme (control) was added to the filter in 98 μl of PNGase digest buffer. Samples were incubated at 50 °C for 2 hours, and, working quickly, an additional 2 μl of enzyme or buffer (control) was added. Samples were vortexed lightly for 1–2 seconds and incubated at 50 °C for an additional 2 hours. Glycans were then eluted by centrifuging the sample at 14,000 x g for 20 min at 20 °C. Two washes, collected in the same vial as the eluent, were performed with 100 μl of PNGase digest buffer, centrifuging at 14,000 x g for 20 min at 20 °C. Glycan recovered from Glycosidase A and PNGase F replicates were pooled into a single sample, stored in the −80°C freezer until frozen (30 – 60 min) and then dried to completion under vacuum.
For glycoproteomics, the filter was then removed and placed in new collection vial. The filter containing the deamidated glycoproteins, was washed twice with 400 μl of ammonium bicarbonate at 14,000 x g for 15 min, discarding the flow-through. After transferring the filter to a new collection via, 50 μl porcine modified trypsin reconstituted in ammonium bicarbonate pH 7.5 was added in a 1:50 enzyme-to-protein ratio (5μg) and lightly vortexed. After incubating at 37 °C for 2 hours, an additional 50 μl of trypsin at the 1:50 ratio was added. The samples were lightly vortexed and then incubated at 50 °C for an additional 2 hours. The peptides were eluted by spinning at 14,000 x g for 15 min. The remaining peptides from the filter were washed with 400 μl of quench buffer containing 1% formic acid and 0.001% zwittergent 3–16 and eluted off the filter by spinning at 14,000 x g for 15 min.
Dried INLIGHT™ reagents (Cambridge Isotope Labs, Andover, MA) were reconstituted in 1 ml of 75% methanol/12.5% acetic acid/12.5% H2O derivatization solution (0.25 mg/ml). Reagents were allowed 10 min to fully solubilize, extensively vortexing to ensure complete solubilization. Sample N-glycans were tagged with 200 μl (50 μg) of either Heavy (SIL) or Light (NAT) INLIGHT™ reagent, pipetting up and down to resuspend dried glycans. Samples were vortexed and then spun down samples for ~5 sec on a bench-top centrifuge. Sample glycans were reacted with INLIGHT™ reagent for 3 hours at 56° C. Samples were dried to completion in a vacuum concentrator at 55°C. Tagged N-glycans were resuspended in 30 μl of H2O, pipetting up and down to ensure N-glycans were fully solubilized. To reduce excess tag, samples were centrifuged at 14,000 x g for 5 min. NAT and SIL sample supernatant were then combined immediately after reconstituting and prior to LC-MS analysis.
LC-MS/MS Analysis and Bioinformatics
Samples were analyzed using an Easy nLC-1000 configured with a Q Exactive™ High Field mass spectrometer (Thermo Scientific, San Jose, CA). For peptide analysis, samples were reconstituted in 250μl of 0.001% zwittergent 3–16 (Calbiochem, La Jolla, CA) and 5μl was injected onto a 25 cm × 75μm column packed with C18 2.6μm, 100 Å resin (Phenomenex, Torrance, CA) in a one column direct inject configuration with a maximum pressure of 600 bar. A 4 hr. gradient was run at 300 nL/min going from 5–30% B (mobile phase B was 98% acetonitrile, 2% H2O, 0.1% formic acid and mobile phase A was 98% H2O, 2% acetonitrile, 0.1% formic acid). Proteins were ionized under the following MS1 conditions: 400–1600 m/z, 120,000 resolution, 3 × 106 AGC. 50 ms injection time, 65 RF S-lens, 325 °C capillary, and 1.75 kV spray voltage and a lock mass of 445.12003 m/z. MS2 conditions consisted of 15,000 resolution, 1 × 105 AGC, 30 ms injection time, 1% underfill ratio, 2.0 Th isolation window, charge state 1 exclusion, top 20 data dependent acquisition, 20 s exclusion window, and 27% normalized collision energy.
For glycan analysis, 2μl of resuspended, INLIGHT™ tagged NAT/SIL equimolar sample was injected onto the column. A 60 min gradient was run at 300 nL/min under the following conditions: maximum injection pressure of 600 bar, 5–30%B (1 min), 30–40%B (40 min), 40–63%B (5 min), 63–90%B (1 min), 90%B (8 min), 90–5%B (1 min), 5%B (10 min). Glycans were ionized according to the previously optimized MS1 conditions: 600–1900 m/z, 60,000 resolution, 5×105 AGC, 64 ms injection time, 65 RF S-lens, 325 °C capillary, and 1.75 kV spray voltage and MS2 conditions: 15,000 resolution, 5×104 AGC, 100 ms injection time, 1% underfill ratio, 1.4 Th window, 125 Th fixed first mass, top 12 data dependent acquisition, 15 s exclusion window, peptide match preferred and 10/20/30 stepped normalized collision energy. A theoretical database was curated based on our previous work in human plasma [22, 27] with the variable addition of a single xylose for a total of 1573 potential glycans. Raw files were converted to mzXML using ProteoWizard [28]. Peak peaking of raw data was performed using Hardklor [29, 30] with the Patterson algorithm, Boxcar avg. (5 scans), Scan filter (3 scans) and a 0.90 correlation (ESM1). Monoisotopic masses were then used to search the theoretical glycan database using in-house developed software which identifies glycans based on intact mass (within 5ppm) and presence of NAT and SIL labelled glycans. This list was then imported into Skyline V3.5 [31] in order to confirm co-elution of NAT and SIL peak pairs.
Proteins were identified using raw files in Proteome Discover. Data was searched using the P. trichocarpa JGI v2.2 protein database [32] containing 45,215 sequences and P. trichocarpa Uniprot database containing 47,350 sequences. The following search rules were implemented: full tryptic peptides, a minimum of 5 amino acids, a max of 2 missed cleavages, fixed carbamidomethyl (C) modification, variable deamidation (N/Q) and oxidation (M) modifications, a maximum of 4 modifications/peptide, a 5 ppm precursor mass tolerance, and 0.02 Da fragment mass tolerance for accurate deamidation identification. Peptides were filtered at a 1% FDR using percolator and proteins were grouped according to strict parsimony principle (minimum one unique peptide/protein group).
Results and Discussion
Comparative analysis of PNGase F, Glycosidase A and Control Deamidation
The P. trichocarpa datasets were filtered for peptides containing deamidated asparagine residues. Across PNGase F and Glycosidase A samples, 3,450 peptides contained deamidated asparagine residues (see Electronic Supplementary Material (ESM), Fig. S1), and 37.2% of these peptides were identified in control samples as well. Of the deamidated peptides exclusively found in treated samples, only ~10% overlapped between Glycosidase A treated and PNGase F treated samples, highlighting the different preferences of the enzymes for cleaving glycosylation sites. A closer inspection of the treated and control deamidated peptides by MotifX [33, 34] yielded a list of enriched motifs (Figure 1A). Many of the leucine containing motifs were shared among treated and control samples, suggesting that these could be in vivo deamidation motifs or motifs prone to chemical deamidation. Motifs with alanine in the −2 position and valine in either the +2 or −2 position were unique to the treated samples, suggesting these motifs could be related glycosylation. Previous studies have found asparagine residues with nearby hydrophobic residues in the X position of the NXS/T motif are less prone to chemical deamidation [15] than small, hydrophilic residues in the X position. Moreover, the NXV motif observed in our data has previously been demonstrated to be a non-consensus glycosylation motif [2].
Figure 1.

A) XXNXX Motifs for all deamidated peptides in treated vs. control samples.
B) XXNXS/T motifs enriched for in deamidated peptides identified in PNGase F and Glycosidase A treated samples. Enriched motifs obtained from MotifX (motif-x.med.harvard.edu) using P. trichocarpa proteome background.
Filtering the deamidated peptides for the known NXS/T glycosylation motif resulted in 858 deamidation sites, a ~75% reduction of results in the enzyme-treated samples. This number represents 6.5% of the total number of motifs present in the P. trichocarpa proteome. We sampled fewer than 10,000 proteins (<20% of the proteome) which is reflected in the number of deamidation sites identified. For motif containing deamidation sites similar trends were observed, with 32.7% of the enzyme-treated pool overlapping with the control peptides and 32.9% of the treated peptides overlapping between Glycosidase A and PNGase F treated samples. This data suggests that filtering based on the motif may not reduce the number of false positives due to background chemical deamidation. The FANGS-INLIGHT protocol that controls for non-specific deamidation was developed in mammalian plasma [12]. Tissue, or perhaps plant, samples contain different levels of extracted metabolites/enzymes that can promote deamidation, and this study indicates that the FANGS-INLIGHT protocol parameters must be re-evaluated for tissue analysis. Moreover, the >1500 deamidated peptides filtered out based on absence of the NXS/T motif could also be indicative of the presence of other motifs in the P. trichocarpa N-linked glycome.
The NXS/T motif-containing deamidated peptides were further examined by MotifX. A series of specific motifs were enriched with respect to the proteome within the treated, XXNXS/T motif deamidated peptides but not in the control samples (Figure 1B). Amino acids most overrepresented within this motif included L, V, G, and S. G and S are small/polar residues that are prone to background chemical deamidation, potentially explaining the overlap between treated and control peptides despite filtering based on the motif. In peptides exclusive to treated samples, leucine could be found in the −2, −1 or +1 position in 26%, 48% and 49% of the NXS/T motifs (141 total), respectively. Valine was found in the +1 and −2 positions at 22% and 15% of NXS/T motifs, respectively. This suggests that these hydrophobic residues could play a role in motif specific glycosylation, possibly in more than simply the +1 position. Potentially relevant but occurring with lesser frequency (<10%) in the +1 position were isoleucine, asparagine, phenylalanine, tyrosine and threonine.
Exclusive to the treated samples, 502 protein groups were identified that contained deamidated peptides (see ESM) and annotated using DAVIDGO algorithm [35] to elucidate pathways and processes which may be regulated by glycosylation (Table 1). Several important pathways were found to be enriched in PNGase F and Glycosidase A treated samples. Processes associated with deglycosylated proteins included: fundamental metabolic pathways (TCA, Pyruvate, Tryptophan, Carbon fixation, Carbohydrate metabolism), protein localization, modification of proteins, protein catabolism and phenylpropanoid (such as lignin) metabolism.
Table 1.
DAVIDGO algorithm pathways and biological processes enriched based on Uniprot Poplar proteins identified by motif containing, deamidated peptides found exclusively in the PNGase F/Glycosidase A treated samples. Annotations obtained from DAVIDGO algorithm (david.ncifcrf.gov).
| Pathways and Processes Enriched | Count | P-Value |
|---|---|---|
| Methane metabolism | 3 | 3.30E-02 |
| Biosynthesis of alkaloids derived from shikimate pathway | 9 | 4.10E-02 |
| Carbon fixation in photosynthetic organisms | 6 | 4.40E-02 |
| Biosynthesis of plant hormones | 12 | 4.40E-02 |
| Other glycan degradation | 3 | 4.60E-02 |
| Glyoxylate and dicarboxylate metabolism | 4 | 5.50E-02 |
| Citrate cycle (TCA cycle) | 5 | 6.70E-02 |
| Pyruvate metabolism | 5 | 6.70E-02 |
| Tryptophan metabolism | 3 | 8.70E-02 |
| Posttranslational modification, protein turnover, chaperones | 13 | 1.20E-03 |
| Intracellular trafficking and secretion | 5 | 3.20E-02 |
| Carbohydrate metabolic process | 37 | 7.80E-07 |
| Phenylpropanoid metabolic process | 4 | 4.70E-02 |
| Protein localization in nucleus | 4 | 9.50E-03 |
| Modification-dependent protein catabolic process | 9 | 1.60E-02 |
| Microtubule-based movement | 10 | 3.20E-05 |
Unique, NXS/T motif containing deamidated peptides were used to identify 13 monolignol proteins containing a total of 14 deamidation sites (Table 2). Peptide spectral matches (PSMs) and PSM occupancy were calculated for each peptide according to Equation 1 to reveal information regarding the absolute and relative abundance of the deamidated peptide with respect to the unmodified form (Table 2). Deamidation sites exclusive to enzymatically treated samples are most likely to be true glycosylation sites. The peptides associated with lignin synthesis and with deamidation confined to the enzymatically treated samples are highlighted in Figure 2. These eight enzymes are known to be important and tightly regulated in wild-type to maintain lignin biosynthetic flux, making them important putative glycosylation identifications. Interestingly, C3H, which catalizes the reaction of p-coumaryl shikimic acid to caffeoyl shikimic acid, contains both the classic NXS/T motif and the more recently proposed NXC glycosylation motif. PO42, the most abundantly expressed class III peroxidase in differentiating xylem of P. trichocarpa [36], was the most confident glycoprotein identification, supported by multiple highly-occupied and unique deamidated peptides.
Table 2.
List of monolignol proteins identified by a motif containing deamidated peptide in PNGase F/Glycosidase A treated samples are shown. Additional information regarding the peptides presence in control samples and site occupancy based on peptide spectral matches (PSMs) yields information regarding confidence in these as possible glycosylation sites.
| Name | In Control | Peptides containing motif | PSM Occupancy | PSMs |
|---|---|---|---|---|
| CAld5H2 | No | KQNNFSEDAETDMVDDMLAFYSEEAR (Glyc A only) | 43% | 3 |
| HCT1 | No | AKEDGNNISYSSYEMLAAHVWR (PNGase F only) | 50% | 2 |
| PAL1 | No | FLNAGIFGNGTETCHTLPHSATR (PNGase F only) | 13% | 1 |
| PAL3 | No | FLNAGIFGNGTETCHTLPHSATR (PNGase F only) | 13% | 1 |
| PO42 | No | DFNNTTVLDIR (GlycA only) | Deamidated only | 13 |
| No | DFNNTTVLDIR (GlycA only) | Deamidated only | 5 | |
| No | DFNNTTVLDIR (GlycA only) | Deamidated only | 3 | |
| No | FATQNETLDNLPPPFANADTILSSLATK | 98% | 53 | |
| C3H | No | LEALRPIREDEVAAMVESIFNDCTNPENNGK | 27% | 3 |
| LIM2 | No | EKGNLSQLEGDIEK | Deamidated only | 1 |
| IRX10-2 | No | SAIQLLSSNWPYWNR | Deamidated only | 6 |
| CAD2 | Yes | VGVGCLVGACHSCESCASDLENYCPK (PNGase F only) | 33% | 2 |
| CCR | Yes | EAIQGCDGVFHTASPVTDDPEEMVEPAVNGTK | 13% | 1 |
| CesA17 | Yes | VSAVLTNAPFMLNLDCDHYVNNSK | 40% | 2 |
| 4CL3 | Yes | NLPLHSYVLENLSK (Glyc A only) | Deamidated only | 1 |
| CCoAOMT1 | Yes | VGGLIGYDNTLWNGSVVAPPDAPMRK | 17% | 18 |
| CCoAOMT2 | Yes | VGGLIGYDNTLWNGSVVAPADAPMRK | 3% (N9) and 35% (N13) | 2 (N9), 22 (N13) |
| PAL4 | Yes | FLNAGIFGNGTESSHTLPR | 29% | 9 |
| PAL5 | Yes | FLNAGIFGNGTESSHTLPR | 29% | 9 |
Figure 2.

Monolignol biosynthetic pathway with putative deamidation/glycosylated enzymes highlighted in red. Adapted from Wang et al. Plant Cell 2014, 3: 894–914 and Shuford et al. J. Proteome Res. 2012, 11:3390–3404.
Many of the motif-containing deamidated peptides mapping to monolignol enzymes were also identified in the control. Among these PAL4|5 was identified in the control but only with 1 spectral count (vs. 9 in treated), which would suggest it may be a glycosylation site. Whereas, CoAOMT1 and CoAOMT2 both appeared to be similar in abundance in control (15 and 28 spectral counts respectively) to treated samples, suggesting it is a product of in vivo or chemical deamidation. CAD2, CCR and 4CL3 were all identified with low numbers of spectral counts which suggests these also may be in vivo or chemical deamidation. Additional experiments would be necessary to rule these out as glycosylated sites on these proteins.
Equation 1. Glycosylation occupancy calculations.
A New Workflow for Discovery-Based Glycomics in P. trichocarpa
The N-glycome of P. trichocarpa has yet to be investigated, and minimal work has been completed on Arabidopsis, its most well-studied homologue, lending itself to a discovery-based approach. A theoretical composition database with the maximum number of saccharides set to N9, H11, A3, F3, and X1, yielded 1573 possibilities. This composition space was too large to manually search, and thus a semi-automated workflow was created (Figure 3). Peak picking was completed by Hardklor [29, 30], and then the neutral masses of each peak were automatically searched using an in-house program, MIsearch (ESM2). The program retained peaks that were within a 5 ppm mass measurement accuracy (MMA) to the theoretical compositions, matched in charge state, and were present in the NAT and SIL labeled forms. The Hardklor peak parameters were selected by iteratively testing them on a dataset from a previously published study on human plasma, in which the glycome was both well-characterized in the literature and well-defined manually [27]. In that dataset, 72 glycans were detected, and the Hardklor parameters were adjusted until zero false negatives minimal false positives were observed with MIsearch. The key peak parameter settings that significantly impacted the results were algorithm type, boxcar averaging, boxcar filter, and correlation. The N-glycans were then manually validated based on their relative retention times (ESM1 Fig. S3), isotope features, and MS/MS data.
Figure 3.

Workflow for processing any RAW data file type through Hardklor for peak picking, MI search for glycan filtering, and manual validation in Skyline.
When applied to the poplar tree, the workflow outlined had a 3.6% false positive detection rate and identified 27 unique compositions (Table 3). Of these, three (H10N2, H6N5F2, and H5N4F1X1) of the 27 were lacking in MS/MS confirmation of their structure due low abundance relative to other co-eluting glycans or contaminants (ESM1 and Table 3). The glycans identified were primarily of the high mannose type, contained core fucoses, or were modified with xyloses. The compositions were searched in the consortium for functional glycomics (CFG) molecular database for cross-referencing [37], and H5N4F2X1 was determined to be a novel species. To support the MS1 assignment, a thorough MS/MS assignment is provided in Figure 4, and as noted throughout the spectrum, there is excellent coverage of the overall structure.
Table 3.
| Composition | NAT | SIL | Charge |
|---|---|---|---|
| H3N2X1 | 640.2585 | 643.2686 | 2 |
| N2 | 661.3085 | 667.3293 | 1 |
| H5N2 | 736.2902 | 739.3003 | 2 |
| H3N3X1 | 741.7982 | 744.8083 | 2 |
| H0N2F1 | 807.3664 | 813.3873 | 1 |
| H3N3F1X1 | 814.8272 | 817.8372 | 2 |
| H6N2 | 817.3166 | 820.3267 | 2 |
| H4N3X1 | 822.8246 | 825.8347 | 2 |
| H3N4X1 | 843.3379 | 846.348 | 2 |
| H4N3F1X1 | 895.8536 | 898.8636 | 2 |
| H7N2 | 898.343 | 901.3531 | 2 |
| H3N4F1X1 | 916.3669 | 919.3769 | 2 |
| H8N2 | 979.3694 | 982.3795 | 2 |
| H4N4F1X1 | 997.3933 | 1000.403 | 2 |
| H9N2 | 1060.396 | 1063.406 | 2 |
| H5N4F1X1 | 1078.42 | 1081.43 | 2 |
| H10N2 | 1141.422 | 1144.432 | 2 |
| H5N4F2X1 | 1151.449 | 1154.459 | 2 |
| H5N4F3X1 | 1224.478 | 1227.488 | 2 |
| H3N2F1X1 | 713.2875 | 716.2976 | 2 |
| H2N2F1X1 | 632.2611 | 635.2711 | 2 |
| H1N2 | 823.3613 | 829.3814 | 1 |
| H1N2F1 | 969.4192 | 975.4393 | 1 |
| H2N2F1 | 1131.472 | 1137.492 | 1 |
| H3N2 | 1147.467 | 1153.487 | 1 |
| H5N5F2 | 1186.967 | 1189.977 | 2 |
| H6N5F2 | 1267.994 | 1271.004 | 2 |
Figure 4.

(A) The XICs of the NAT and SIL species of H5N4F2X1, an N-glycan identified for the first time across any species, co-elute (16.1 – 16.5 min) and are observed in high abundance.
(B) An annotated, example MS/MS spectrum from NAT tagged H5N4F2X1, with the regions from 1000–2000 m/z and 500–1000 m/z highlighted, is given. The assignments across all H5N4F2X1 MS/MS spectra observed are provided in Table S1.
Of the remaining glycans, 23 were found to be ubiquitious across plants, with the non-xylose glycans common across many mammalian species. H4N3F0X1, H4N3F1X1, and H5N4F1X1 was solely identified in Nicotiana alata (jasmine tobacco), Phaseolus vulgaris (common bean), and Helix pomatia (the Roman snail), respectively, and P. trichocarpa shares important commonalities with each of these species. In the jasmine tobacco plant, S-class glycoproteins are involved in self-pollination in plants of the Solanaceae family [38, 39], yet they are also predicted to be found in P. trichocarpa (UniProt), though its function would necessarily be different. In the case of the common bean, the two species both express purple acid phosphatase proteins, which are involved in scavenging phosphate from soil and the conversion of phosphate esters (Blastp, nr database, ID 4KPB, organism P. trichocharpa). Thirdly, H5N4F1X1 was identified on A-type hemocyanin in the snail, which is a metalloprotein that transports oxygen in invertebrates such as mollusks [40, 41]. Its amino acid sequence is a homologue to a tyrosinase (EC: 1.14.18.1), which in P. trichocarpa plays a critical role a variety of metabolic pathways, including riboflavin [42], to convert quinone to hydroquinone, and isoquinoline alkaloid biosynthesis [43], catalyzing tyramine to dopamine. The N-glycans identified represent a diverse group overall that clearly play critical roles in key plant physiological pathways.
Within the monolignol pathway, putative glycoprotein assignments were made with localization to the cell wall (PO42), membrane (CAld5H, C3H) or cytosol (PAL1|3 and HCT1) [36, 44, 45]. Without follow-up studies, it is not clear which glycans are providing functionality to each of these proteins, though all likely contribute to key functional activities [46]. Glycosylation of these proteins could provide a potential mechanism for regulating the localization, stability, functional activity as well the ability of these proteins to participate in multienzyme complexes (C3H) [47] which may control monolignol biosynthetic flux.
Assessing a Targeted Glycan Workflow for Quantitative and Qualitative Analysis
The ability to perform high-throughput and standardized discovery-type analysis has been a limiting factor in the expansion of the glycomics. Even in the semi-automated workflow detailed above, the monoisotopic masses of the parsed glycan list, of which 3.6% (1 of 28) were determined to be false positives, had to be manually inputted into the XCalibur Quan browser. The advent of a targeted analyte workflow in Skyline offers a new software available in which to perform manual analysis of N-glycans. This interface is amenable to easily importing m/z transition lists, peak picking based on the chemical formula, and viewing NAT and SIL spectra concurrently. The Skyline targeted workflow is a new feature and has yet to be used in the field of glycomics, which introduces new and unique challenges, and characterization of the program is needed.
The Skyline targeted method was vetted against the current integration method, performed in the XQuan browser of XCalibur. The log2 of the normalized relative abundances of the NAT to SIL species were assessed in each method, and there was no significant difference in the mean across all ratios (matched t-test, p > 0.05). However, the variability between the two methods was significantly different (Levene’s test, p < 0.05), with Skyline producing a tighter distribution (ESM1 Fig. S2). A review of the XIC traces suggested that the largest differences in variability came from N-glycans that were low abundant and had poor Gaussian peak shapes. This difference was hypothesized to be a function of the data smoothing applied in the XCalibur analysis (Guassian integration, N = 15), however, when the data was reevaluated with smoothing removed in XCalibur, the results remained unchanged.
Eight N-glycans, while not statistically significant, appeared to be major contributors to the variance, yielding ratios greater than or less than ±0.98 in at least one XCalibur run, while their counterpart ratio in Skyline was centered closer on zero. A qualitative comparison of the integration bounds used between Skyline and XCalibur showed consistency. We then supposed that differences could occur if integration was performed on a different pool of spectra, resulting from differences in the generation of XICs between the programs. This type of difference would not be overcome by post-processing procedures, such as smoothing, and rather only effected by pre-processing peak picking parameters. The number of spectra integrated for each glycan was normalized to the elution window (scan/RT), and for each of the eight glycans, the XCalibur data had significantly higher scan/RT ratio (matched t-test, 1-sided, p < 0.05). When the data was pooled across these eight glycans, the difference became significantly more pronounced (p = 5.4 × 10−14), reflecting a global trend (ESM1 Fig. S2). Skyline applies various data filters such as baseline subtraction to each chromatogram, selects peaks based on a variable MMA (60,000 RP at 200 m/z versus standardized 5 ppm window), and permits analysis of the isotopic envelope versus solely the monoisotopic mass. It may be concluded that the application of such features in Skyline leads to more consistent quantitative results.
Conclusion
Through the integrated FANGS-INLIGHT protocol we were able to identify over 500 potential glycosylated proteins in the xylem proteome of Populus trichocarpa, 13 of which belonged to lignin biosynthetic proteins. In parallel we were able to identify 27 specific glycan structures which are potentially associated with these glycosylation sites. This study contributes to a greater understanding of the role of glycosylation in differentiating xylem. Moreover, the glycosite and glycan structural information has the potential to understand glycosylation as a regulatory mechanism for lignin biosynthesis during wood formation. From an analytical perspective we were able to develop novel software workflow for identification and relative quantification of glycans using a combination of monoisotopic peak picking, NAT/SIL peak pair identification from a theoretical library of glycans and manual validation of a parsed list within Skyline. The false positive rate for automated identification through this workflow was demonstrated to be <5%. Comparative analysis for relative quantification demonstrated Skyline performed more accurately and precisely than Xcalibur processing and that this was likely due to its pre-processing, peak picking algorithm. This approach will improve throughput for discovery-based and robustness of quantitative experiments. This improves our overall understanding of Skyline as a tool for the glycomics community and will lead to further applications and development for analysis of glycans.
Supplementary Material
Fig. S1 (A) Venn diagrams showing the overlap of deamidated peptides identified by control (no enzymatic treatment) samples and treated (PNGase F or Glycosidase A) samples. (B) The number of deamidated peptides in the treated is further reduced by filtering out peptides which do not contain the NXS/T glycosylation motif
Oliveros, J.C. (2007–2015) Venny. An interactive tool for comparing lists with Venn’s diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html
Fig. S2 (A) The distribution of the glycan ratios calculated in Skyline or XCalibur are compared. (B) For eight glycans with mean abundance ratios greater than 0.98, the scan/RT ratios were calculated for each method. When pooled, the mean scan/RT ratio was 42.9 and 32.9 for XCalibur and Skyline method, respectively (p = 5.4 × 10−14)
Fig. S3 Confirmation of Glycans by Co-elution of Native/SIL Species in 1-to-1 Abundance, along with accurate mass and isotopic distribution in Skyline
For any complex-type glycan, selected diagnostic fragments are reported. In each MS/MS, the associated NAT or SIL tag was observed. For the sake of brevity, small saccharide ions in the MS/MS spectra that reveal little about complex structure were emitted, as were tables for glycans with whose structure may be completely inferred by biology (e.g. di- and tri- saccharides, N-glycan core structures). H10N2, H4N2F1X1, H6N5F2, and H5N4F1X1 were not selected for MS/MS
Acknowledgments
This material is based on work supported by North Carolina State University and the Chemistry Graduate Assistantship and the NIH/NCSU Molecular Biotechnology Training Program (Grant 5T32GM00-8776-08). We gratefully acknowledge Jack P. Wang and Vincent L. Chiang for providing the xylem used in this study. Finally, we would also like to thank Jon Ziefle for writing MIsearch.py.
Footnotes
Compliance with Ethical Standards
The authors declare no conflict of interest in the present work.
References
- 1.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Bba-Gen Subjects. 1999;1:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 2.Sun S, Zhang H. Identification and Validation of Atypical N-Glycosylation Sites. Anal Chem. 2015;24:11948–11951. doi: 10.1021/acs.analchem.5b03886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lerouge P, Cabanes-Macheteau M, Rayon C, Fischette-Laine AC, Gomord V, Faye L. N-glycoprotein biosynthesis in plants: recent developments and future trends. Plant Mol Biol. 1998;1–2:31–48. [PubMed] [Google Scholar]
- 4.Lukowitz W, Nickle TC, Meinke DW, Last RL, Conklin PL, Somerville CR. Arabidopsis cyt1 mutants are deficient in a mannose-1-phosphate guanylyltransferase and point to a requirement of N-linked glycosylation for cellulose biosynthesis P. Natl Acad Sci USA. 2001;5:2262–2267. doi: 10.1073/pnas.051625798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shuford CM, Li QZ, Sun YH, Chen HC, Wang J, Shi R, Sederoff RR, Chiang VL, Muddiman DC. Comprehensive Quantification of Monolignol-Pathway Enzymes in Populus trichocarpa by Protein Cleavage Isotope Dilution Mass Spectrometry. J Proteome Res. 2012;6:3390–3404. doi: 10.1021/pr300205a. [DOI] [PubMed] [Google Scholar]
- 6.Loziuk PL, Sederoff RR, Chiang VL, Muddiman DC. Establishing ion ratio thresholds based on absolute peak area for absolute protein quantification using protein cleavage isotope dilution mass spectrometry. The Analyst. 2014;21:5439–5450. doi: 10.1039/c4an00567h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loziuk PL, Wang J, Li QZ, Sederoff RR, Chiang VL, Muddiman DC. Understanding the Role of Proteolytic Digestion on Discovery and Targeted Proteomic Measurements Using Liquid Chromatography Tandem Mass Spectrometry and Design of Experiments. J Proteome Res. 2013;12:5820–5829. doi: 10.1021/pr4008442. [DOI] [PubMed] [Google Scholar]
- 8.Loziuk PL, Parker J, Li W, Lin CY, Wang JP, Li QZ, Sederoff RR, Chiang VL, Muddiman DC. Elucidation of Xylem-Specific Transcription Factors and Absolute Quantification of Enzymes Regulating Cellulose Biosynthesis in Populus trichocarpa. J Proteome Res. 2015;10:4158–4168. doi: 10.1021/acs.jproteome.5b00233. [DOI] [PubMed] [Google Scholar]
- 9.Wang JP, Naik PP, Chen HC, Shi R, Lin CY, Liu J, Shuford CM, Li QZ, Sun YH, Tunlaya-Anukit S, et al. Complete Proteomic-Based Enzyme Reaction and Inhibition Kinetics Reveal How Monolignol Biosynthetic Enzyme Families Affect Metabolic Flux and Lignin in Populus trichocarpa. Plant Cell. 2014;3:894–914. doi: 10.1105/tpc.113.120881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang JP, Chuang L, Loziuk PL, Chen H, Lin YC, Shi R, Qu GZ, Muddiman DC, Sederoff RR, Chiang VL. Phosphorylation is an on/off switch for 5-hydroxyconiferaldehyde O-methyltransferase activity in poplar monolignol biosynthesis P. Natl Acad Sci USA. 2015;27:8481–8486. doi: 10.1073/pnas.1510473112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christensen JH, Bauw G, Welinder KG, Van Montagu M, Boerjan W. Purification and characterization of peroxidases correlated with lignification in poplar xylem. Plant Physiol. 1998;1:125–135. doi: 10.1104/pp.118.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hecht ES, McCord JP, Muddiman DC. A Quantitative Glycomics and Proteomics Combined Purification. Strategy. 2016;109:e53735. doi: 10.3791/53735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rath CM, Sweeney M, Schmidt B. Analysis of deamidation in monoclonal antibodies: Comparing peptide mapping using LC/MS and enzymatic isoaspartate detection using strong cation exchange chromatography. Abstr Pap Am Chem S. 2005:U120–U120. [Google Scholar]
- 14.Tretter V, Altmann F, Marz L. Peptide-N4-(N-Acetyl-Beta-Glucosaminyl)Asparagine Amidase-F Cannot Release Glycans with Fucose Attached Alpha-1-]3 to the Asparagine-Linked N-Acetylglucosamine Residue. Eur J Biochem. 1991;3:647–652. doi: 10.1111/j.1432-1033.1991.tb16166.x. [DOI] [PubMed] [Google Scholar]
- 15.Hao P, Ren Y, Alpert AJ, Sze SK. Detection, evaluation and minimization of nonenzymatic deamidation in proteomic sample preparation. Mol Cell Proteomics. 2011;10:O111 009381. doi: 10.1074/mcp.O111.009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao P, Ren Y, Datta A, Tam JP, Sze SK. Evaluation of the effect of trypsin digestion buffers on artificial deamidation. J Proteome Res. 2015;2:1308–1314. doi: 10.1021/pr500903b. [DOI] [PubMed] [Google Scholar]
- 17.Robinson NE, Robinson AB. Molecular clocks P. Natl Acad Sci USA. 2001;3:944–949. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nepomuceno AI, Gibson RJ, Randall SM, Muddiman DC. Accurate identification of deamidated peptides in global proteomics using a quadrupole orbitrap mass spectrometer. J Proteome Res. 2014;2:777–785. doi: 10.1021/pr400848n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez J, Takao T, Hori H, Besada V, Rodriguez R, Padron G, Shimonishi Y. A Method for Determination of N-Glycosylation Sites in Glycoproteins by Collision-Induced Dissociation Analysis in Fast-Atom-Bombardment Mass-Spectrometry - Identification of the Positions of Carbohydrate-Linked Asparagine in Recombinant Alpha-Amylase by Treatment with Peptide-N-Glycosidase-F in O-18-Labeled Water. Anal Biochem. 1992;1:151–158. doi: 10.1016/0003-2697(92)90592-u. [DOI] [PubMed] [Google Scholar]
- 20.Shuford CM, Muddiman DC. Capitalizing on the hydrophobic bias of electrospray ionization through chemical modification in mass spectrometry-based proteomics. Expert Rev Proteomics. 2011;3:317–323. doi: 10.1586/epr.11.24. [DOI] [PubMed] [Google Scholar]
- 21.Walker SH, Taylor AD, Muddiman DC. Individuality Normalization when Labeling with Isotopic Glycan Hydrazide Tags (INLIGHT): A Novel Glycan-Relative Quantification Strategy. J Am Soc Mass Spectr. 2013;9:1376–1384. doi: 10.1007/s13361-013-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hecht ES, McCord JP, Muddiman DC. Definitive Screening Design Optimization of Mass Spectrometry Parameters for Sensitive Comparison of Filter and Solid Phase Extraction Purified, INLIGHT Plasma N-Glycans. Anal Chem. 2015;14:7305–7312. doi: 10.1021/acs.analchem.5b01609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meitei NS, Apte A, Snovida SI, Rogers JC, Saba J. Automating mass spectrometry-based quantitative glycomics using aminoxy tandem mass tag reagents with SimGlycan. J Proteomics. 2015:211–222. doi: 10.1016/j.jprot.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 24.Peng B, Ahrends R. Adaptation of Skyline for Targeted Lipidomics. J Proteome Res. 2016;1:291–301. doi: 10.1021/acs.jproteome.5b00841. [DOI] [PubMed] [Google Scholar]
- 25.Liu SS, Chen X, Yan ZH, Qin SS, Xu JH, Lin JP, Yang C, Shui WQ. Exploring skyline for both MSE-based label-free proteomics and HRMS quantitation of small molecules. Proteomics. 2014;2–3:169–180. doi: 10.1002/pmic.201300352. [DOI] [PubMed] [Google Scholar]
- 26.Shi R, Sun YH, Li Q, Heber S, Sederoff R, Chiang VL. Towards a systems approach for lignin biosynthesis in Populus trichocarpa: transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol. 2010;1:144–163. doi: 10.1093/pcp/pcp175. [DOI] [PubMed] [Google Scholar]
- 27.Hecht ES, Scholl EH, Walker SH, Taylor AD, Cliby WA, Motsinger-Reif AA, Muddiman DC. Relative Quantification and Higher-Order Modeling of the Plasma Glycan Cancer Burden Ratio in Ovarian Cancer Case-Control Samples. J Proteome Res. 2015;10:4394–4401. doi: 10.1021/acs.jproteome.5b00703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holman JD, Tabb DL, Mallick P. Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Curr Protoc Bioinformatics. 2014;13(24):11–19. doi: 10.1002/0471250953.bi1324s46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoopmann MR, Finney GL, MacCoss MJ. High-speed data reduction, feature detection and MS/MS spectrum quality assessment of shotgun proteomics data sets using high-resolution mass. Spectrometry Anal Chem. 2007;15:5620–5632. doi: 10.1021/ac0700833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoopmann MR, MacCoss MJ, Moritz RL. Identification of peptide features in precursor spectra using Hardklor and Kronik. Curr Protoc Bioinformatics. 2012;(Unit13):18. doi: 10.1002/0471250953.bi1318s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;7:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Djerbi S, Lindskog M, Arvestad L, Sterky F, Teeri TT. The genome sequence of black cottonwood (Populus trichocarpa) reveals 18 conserved cellulose synthase (CesA) genes. Planta. 2005;5:739–746. doi: 10.1007/s00425-005-1498-4. [DOI] [PubMed] [Google Scholar]
- 33.Chou MF, Schwartz D. Biological sequence motif discovery using motif-x. Curr Protoc Bioinformatics. 2011;(Unit 13):15–24. doi: 10.1002/0471250953.bi1315s35. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz D, Chou MF, Church GM. Predicting protein post-translational modifications using meta-analysis of proteome scale data sets. Mol Cell Proteomics. 2009;2:365–379. doi: 10.1074/mcp.M800332-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;9:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin CY, Li QZ, Tunlaya-Anukit S, Shi R, Sun YH, Wang JP, Liu J, Loziuk P, Edmunds CW, Miller ZD, et al. A cell wall-bound anionic peroxidase, PtrPO21, is involved in lignin polymerization in Populus trichocarpa. Tree Genet Genomes. 2016;2 [Google Scholar]
- 37.CFG: Functional Glycomics Gateway: Consortium for Functional Glycomics. 2016. Glycan Structures Database. [Google Scholar]
- 38.Woodward JR, Craik D, Dell A, Khoo K-H, Munro SLA, Clarke AE, Bacic A. Structural analysis of the N-linked glycan chains from a stylar glycoprotein associated with expression of self-incompatibility in Nicotiana alata. Glycobiology. 1992;3:241–250. doi: 10.1093/glycob/2.3.241. [DOI] [PubMed] [Google Scholar]
- 39.Oxley D, Bacic A. Microheterogeneity of N-glycosylation on a stylar self-incompatibility glycoprotein of Nicotiana alata. Glycobiology. 1995;5:517–523. doi: 10.1093/glycob/5.5.517. [DOI] [PubMed] [Google Scholar]
- 40.Lommerse JPM, Thomas-Oates JE, Gielens C, Préaux G, Kamerling JP, Vliegenthart JFG. Primary Structure of 21 Novel Monoantennary and Diantennary N-Linked Carbohydrate Chains from αD-Hemocyanin of Helix Pomatia. Eur J Biochem. 1997;1:195–222. doi: 10.1111/j.1432-1033.1997.00195.x. [DOI] [PubMed] [Google Scholar]
- 41.Aguilera F, McDougall C, Degnan BM. Origin, evolution and classification of type-3 copper proteins: lineage-specific gene expansions and losses across the Metazoa. BMC Evol Biol. 2013;1:1–12. doi: 10.1186/1471-2148-13-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riboflavin Metabolism. 2015 http://www.genome.jp/kegg-bin/show_pathway?org_name=pop&mapno=00740&mapscale=&show_description=hide: KEGG Pathway 00740.
- 43.Isoquinoline alkaloid biosynthesis. 2014 http://www.genome.jp/kegg-bin/show_pathway?org_name=pop&mapno=00950&mapscale=&show_description=hide: KEGG Pathway 00950.
- 44.Ro DK, Mah N, Ellis BE, Douglas CJ. Functional characterization and subcellular localization of poplar (Populus trichocarpa × Populus deltoides) cinnamate 4-hydroxylase. Plant Physiol. 2001;1:317–329. doi: 10.1104/pp.126.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffmann L, Besseau S, Geoffroy P, Ritzenthaler C, Meyer D, Lapierre C, Pollet B, Legrand M. Silencing of hydroxycinnamoyl-coenzyme A shikimate/quinate hydroxycinnamoyltransferase affects phenylpropanoid biosynthesis. Plant Cell. 2004;6:1446–1465. doi: 10.1105/tpc.020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ceriotti A, Duranti M, Bollini R. Effects of N-glycosylation on the folding and structure of plant proteins. J Exp Bot. 1998;324:1091–1103. [Google Scholar]
- 47.Chen HC, Li QZ, Shuford CM, Liu J, Muddiman DC, Sederoff RR, Chiang VL. Membrane protein complexes catalyze both 4-and 3-hydroxylation of cinnamic acid derivatives in monolignol biosynthesis P. Natl Acad Sci USA. 2011;52:21253–21258. doi: 10.1073/pnas.1116416109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 (A) Venn diagrams showing the overlap of deamidated peptides identified by control (no enzymatic treatment) samples and treated (PNGase F or Glycosidase A) samples. (B) The number of deamidated peptides in the treated is further reduced by filtering out peptides which do not contain the NXS/T glycosylation motif
Oliveros, J.C. (2007–2015) Venny. An interactive tool for comparing lists with Venn’s diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html
Fig. S2 (A) The distribution of the glycan ratios calculated in Skyline or XCalibur are compared. (B) For eight glycans with mean abundance ratios greater than 0.98, the scan/RT ratios were calculated for each method. When pooled, the mean scan/RT ratio was 42.9 and 32.9 for XCalibur and Skyline method, respectively (p = 5.4 × 10−14)
Fig. S3 Confirmation of Glycans by Co-elution of Native/SIL Species in 1-to-1 Abundance, along with accurate mass and isotopic distribution in Skyline
For any complex-type glycan, selected diagnostic fragments are reported. In each MS/MS, the associated NAT or SIL tag was observed. For the sake of brevity, small saccharide ions in the MS/MS spectra that reveal little about complex structure were emitted, as were tables for glycans with whose structure may be completely inferred by biology (e.g. di- and tri- saccharides, N-glycan core structures). H10N2, H4N2F1X1, H6N5F2, and H5N4F1X1 were not selected for MS/MS