

Abstract Abstract
Rapid access to lung-derived cells from stable subjects is a major challenge in the pulmonary hypertension field, given the relative contraindication of lung biopsy. In these studies, we sought to demonstrate the importance of evaluating a cell type that actively participates in disease processes, as well as the potential to translate these findings to vascular beds in other nonlung tissues, in this instance perivascular skin mesenchymal cells (MCs). We utilized posttransplant or autopsy lung explant–derived cells (ABCG2-expressing mesenchymal progenitor cells [MPCs], fibroblasts) and skin-derived MCs to test the hypothesis that perivascular ABCG2 MPCs derived from pulmonary arterial hypertension (PAH) patient lung and skin would express a gene profile reflective of ongoing vascular dysfunction. By analyzing the genetic signatures and pathways associated with abnormal ABCG2 lung MPC phenotypes during PAH and evaluating them in lung- and skin-derived MCs, we have identified potential predictor genes for detection of PAH as well as a targetable mechanism to restore MPCs and microvascular function. These studies are the first to explore the utility of expanding the study of ABCG2 MPC regulation of the pulmonary microvasculature to the epidermis, in order to identify potential markers for adult lung vascular disease, such as PAH.
Keywords: mesenchymal progenitor cells, skin, lung, microvascular, pulmonary hypertension, idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease, BMPR2, Wnt signaling, LRP6, DKK1
Pulmonary vascular dysfunction or disease (PVD) is characterized by altered lung vascular structure and function. A significant loss of vascular-bed function, as seen in PVD, is thought to precede the clinical presentation of pulmonary hypertension (PH).1 PH is associated with a wide array of comorbid conditions, such as systemic sclerosis, chronic obstructive pulmonary disease (COPD), and pulmonary fibrosis, and also occurs as a primary PVD known as either idiopathic pulmonary arterial hypertension (IPAH) or heritable pulmonary arterial hypertension (HPAH).2-4 PH is characterized by elevated pulmonary artery pressures and widespread vascular remodeling, including endothelial cell dysfunction and occlusion or rarefaction of the peripheral pulmonary microvasculature.5-7 All forms of PH have a high mortality rate despite current therapeutic options. The current limited understanding of PVD as a predecessor to PH and lack of diagnostic approaches or criteria specific to preclinical PVD have hampered the study of the early stages of PVD in both rodent models and the clinical setting.
Approximately 80% of HPAH patients have a known mutation in the gene bone morphogenetic protein receptor type 2 (BMPR2). BMPR2 mutation–associated PAH is an autosomal dominant disease with low penetrance (approx. 20%); hence, not all mutation carriers develop PAH. In addition, approximately 20% of patients initially labeled as having IPAH also have a mutation in BMPR2 and thus heritable disease.8 In addition to genetic mutations, dysregulated BMPR2 signaling is strongly associated with the development of IPAH and other forms of PAH.9,10 Thus, impaired BMPR2 signaling is a common feature in PAH pathogenesis, although not the only feature; for example, mutations in caveolin 1 (CAV1), KCNK3, and other genes have also been identified.11 While BMPR2 is clearly related to PAH, other factors influence the disease onset, progression, and symptoms.
To date, the exact molecular mechanisms through which BMPR2 derangement promotes PVD and PH are unknown. Unfortunately, most genetic rodent models of PAH do not precisely recapitulate the disease pathology, displaying less substantial pulmonary vascular remodeling and inflammation.12 Alternative animal models have been used, such as monocrotaline injection, hypoxia, or the combination of a vascular endothelial growth factor (VEGF) receptor antibody and hypoxia. These toxin- or pharmacologically induced rodent models of PAH display substantial remodeling but are most likely the result of nonspecific activation of signaling networks by mechanisms that are not representative of the underlying causes of PAH, and they are complicated by the fact that the animal models will recover from these injuries.12 Because of the limitations of animal models, predictive biomarker and drug discovery efforts have thus far been of limited success.
Our previous work demonstrated that ABCG2-expressing mesenchymal progenitor cells (MPCs) are well poised to mediate vascular homeostasis, repair, and injury response in murine models of PAH and PH associated with fibrosis, as defined by leak, vessel loss, or muscularization.13-15 We have identified ABCG2 MPCs as a noncontractile pericyte precursor population. These MPCs support microvessels during homeostasis and contribute to remodeled microvasculature and parenchyma after injury.10,13,15 On the basis of this work, we theorize that during disease, ABCG2 MPCs influence microvessel function and remodeling. We therefore propose that ABCG2 lung MPCs are a target to identify underlying processes involved in the development of PVD and PAH/PH.
Rapid access to lung-derived cells from stable subjects is a major challenge in the PH field, given the relative contraindication of lung biopsy. To address this issue, we utilized lung explant-derived mesenchymal cells (ABCG2 MPCs, fibroblasts) and skin-derived mesenchymal cells (MCs). Our current approach allowed us to test the hypothesis that perivascular ABCG2 MPCs derived from PAH-patient lung and skin would express a gene profile reflective of deregulated bone morphogenetic protein (BMP) signaling and ongoing vascular dysfunction (Fig. 1). In these studies, we sought to demonstrate the importance of evaluating a cell type that actively participates in disease processes versus a bystander, as well as the potential to translate these findings to vascular beds in other nonlung tissues, in this instance perivascular skin MCs. We chose to evaluate PAH MPCs because of the association of both IPAH and HPAH with deregulated BMPR2 signaling. By analyzing the genetic signatures and pathways associated with abnormal ABCG2 lung MPC phenotypes during PH and evaluating them in lung- and skin-derived MCs, we have identified genes associated with PVD. These genes may next be evaluated in more accessible tissue, such as plasma, for their utility as biomarkers or predictor genes. In addition, these studies have highlighted DKK1-LRP6 signaling as a targetable mechanism to restore MPC and microvascular function.
Figure 1.
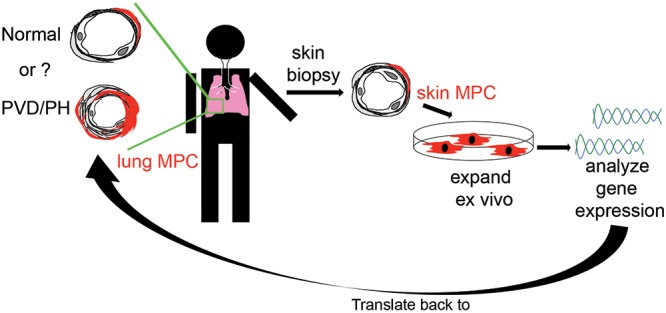
Schematic hypothesis. Perivascular ABCG2-expressing MPCs derived from PAH patient lung and skin express a common gene signature reflective of ongoing vascular dysfunction. MPCs: mesenchymal progenitor cells; PAH: pulmonary arterial hypertension; PH: pulmonary hypertension; PVD: pulmonary vascular disease.
Methods
Isolation and characterization of primary ABCG2 lung and skin MPCs
Human lung plastic adherent cells were isolated from explant lung tissue after autopsy or transplant by collagenase digest (Vanderbilt Institutional Review Board [IRB] protocol 9401) to form a suspension. The cells were stained with antibodies to sort CD45negative ABCG2 cells (lung MPCs) in a BD FACSAria III (BD Biosciences, San Jose, CA). Fluorescent minus one (FMO) and IgG2b isotype (eBioscience, San Diego, CA, catalog no. 12–8888–82) controls were used to set the ABCG2-PE gates. DAPI (4′,6-diamidino-2-phenylindole) was used to exclude dead cells. The compensation controls were established as cells only, cells + DAPI, cells + APC-CD45 antibody, and cells + PE-ABCG2 antibody; alternatively, compensation beads were used. The gating strategy routinely included forward-scatter/side-scatter (FSC/SSC), single cells gated by SSC-Width (SSC-W)/SSC-Height (SSC-H), FSC-W/FSC-H, DAPI+Ter119 to gate out dead and red blood cells followed by gating on the CD45negative population. The sort sample consisted of cells + DAPI + APC-CD45 antibody + PE-ABCG2 antibody. A summary of human MPC lines and characterization is presented in Table 1.
Table 1.
Cell surface determinant expression by human lung and skin MPCs
| MPC | Age, years | Sex | CD44 | CD73 | CD105 | CD106 | CD146 | CD140b | CD14 | CD31 | CD34 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Human lung MPCs | |||||||||||
| Control 1 | 57 | M | 99.0 | 99.4 | 98.6 | 6.5 | 53.2 | 90.4 | 0 | 0.1 | 0 |
| Control 2 | Unknown | M | 99.8 | 100 | 99.9 | 0.44 | 33.1 | 99.9 | 0 | 0 | 0 |
| Control 3 | 66 | F | 98.9 | 99.9 | 99.9 | 0.46 | 31.3 | 99.5 | 0 | 0 | 0 |
| Control 4 | 35 | M | 100 | 100 | 100 | 3.73 | 5.31 | 95 | 0 | 0 | 0 |
| HPAH | 12 | F | 100 | 100 | 100 | 7.64 | 63.1 | 90.2 | 0 | 0 | 0 |
| IPAH 1 | 32 | F | 99.1 | 99.7 | 83.6 | 0.72 | 53.8 | 97 | 0 | 0.025 | 0.024 |
| IPAH 2 | Unknown | F | 99.9 | 99.6 | 96.9 | 9.74 | 72.3 | 93.2 | 0.015 | 0 | 0.015 |
| Human skin MPCs | |||||||||||
| Control 1 | 45 | F | 99.9 | 100 | 100 | 0.19 | 46.9 | 98.7 | 0 | 0 | 0.4 |
| Control 2 | 40 | F | 99.9 | 95.4 | 100 | 0.13 | 48.5 | 99.9 | 0.1 | 0 | 0.1 |
Unless otherwise specified, data are percentages. HPAH: heritable pulmonary arterial hypertension; IPAH: idiopathic pulmonary arterial hypertension; MPC: mesenchymal progenitor cell.
Identification and localization of ABCG2 skin MPCs
All procedures and protocols were approved by the Institutional Animal Care and Use Committee at Vanderbilt University. Mice were on a C57Bl6/B129 background. ABCG2-CreERT2 mice, obtained in collaboration with B. P. Sorrentino,16 were crossed to a fluorescent eGFP (enhanced green fluorescent protein) reporter ((Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP) labeled as Rosa26mtomato/mGFPlox-stop [reporter mice; Jackson Laboratory, stock no. 007676], or Rosa26mT/mG) strain to facilitate lineage-tracing analysis. Mice were injected intraperitoneally at 8–10 weeks of age with 0.5 mg tamoxifen in a single dose, as previously described.15
Patient skin fibroblast isolation and identification and characterization of the BMPR2 mutation
The subjects were recruited through the Vanderbilt Pulmonary Hypertension Center. The Vanderbilt University Medical Center IRB approved all study protocols (protocol 9401). All participants gave informed written consent to participate in genetic and clinical studies and underwent genetic counseling in accordance with the guidelines of the American College of Chest Physicians.17 The PAH phenotype was defined according to accepted international standards of diagnosis, as previously described.10,18,19 Skin biopsy specimens were obtained via a sterile 3-mm punch skin biopsy. Primary skin fibroblasts were cultured and sequenced as previously reported.10,20 A summary of human skin mesenchymal lines and characterization is presented in Tables 1 and 2.
Table 2.
Characteristics of skin mesenchymal cells
| Human skin FBs | Age, years | Sex | Mutation |
|---|---|---|---|
| Control 1 | 35 | M | None |
| Control 2 | 26 | F | None |
| Control 3 | 64 | M | None |
| Control 4 | 46 | F | None |
| Control 5 | 40 | F | None |
| Unaffected BMPR2 mutant 1 | 41 | M | c.3354T>G p.C118W |
| Unaffected BMPR2 mutant 2 | 62 | F | c.2504delC p.T835fs |
| Unaffected BMPR2 mutant 3 | 58 | M | BMPR2 c.G350A |
| Unaffected BMPR2 mutant 4 | <1 | ND | BMPR2 c.354T>G |
| HPAH BMPR2 mutant 1 | 18 | F | c.3354T>G p.C118W |
| HPAH BMPR2 mutant 2 | 41 | M | BMPR2 c.354T>G |
| HPAH BMPR2 mutant 3 | 32 | F | BMPR2 cG350A |
| IPAH 1 | 30 | F | None detected |
| IPAH 2 | 64 | F | None detected |
| IPAH 3 | 43 | F | None detected |
| HPAH CAV1 mutant 1 | 72 | M | CAV1 c.474delA p.P158Pfsx22 |
| HPAH CAV1 mutant 2 | 71 | F | CAV1 c.474delA p.P158Pfsx22 |
| HPAH CAV1 mutant 3 | 25 | M | CAV1 c.474delA p.P158Pfsx22 |
FB: fibroblast; HPAH: heritable pulmonary arterial hypertension; IPAH: idiopathic pulmonary arterial hypertension; mut: mutant; ND: not determined.
Transcriptome analysis
Array analysis and qRT-PCR (quantitative reverse transcription polymerase chain reaction) validation were performed as described previously.13,14 Briefly, total RNA was prepared with Qiagen RNA Isolation Kit reagents (Qiagen, Valencia, CA) for total RNA isolation and analysis of gene expression. The qRT-PCR assays were performed in triplicate, and levels of analyzed genes were normalized to hypoxanthine phosphoribosyltransferase (HPRT) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) abundance (the primer list is provided in Table S1). Complimentary DNA (cDNA) generated from amplified RNA was hybridized to duplicate Affymetrix (Santa Clara, CA) Human Gene 1.0 ST chips.
Gene ontology groups were analyzed and compiled with Webgestalt (Vanderbilt University); heat maps with JMP, version 9 (JMP9); and the correlation plots with Microsoft Excel. Statistics were calculated with JMP9.
Western blot analysis
Total-protein extracts were made by scraping cells in radioimmunoprecipitation assay buffer (Cell Signaling, Boston; catalog no. 9806S) containing protease and phosphatase inhibitors (ThermoFisher Scientific, Waltham, MA; catalog no. 78444). After determination of protein concentrations and standardization, cell lysates were mixed with reducing agent and an equal volume of Laemmli sodium dodecyl sulfate (SDS) loading buffer, resolved on 4%–12% polyacrylamide-SDS gels, and transferred to polyvinylidene difluoride membranes. The blots were blocked with Tris-buffered saline containing 5% fetal bovine serum and 0.1% Tween 20 and then treated with antibodies that detect the target proteins, as labeled in Figures 5 and 7, overnight at 4°C. The blots were washed and subsequently treated with appropriate secondary antibodies conjugated to horseradish peroxidase. After the blots were washed, specific immune complexes were visualized with SuperSignal West Pico chemiluminescent substrate (Table S1). Where membrane and cytoplasmic fractions were probed, protein extracts were made with the Mem-PER Plus kit (ThermoFisher Scientific, catalog no. 89842).
Figure 5.

DKK1 regulates gene expression in isolated PAH patient BMPR2-mutant cells. A, Western blot analysis was performed with cell lysates from lung MPCs to detect DKK1 and LRP6 levels. B, C, Densitometry was performed and normalized to β-actin housekeeping levels. Three control, 2 BMPR2-mutant HPAH, and 4 IPAH patient lung MPC lines were analyzed per group. D, E, Modulation of DKK1 signaling was performed by stimulation with DKK1 treatment (100 ng/mL) or DKK1 inhibition with the antagonist gallocyanine (NCI8642; 100 μm). The resulting gene expression was analyzed after 48 hours. A qRT-PCR analysis was performed to analyze changes in gene expression by control or BMPR2-mutant HPAH lung MPCs. In addition to that of LRP6 and DKK1, expression of genes associated with pericyte differentiation (ACTA2, CSPG4) and matrix production (COL1A1, COL3A1, FN1) was also analyzed. All amplification was normalized to a housekeeping gene, and the results are presented as mean fold change over untreated controls ± standard error. *P < 0.5; **P < 0.01. F, Schematic representation of the relationship between DKK1 and LRP6. HPAH: heritable PAH; HuLung: human lung; IPAH: idiopathic PAH; MPC: mesenchymal progenitor cell; mut: mutant; PAH: pulmonary arterial hypertension; qRT-PCR: quantitative reverse transcription polymerase chain reaction; UT: untreated; Veh: gallocyanine vehicle 1N NH4OH.
Figure 7.

Analysis of targeted gene expression profiles of lung MPCs from COPD and IPF patients highlight that the DKK1 pathway is affected in multiple chronic pulmonary diseases. A. A qRT-PCR analysis was performed to evaluate the patterns of representative genes expressed by lung MPCs from adult chronic lung disease, including COPD and IPF, relative to control and PAH samples; n = 3 patients for each group. All amplification was normalized to a housekeeping gene, and the results presented as mean fold change over control ± standard error. *P < 0.5. B–F, Representative bright-field images of immunohistochemical localization of DKK1. Scale bar: 100 μm. G, Western blot analysis using cell lysates from lung MPC to detect DKK1 levels. Densitometry was performed and normalized to β-actin housekeeping levels. Sample: 3 control and 3 IPF MPCs. Data presented as the mean ± standard error. COPD: chronic obstructive pulmonary disease; IPF: idiopathic pulmonary fibrosis; MPCs: mesenchymal progenitor cells; PAH: pulmonary arterial hypertension; qRT-PCR: quantitative reverse transcription polymerase chain reaction.
Modulation of DKK1 activity in isolated human lung MPCs
Modulation of DKK1 signaling was performed by stimulation with DKK1 treatment or DKK1 inhibition with gallocyanine. Control and PAH human lung MSCs were plated at 50,000 cells per well with the following treatment conditions: untreated, 1N ammonium hydroxide (Sigma-Aldrich, St. Louis, MO, catalog no. 318612), 100 μM gallocyanine (Sigma-Aldrich, catalog no. 124508), and 100 ng/mL DKK1 (OriGene, Rockville, MD, catalog no. TP723065). After 48 hours, cells were washed with 1× Dulbecco’s phosphate-buffered saline (Gibco, by Life Technologies, Carlsbad, CA, catalog no. 14190–144) and lysates were collected. RNA was isolated from the cell lysates with the RNeasy Mini Kit (Qiagen, catalog no. 74106). The cDNA was synthesized from total RNA with the QuantiTect Reverse Transcription Kit (Qiagen, catalog no. 205311). The qRT-PCR validation was performed, and the level of gene expression of analyzed genes compared to HPRT or GAPDH was assessed (Table S1).
Detection of DKK1 in human lung tissue specimens
Human tissue was obtained from PAH patients after approval from the Vanderbilt University IRB (3 controls, 3 PAH patients with different mutations, and 3 IPAH patients). These samples originated from postautopsy specimens. Sections of patient lung tissue were evaluated by antibody staining for the presence of the secreted Wnt inhibitor DKK1, with DAB (diaminobenzidine) detection. Images were captured with a Nikon Eclipse 90i/DSFi-1 using NIS Elements software.
Statistical analysis
Data were analyzed by 1-way ANOVA followed by Tukey HSD (honest significant difference) post-hoc analysis in JMP, version 5.0.12. Murine qRT-PCR data were analyzed by 1-way ANOVA and a Tukey post hoc test with JMP, version 5.0.12. Patient samples were analyzed with the nonparametric Wilcoxon/Kruskal-Wallis test and a χ2 approximation. Significance was defined as P < 0.05; results with P < 0.01 are also indicated in the figures.
Results
Identification of a diagnostic pattern of gene expression in lung ABCG2 MPCs during PVD/PAH
We are focused on identifying underlying mechanisms by which MPCs regulate pulmonary microvascular stability and dysfunction leading to the development of PVD associated with chronic diseases, including PAH. The novel regulatory factors and pathways involved in this process may then be evaluated as potential biomarkers to enhance diagnosis of disease before the onset of clinical symptoms, when the pulmonary vascular bed has been significantly compromised. We initially compared the global gene expression patterns of primary ABCG2 MPCs isolated from control and PAH patients (Fig. 2A; Table S2). Genes were selected for validation by qRT-PCR on the basis of a 2-fold or greater change in expression, compared to controls, and their gene ontology association with vascular processes, as well as the BMPR2 or Wnt signaling pathways (Fig. 2B; Table 3). We analyzed the additional targets WISP1 and LRP6 because they are a Wnt target gene that regulates BMP2 activity and a Wnt coreceptor, respectively.31,32 NEO1 demonstrated increased expression, along with PEAR1, WISP1, LRP6, and SPON2.21,22 DKK1, PDK4, RGS5, and PTGS2 showed decreased expression. We have validated a set of genes with differential expression between control and PAH ABCG2 lung MPCs.
Figure 2.

Global gene expression profile of PAH-patient MPCs relative to those of controls. A, Heat map analysis of gene segregation; red and green indicate high and low levels of expression, respectively. B, C, A qRT-PCR analysis was performed to validate the patterns of representative genes expressed by ABCG2-expressing lung MPCs (B) and lung fibroblasts (C). Samples: 3 control and 3 PAH patients. All amplification was normalized to a housekeeping gene, and the results are presented as mean fold change over control ± standard error. FB: fibroblasts; hPAH/HPAH: heritable PAH; iPAH/IPAH: idiopathic PAH; Hu: human; MPCs: mesenchymal progenitor cells; PAH: pulmonary arterial hypertension; qRT-PCR: quantitative reverse transcription polymerase chain reaction; PVOD: pulmonary veno-occlusive disease; WT: wild-type control. *P < 0.5; **P < 0.01.
Table 3.
Significance of changes in gene expression in PAH MPCs versus control
| PAH versus control | |||
|---|---|---|---|
| Gene symbol | Adjusted P value | Fold change | Comments |
| SPON2 | 0.0386202 | 2.16564 | LRP6 ligand, canonical Wnt activator21,22 |
| PEAR1 | 0.00635016 | 2.12031 | Platelet endothelial aggregation receptor 1, platelet aggregation23 |
| TNC | 0.0348311 | 2.2882 | Matrix, increased in PAH24 |
| NEO1 | 0.0329007 | 2.37015 | BMP receptor, neogenin25 |
| DKK1 | 0.0201554 | −2.27936 | Dickkopf1, a canonical Wnt inhibitor and LRP6 ligand26 |
| PDK4 | 4.00E−05 | −2.98576 | Wnt gene target and regulator of metabolism, pyruvate dehydrogenase kinase-isozyme 427,28 |
| WLS | 0.0328988 | −2.07564 | Members or targets of Wnt signaling |
| RGS5 | 0.00629714 | −3.45307 | Regulator of G-protein signaling 5, vascular tone29,30 |
| PTGS2 | 0.040118 | −5.5725 | Cyclooxygenase-2/cox2, vascular tone29,30 |
BMP: bone morphogenetic protein; PAH: pulmonary arterial hypertension.
Previous gene array studies have utilized mixed and heterogenous populations of lung cells/tissue in which subtle but important genetic changes may be masked. In order to identify whether the selected gene expression patterns identified were universal to all pulmonary MCs, we compared gene expression of heterogenous lung fibroblasts to the specific and enriched subpopulation of ABCG2 MPCs isolated from the same patient. WISP1, NEO1, SPON2, and PDK4 exhibited the same trends in gene expression as in the MPCs (Fig. 2C). However, RGS5, LRP6, and DKK1 were unchanged and did not resemble the MPC profile. These data illustrate differences in gene expression profiles between mesenchymal subpopulations within the lung and the importance of studying subpopulations to understand signaling mechanisms unique to the specific function of each population.
The diagnostic pattern of gene expression in ABCG2 MPCs extends to skin
Our previous studies have demonstrated that ABCG2 lung MPCs are a perivascular pericyte precursor in the alveolar-capillary network of both murine and human lung.13-15 We recently reported that induced pluripotent stem cell–derived cells from PAH patients, as well as skin fibroblasts (FBs) and lung MPCs, exhibited common genetic signatures associated with PAH.10 In addition, unlike lung FBs, skin MCs form colony-forming unit FBs, a characteristic of MPCs. This may be attributed to the skin FB isolation procedure that enriches for cells that migrate from biopsies to plastic. Skin FBs are an accessible patient source of perivascular and other MCs, ideal for cell isolation and gene expression studies. Microvascular pathology in both lung and skin is also present in systemic sclerosis, which suggests that the skin-derived MCs may share similar gene signatures with lung MPCs and therefore may be exploited to identify changes in the pulmonary vascular bed as well as tissue-specific patterns of protein expression.4,33-36
In order to identify whether the selected gene expression patterns identified were recapitulated in skin MCs, we analyzed gene expression of human skin FBs. Cells were isolated from 5 groups: controls, patients with known BMPR2 mutations but not PAH, patients with known BMPR2 mutations and PAH, patients with IPAH, and patients with CAV1 mutations and PAH. Gene expression trends for LRP6 were the same as those described for lung MPCs (Fig. 3A); however, while DKK1 expression in the CAV1 mutants resembled that in lung MPCs, IPAH cells demonstrated increased levels of expression. WISP1 and RGS5 expression did not change in the known-BMPR2-mutation-with-PAH group versus controls. WISP1 expression did, however, significantly increase in the CAV1-mutation PAH group. NEO1 expression was variable in the skin cells. Interestingly, PDK4 gene expression was significantly increased in the known-BMPR2-mutation-with-PAH group versus controls, opposite of the lung MPCs. Immunofluorescent staining to detect DKK1 and PDK4 in control versus HPAH skin and lung cells suggested differences in protein localization, with the HPAH cells exhibiting increased perinuclear DKK1 and PDK4 protein (Fig. 3B).
Figure 3.

Analysis of targeted gene expression profiles of skin mesenchymal cells from PAH BMPR2 and CAV1 mutation carriers relative to wild-type individuals (i.e., noncarriers of mutations) and unaffected mutation carriers. A, A qRT-PCR analysis was performed to validate the patterns of representative genes expressed by skin MCs; n = 3 patients for each group. The mean of combined patient samples per group and results for individual samples are presented. All amplification was normalized to a housekeeping gene, and the results are presented as mean fold change over control ± standard error. B, Representative immunofluorescent images of DKK1 and PDK4 localization in skin MCs and lung MPCs. Scale bar: 100 μm. DAPI: 4′,6-diamidino-2-phenylindole; HPAH: heritable PAH; IPAH: idiopathic PAH; MCs: mesenchymal cells; MPCs: mesenchymal progenitor cells; mut: mutant; PAH: pulmonary arterial hypertension; qRT-PCR: quantitative reverse transcription polymerase chain reaction.
Skin MCs, similar to those in the lung, can be further enriched with ABCG2 to isolate MPCs with clonogenic potential similar to that of lung MPCs (Fig. 4A, 4B; Tables 1, 2). Lineage analysis shows that murine skin ABCG2 MPCs, like murine lung MPCs, localize to a perivascular niche (Fig. 4C–4K). Global gene expression analysis was performed to characterize lung versus skin MPC tissue-specific patterns of gene expression. To identify similarities between lung and skin MPCs, MPC samples were compared against lung FBs. Fold change of lung MPCs was plotted against that of skin MPCs. A Pearson correlation value of r2 = 0.7599417 was obtained for the values of the 356 genes that identified similarities between the MPC populations (Fig. 4L; Table S3). Hierarchical clustering of gene expression, selecting differences of more than 1.5-fold with a P value of 0.05, identified 88 differentially expressed genes (Fig. 4L; Table S4). Tissue-specific differences in genes associated with BMP and Wnt signaling, as well as with angiogenesis, were also identified (Fig. 4M–4O). Both DKK1 and LRP6 were differentially expressed.
Figure 4.

Identification of a perivascular skin MPC population. A, Lung fibroblasts were isolated from explanted human lung or skin tissue via collagenase digest to form a cell suspension. Adherent cells were plated and expanded for 2 passages. At this time, flow cytometric analysis was performed on single-cell suspensions of human lung tissue to detect CD45negative ABCG2-expressing (horizontal axis) cells. B, Representative Giemsa-stained colony-forming unit fibroblast analysis confirmed the clonogenic potential of the ABCG2-enriched populations. C–K, Lung or epidermal ear and dorsal ABCG2 MPCs targeted recombination in vivo. Recombination following administration of low-dose tamoxifen (0.5 mg) resulted in the appearance of eGFP expression, detectable by immunofluorescent staining (green). The eGFP-expressing MPCs were localized in or adjacent to SMA-positive microvessel layers (red). Scale bar: 50 mm. L, Heat map analysis of gene similarities and segregation between control skin and lung MPCs; yellow and red indicate high and low levels of expression, respectively. M–O, Probes belonging to gene ontology functional categories were selected from the whole expression data of the lung and skin MPC groups, and corresponding heat maps of expression data were constructed. A self-contained gene set test, ROAST, was employed to test for significant change in expression of sets (BMP signaling: P < 0.004; Wnt signaling: P < 0.006; angiogenesis: P < 0.01). Representative gene changes of >1.5-fold with P < 0.05 are depicted. eGFP: enhanced green fluorescent protein; FB: fibroblasts; huLung: human lung; huSkin: human skin; MC: mesenchymal cells; MPC: mesenchymal progenitor cells; SMA: smooth muscle actin; SSC-A: side-scatter.
Targeting of the DKK1-LRP6 axis in BMPR2-mutant PAH MPCs
Wnt signaling is regulated by BMP signaling during lung development and disease.13,37-40 Our recently published work showed that decreased BMPR2 signaling increased Wnt/β-catenin signaling in human PAH MPCs as well as in murine control and human wild-type (i.e., nonmutant) MPCs after BMPR signaling inhibition.10 DKK1 is a potent canonical Wnt signaling inhibitor as well as an activator of noncanonical Wnt.32,41 Because DKK1’s differential expression, as well as that of its receptor LRP6, was maintained in MPCs from both lung and skin, we next analyzed protein expression as well as targeted activation or inhibition of DKK1. Quantitative Western blot analysis detected increased levels of DKK1 and decreased levels of LRP6 protein in lung MPC, in a reciprocal trend likely attributable to LRP6 internalization in the presence of DKK1 (Fig. 5A–5C).
To examine the effects of DKK1 on MPCs, we exposed MPCs to recombinant protein and analyzed the gene expression levels of DKK1, LRP6, and the pericyte/VSMC (vascular smooth muscle cell) markers ACTA2 (smooth muscle alpha actin) and CSPG4 (NG2) as well as the matrix protein genes COL1A1, COL3A1, and FN1. Interestingly, treatment of control or BMPR2-mutant PAH MPCs with recombinant DKK1 revealed that only the PAH MPCs responded with increased gene expression characteristic of a remodeling or a myofibroblast transition (Fig. 5D).
To test the effect of the converse, we treated the control or BMPR2-mutant PAH MPCs with a DKK1 antagonist, gallocyanine,32 and decreased the expression of all the genes, with the exception of LRP6. Taken together these results suggest that decreased BMPR2 signaling inherently alters the PAH MPCs, driving an abnormal response to DKK1, and that this pathway is targetable.
Immunostaining was performed to detect and localize DKK1 in PAH patient lung tissue sections. DKK1 localized to alveolar epithelium, while the smooth muscle layer of the vasculature appeared negative in control lung tissue samples (Fig. 6A–6D). PAH lung tissue exhibited increased levels of DKK1 localization to the smooth muscle and perivascular regions, independent of BMPR2 mutation type (Fig. 6E–6J).
Figure 6.

Representative bright-field images of immunohistochemical localization of DKK1. DKK1 localization increased in the vasculature of PAH patients, independent of mutation status. Patient lung tissue was analyzed by immunostaining of paraffin-embedded lung sections with DAB detection (black). A–D, DKK1 localized to alveolar epithelium and a few endothelial cells in control sections. E–J, DKK1 was present with increasing intensity in the smooth muscle layers of PAH tissue relative to control. Sample: 3 controls, 3 HPAH patients, 3 IPAH patients. Scale bar: 100 μm. DAB: diaminobenzidine; HPAH: heritable PAH; IPAH: idiopathic PAH; mut: mutant; PAH: pulmonary arterial hypertension.
Pathological changes in the DKK1-LRP6 axis are present in human PVD associated with chronic lung disease
PH complicates many adult chronic lung diseases, so we next analyzed whether lung MPCs from COPD and idiopathic pulmonary fibrosis (IPF) tissue displayed trends in gene expression similar to those in the PAH MPCs. Interestingly, PDK4 and RGS5 expression was decreased in all MPCs evaluated (Fig. 7A). DKK1 levels were decreased in IPF and PAH but increased in COPD tissue. LRP6 and SPON2 demonstrated increasing trends in expression relative to controls. DKK1 expression and localization were examined by immunostaining. Similar to the finding in PAH tissues, DKK1 localized to the alveolar epithelium, and increased levels of DKK1 staining intensity were detected as well as increased localization to the smooth muscle and perivascular regions (Fig. 7B–7F). Western blot analysis of IPF lung MPCs suggest a trend in increased DKK1 protein levels with disease (Fig. 7G). These results suggest that the DKK1-LRP6 pathway may be affected in PVD associated with multiple adult chronic lung diseases.
Discussion
Our studies utilizing primary human ABCG2 lung MPCs successfully identified gene changes characteristic of PAH that can also be detected in lung and skin MCs. The underlying concept is that there are common gene expression patterns and signaling pathways during lung vascular disease that are echoed throughout the body, common to specific cell types (Fig. 1). The altered gene expression patterns of DKK1 and LRP6 were highlighted as potential markers of PVD and PAH in skin MCs. Manipulation of the DKK1-LRP6 interaction in vitro demonstrated that increasing DKK1 exacerbated a pro-remodeling or myofibroblast-like lung MPC phenotype only in the presence of BMPR2 mutation. These studies provide the first documentation that these novel targets, DKK1 and LRP6, may be useful as markers of PVD and that their manipulation may be useful for restoration of ABCG2 lung MPCs as well as pulmonary microvascular function in vivo.
Our data show that the differential expression of a few genes by perivascular lung MPCs associated with PVD could also be detected in perivascular skin MCs. In light of these results, it is interesting to consider the possibility of utilizing skin cells as a reservoir for information regarding the function of the pulmonary vasculature if plasma samples do not recapitulate a vascular-associated gene expression profile. DKK1 gene expression was decreased in both lung MPCs and skin MCs (BMPR2 and CAV1 mutants), while protein levels were increased during PAH. The opposite was detected for LRP6, the Wnt ligand coreceptor and receptor for DKK1. The expression trend for LRP6 was recapitulated in IPAH and CAV1-mutant skin MCs. The apparent difference between the DKK1 gene expression in IPAH relative to that in HPAH and CAV1-mutant cells may be attributed to differences in the molecular mechanisms of disease. For example, differences between PAH samples may be attributed to patient-specific genetic modifiers, inherent differences in Wnt ligand–mediated canonical or noncanonical signaling activity, LRP6 activity, and subcellular localization as well as variable levels of BMPR2 signaling and SMAD activity. BMPR signaling through SMADs 1/5/8 has been shown to regulate transcription of Dkk1 during patterning in development.41-46 PDK4 can also directly phosphorylate SMADs 1/5/8 and regulate their activity.47 Because deregulated or reduced BMP signaling is a common feature of PAH,8,48 it is not surprising that the gene expression trends would be similar in the skin MCs from BMPR2-mutant, CAV1-mutant, and IPAH patients.
DKK1 is also a TCF/β-catenin target gene that inhibits Wnt signaling via binding the Wnt/frizzled coreceptor LRP6, preventing ligand activation of the canonical/β-catenin pathway.26,32,41,49 DKK1 binding to LRP6 may also decrease Wnt/β-catenin signaling via stimulation of receptor internalization,26,32,49,50 which is regulated by CAV1, a mutation associated with HPAH.51,52 Interestingly, DKK activity is also regulated by hormone levels, including aldosterone, estrogen, and parathyroid hormone, which is particularly important, given that PAH affects predominantly females. Aldosterone levels regulate DKK1 expression, which was then identified as a modifier gene for potassium channel function, specifically KCNK3, during the development of endocrine secondary arterial hypertension.53 KCNK3 mutation is also associated with the development of HPAH.8 Estrogen levels regulate Dkk1 expression, important in instances of neuroprotection and bone resorption,54,55 possibly through an interaction between estrogen receptor/β-catenin nuclear complex.56 In support of our results, DKK1 expression has been linked to mutations associated with the development of HPAH.
Another important observation from these studies is the increased localization of DKK1 to lung microvessels and its decreased gene expression during PAH as well as IPF and COPD. In terms of the vasculature, DKK1 negatively regulates angiogenesis and neovascularization during lung development, in normal tissue repair, and in tumors.57-59 Our results further define the importance of DKK1 in PAH by revealing that only in the presence of BMPR2 mutation did DKK1 treatment increase gene expression characteristic of the pro-remodeling lung MPCs, including increased expression of ACTA2 and matrix proteins. DKK1 also controls differentiation of adipocytes and influences metabolism via upregulation of peroxisome proliferator–activated receptor γ (PPARγ) and C/EBPα and inhibition of β-catenin.60 We found that modulation of DKK1 levels regulated the expression of PDK4 in BMPR2-mutant lung MPCs. PDK4 maintains energy homeostasis by regulating the transition between glucose and fatty acid utilization.27 Its expression also regulates blood glucose levels and glucose tolerance,61 which are altered in patients with PAH.62 Gene expression of PDK4 was decreased in lung MPCs from PAH, IPF, and COPD patients and was increased in BMPR2-mutant skin MCs, suggesting a lung-specific vascular metabolic phenotype linked to altered BMPR2 signaling. Taken together, abnormal DKK1 expression has been associated with vascular disease and pathology as well as altered metabolism, features characteristic of PAH.
In this regard, it is interesting to note that inhibition of DKK1 signaling reversed the BMPR2-mutant lung MPC pro-remodeling phenotype by downregulation of gene expression, demonstrating that this pathway is targetable in PAH. The small molecule NCI8642/gallocyanine efficiently displaces DKK1 from LRP6 and blocks DK1 inhibition of canonical Wnt signaling.32 Interestingly, PAH lung MPCs, but not skin MPCs, expressed increased levels of the DKK1 inhibitor RSPO2. RSPO2, or r-spondin 2, also activates canonical Wnt signaling by inhibiting the DKK1-LRP6 interaction.21 The elevated expression of RSPO2 has been associated with transformation of cells in cancer.63 Increased DKK1 and LRP6 activity has been associated with the development and poor prognosis of cancers.32,64 Both molecules are currently under study as therapeutic targets. Current systemic inhibition of Wnt signaling has negative side effects, highlighting the need for the identification and development of novel targets and inhibitors as well as an understanding of the cell-specific variations in DKK1-LRP6-regulated Wnt signaling, as a result of decreased BMPR2 signaling, even within the pulmonary vasculature.
Cell-specific variations in signaling and contradictory reports as to the effects of Wnt signaling on cell phenotype and function are likely the products of cellular and environmental context as well as the multifunctional roles many proteins exert.65,66 DKK1 and LRP6 are regulators of canonical Wnt signaling, but they may also induce noncanonical Wnt signaling pathways,67,68 both being implicated in the pathology of PAH.10,13,15,69,70 To date, we do not understand why carriers of mutations in BMPR2 and other genes (e.g., CAV1) develop PAH; however, a common underlying feature is decreased BMPR2 signaling activity.8 The intimate associations between the BMPR2 and Wnt signaling pathways, as well as those between BMPR2, DKK1, and LRP6 in PVD, cancer, and development, are an indication that they are likely important in both microvascular repair and the transition to a disease state. However interesting, these studies have been limited by the number and diversity of available patient samples. Ideally, future studies will test whether peripheral blood mononuclear cell expression of PDK4 and DKK1 or circulating plasma DKK1 levels may be used as a biomarker for PVD in PAH, as well as other chronic lung diseases, in a large cohort of patient samples.
In summary, by analyzing the genetic signatures and pathways associated with abnormal ABCG2 lung MPC phenotypes during PAH and evaluating them in lung- and skin-derived MCs, we have identified potential predictor genes for detection of PAH: DKK1, LRP6, and PDK4. We have also identified the DKK1-LRP6 interaction as a targetable mechanism to restore MPC and microvascular function. Our ability to manipulate the LRP6-DKK1 axis in lung MPCs, both in vitro and in vivo, will allow us to substantiate this hypothesis. Broadly, understanding how the elements of MPC and microvascular dysfunction in mutation carriers and idiopathic patients are regulated may have direct relevance to the identification and validation of predictive markers of disease. For example, circulating DKK1 levels have previously been evaluated as biomarkers for cancer as well as coronary atherosclerosis.71 In addition, the knowledge gained from these studies may also lead to the identification of potential routes for intervention in PVD, before the development of PAH and PH associated with chronic lung diseases, including IPF and COPD.
Acknowledgments
The contents of this article are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health (NIH). Experiments were performed with the University of Colorado Cancer Center Microarray core (NCI P30 CA 46934–14).
Appendix.
Table S1.
Reagent information
| Gene/protein | Human qRT-PCR primer: forward | Human qRT-PCR primer: reverse | Antibody |
|---|---|---|---|
| DKK1 | Hs00183740_m1 | (Taqman) | |
| WISP1 | AGGAACTGCATAGCCTACACA | TGGTACACAGCCAGACACTTC | |
| TNC | TGCGAAGAAGGCTTCACA | TACACATTTGCCCTCGACAC | |
| NEO1 | GGAGCCGGTGGATACACTCT | TGGCGTCGATCATCTGATACTA | |
| RGS5 | CGGAGGCTCCTAAAGAGGTG | CAAAGCGAGGCAGAGAATCC | |
| SPON2 | CGGCCAAATACAGCATCACC | CCCAGCAGCGAAGACCACT | |
| WLS | ATGAGGGCCGTTACTATGAATGT | CCTTGGTGAAGCCTCCATTTTG | |
| LRP6 | ACGATTGTAGTTGGAGGCTTG | ATGGCTTCTTCGCTGACATCA | |
| PTGS2 | Hs00153133_m1 | (Taqman) | |
| PEAR1 | Hs01378394_m1 | (Taqman) | |
| PDK4 | Hs01037712_m1 | (Taqman) | |
| LRP6 | Santa Cruz SC-25317 | ||
| DKK1 | Santa Cruz SC-374574 |
qRT-PCR: quantitative reverse transcription polymerase chain reaction.
Table S2.
Differential gene expression between PAH and control MPCs
| PAH versus control | PAH versus control | ||||
|---|---|---|---|---|---|
| Gene | Fold change | P value | Gene | Fold change | P value |
| ABCA8 | −5.74566 | 0.000426918 | P4HA3 | −3.71075 | 0.00359561 |
| ACAD11 | −3.02428 | 0.0115302 | PAR4 | −2.29911 | 0.0279132 |
| ACSS3 | −2.30662 | 0.00832764 | PARP8 | −2.05851 | 0.0108406 |
| ADAM23 | −2.53986 | 0.0426075 | PARP9 | 2.26876 | 0.00347152 |
| ADAMTS19 | −2.26564 | 0.00381684 | PCDHB2 | −2.28736 | 0.0145255 |
| AP1S3 | −2.1376 | 0.0105606 | PCDHB5 | −2.2907 | 0.00488807 |
| ARNTL2 | 2.01563 | 0.030135 | PCOLCE2 | −2.75813 | 0.0368619 |
| ASAH1 | −2.04675 | 0.0021534 | PDK4 | −2.98576 | 4.00E−05 |
| BGN | 2.44454 | 0.0365964 | PEAR1 | 2.12031 | 0.00635016 |
| BMP6 | −2.22251 | 0.00389416 | PGM2L1 | −2.99221 | 0.00011248 |
| BRIP1 | 2.32996 | 0.0309359 | PLAT | −4.53043 | 0.025726 |
| CCL20 | −3.3151 | 0.00378156 | PPP1R3C | −2.64459 | 0.034272 |
| CCND2 | −5.5355 | 0.0155638 | PTGS2 | −5.5725 | 0.040118 |
| CENPI | 2.9553 | 0.0338275 | PTHLH | −2.18944 | 0.00901834 |
| CHN1 | 2.40679 | 0.0194867 | PTTG1 | 2.69096 | 0.0355188 |
| CPE | −2.1992 | 0.0392921 | RELN | −4.75644 | 0.0329863 |
| CTSC | 2.19773 | 0.020409 | RGS5 | −3.45307 | 0.00629714 |
| CXCL10 | 2.5709 | 0.00461782 | ROBO2 | −2.38973 | 0.000585671 |
| DKK1 | −2.27936 | 0.0201554 | SAT1 | −2.70967 | 0.0129793 |
| DOCK10 | 2.32133 | 0.00869289 | SATB1 | −2.99009 | 5.53E−06 |
| DTX3L | 2.25824 | 0.00192234 | SCG2 | −3.14705 | 0.000159118 |
| EPGN | −2.73003 | 0.0125658 | SCG5 | −2.91574 | 0.0235747 |
| FAM183B | 2.44479 | 0.00221733 | SERPINE2 | −3.63115 | 0.0132981 |
| FGL2 | −8.19764 | 0.0102414 | SERPINI1 | −2.54572 | 0.00190024 |
| FLRT2 | −2.23673 | 0.0167511 | SESN3 | −5.88877 | 0.000105984 |
| GLB1L | −2.62377 | 0.00348021 | SKA3 | 3.47573 | 0.0438041 |
| H2AFZ | 2.45075 | 0.0338116 | SLC14A1 | 2.97187 | 0.0335797 |
| HAS2 | 3.04969 | 0.036305 | SLC1A1 | −2.66717 | 0.0492348 |
| HIST1H1E | 3.19816 | 0.0428945 | SLC38A11 | −2.39641 | 0.00853225 |
| HIST1H4B | 2.22934 | 0.0499595 | SLC39A8 | −2.01716 | 0.0113132 |
| HIST2H2AB | 3.21531 | 0.0231178 | SLC40A1 | −4.08569 | 0.045058 |
| HIST2H2BE | −2.35754 | 0.0185113 | SNAP25 | −2.17275 | 0.0364734 |
| HTATSF1P2 | 3.61831 | 0.00167649 | SNRPN | −2.88561 | 0.0205426 |
| IFI44 | 2.36029 | 0.0123215 | SPON2 | 2.16564 | 0.0386202 |
| IFIT1 | 2.81063 | 0.0300291 | STEAP1 | 2.01976 | 0.0471408 |
| IFIT3 | 2.14972 | 0.00241179 | TCF19 | 2.35564 | 0.0330562 |
| KIAA0101 | 2.78886 | 0.0499342 | TCF19 | 2.2882 | 0.0348311 |
| KIAA1377 | −2.36545 | 0.0383475 | TNC | 3.29154 | 0.00975554 |
| KLF4 | −2.16152 | 0.04311 | TNFRSF1B | −2.30988 | 0.0317549 |
| LBR | 2.40004 | 0.0060019 | TNXA | −3.03839 | 0.00362081 |
| LGMN | −2.35556 | 0.00652633 | TNXB | −3.07654 | 0.00396426 |
| LIPH | −3.34977 | 0.0131982 | TOX2 | 2.31942 | 1.62E−05 |
| LPHN2 | −3.71932 | 0.0229826 | TRHDE | 2.18208 | 0.0415992 |
| MAOA | −3.75233 | 0.0194706 | TRPV2 | 2.65022 | 0.00301275 |
| MEDAG | −6.02268 | 0.0290316 | TYMS | 3.46746 | 0.0325237 |
| MME | 2.74555 | 0.0151204 | VCAN | −2.77318 | 0.00759963 |
| NEO1 | 2.37015 | 0.0329007 | WLS | −2.07564 | 0.0328988 |
| NRM | 2.15887 | 0.0399557 | XAF1 | 2.22607 | 0.00747729 |
| NUF2 | 3.26221 | 0.047235 | |||
| NXF3 | −2.02669 | 0.0166706 | |||
MPCs: mesenchymal progenitor cells; PAH: pulmonary arterial hypertension.
Table S3.
Gene expression common in lung and skin mesenchymal progenitor cells relative to lung fibroblasts
| Gene | Adjusted P value | Gene | Adjusted P value | Gene | Adjusted P value |
|---|---|---|---|---|---|
| KARS | 0.003754665 | CCND2 | 0.0118961 | SHROOM3 | 0.003670784 |
| EIF3F | 0.000481415 | CRACR2A | 0.013054831 | NFKB1 | 0.00966527 |
| GDI2 | 0.003475123 | GNS | 0.010886852 | USP53 | 0.008091899 |
| HNRNPD | 0.001742548 | EEA1 | 0.003672702 | MSMO1 | 0.00427293 |
| HINT1 | 0.001521358 | AKAP11 | 0.006968455 | SH3D19 | 0.012597167 |
| MCTP2 | 0.002305372 | MIPEP | 0.000413416 | DMXL1 | 0.019848346 |
| TCEB2 | 0.00540237 | FLRT2 | 0.016810132 | PRDM6 | 0.005593715 |
| YY1 | 0.004252223 | GPHB5 | 0.032152095 | CSNK1G3 | 0.008915714 |
| RNPS1 | 0.015067654 | TRIP11 | 0.00427293 | GM2A | 0.02849702 |
| KARS | 0.004654451 | MAP2K5 | 0.019951227 | SQSTM1 | 0.003618116 |
| FNTA | 0.000287202 | SPG11 | 0.013480302 | HMGCS1 | 0.040399444 |
| NPM1 | 0.008771686 | CNOT6L | 0.043797907 | COL4A3BP | 0.008843249 |
| IK | 0.00051101 | VPS13C | 0.011734291 | HOMER1 | 0.017509619 |
| EIF4G2 | 0.007731736 | SLC9A3R2 | 0.018675918 | CCDC112 | 0.003205645 |
| PRPF8 | 0.00427293 | MT1X | 0.012356098 | GNPDA1 | 0.007003414 |
| EIF3D | 0.016390177 | SLC12A4 | 0.005607752 | DPYSL3 | 0.002353872 |
| OAZ1 | 0.014613998 | LCAT | 0.005607752 | DUSP1 | 0.012619233 |
| TAF10 | 0.005327146 | CTNS | 0.003076261 | ZFP62 | 0.004509464 |
| SART3 | 0.013316994 | KRTAP9-1 | 0.01081735 | EDN1 | 0.000368188 |
| SART1 | 0.02965512 | WNK4 | 0.002765411 | ACOT13 | 0.004396663 |
| ILF2 | 0.007557336 | SPHK1 | 0.000307472 | PHF3 | 0.014245617 |
| CBX3 | 0.009759887 | ABR | 0.030489212 | FIG4 | 0.000782556 |
| HINT1 | 0.00328246 | VAT1 | 0.027011742 | GJA1 | 0.013136088 |
| USP22 | 0.007769826 | ARHGAP28 | 0.018510553 | CGA | 0.042434217 |
| EIF4G2 | 0.001317398 | MIB1 | 0.005214734 | GPNMB | 0.041949672 |
| DAD1 | 0.002940223 | CCDC130 | 0.018938761 | EIF4B | 0.007142088 |
| HNRNPK | 0.012513294 | ZNF766 | 0.011302949 | INSIG1 | 0.043790577 |
| NPM1 | 0.001737272 | ZNF845 | 0.002027839 | SEMA3C | 0.00230772 |
| ANAPC5 | 0.017192235 | ZNF135 | 0.018938761 | SLC25A40 | 0.011560029 |
| SEPT2 | 0.004029966 | C3 | 0.035174269 | NAMPT | 0.010432597 |
| GDI2 | 0.000324036 | ZNF850 | 0.005995346 | WNT2 | 0.031797392 |
| CANX | 0.015166734 | ZNF780B | 0.014920479 | SDCBP | 0.011473718 |
| CAPNS1 | 0.03020659 | SERTAD1 | 0.003998191 | OXR1 | 0.027256608 |
| KAT7 | 0.016235184 | TAF1B | 0.016549637 | EMC2 | 0.007226713 |
| GUK1 | 0.002504509 | EHBP1 | 0.012115039 | DENND3 | 0.018085302 |
| SEPT14 | 0.018830105 | FER1L5 | 0.018675918 | LONRF1 | 0.020935755 |
| IFI16 | 0.011560029 | GYPC | 0.003761691 | PSD3 | 0.006978096 |
| DDR2 | 0.003672702 | ACVR2A | 0.036192987 | RB1CC1 | 0.003244456 |
| ZC3H11A | 0.011542988 | ADI1 | 0.00540237 | GEM | 0.001479204 |
| GPR137B | 0.004349162 | NBAS | 0.01179363 | HRSP12 | 0.014240025 |
| RRAGC | 0.018146601 | SMC6 | 0.034521649 | EIF3E | 0.001739213 |
| OMA1 | 0.00911309 | LDAH | 0.002524608 | JAK2 | 0.015096052 |
| ZNHIT6 | 0.003244456 | LRPPRC | 0.001444187 | SCAI | 0.001135774 |
| CDC42SE1 | 0.0118961 | LYPD1 | 0.004571177 | PDK3 | 0.012591566 |
| S100A10 | 0.020134342 | GTDC1 | 0.01584706 | MAOB | 0.026924674 |
| FMOD | 0.011432404 | TGM2 | 0.018692484 | XAGE3 | 0.014986484 |
| LEFTY2 | 0.020300959 | SLC5A3 | 0.007068372 | MORC4 | 0.011396562 |
| CELF2 | 0.004066829 | APOL6 | 0.016328036 | GPC4 | 0.000108718 |
| IFIT2 | 0.000147613 | TCEA1 | 0.010128669 | GPSM3 | 0.011473718 |
| EXOC6 | 0.000118809 | C3orf14 | 0.02523779 | RASA4 | 0.00427293 |
| SCD | 0.004288013 | GXYLT2 | 0.009005018 | MRGPRX3 | 0.008692386 |
| MARCH8 | 0.01999115 | DTX3L | 0.00993769 | SLC25A52 | 0.001521358 |
| JMJD1C | 0.005612215 | CLDN11 | 0.001555164 | SLC25A51 | 0.001521358 |
| PPA1 | 0.001250116 | ARMC10 | 0.003796911 | KARS | 0.003754665 |
| ANO1 | 0.031546574 | CFAP44 | 0.001237906 | EIF3F | 0.000481415 |
| RNF169 | 0.004265815 | BCHE | 0.000813023 | GDI2 | 0.003475123 |
Table S4.
Differential gene expression between ABCG2-expressing skin and lung mesenchymal progenitor cells
| Gene | Fold change | Gene | Fold change |
|---|---|---|---|
| ABCA1 | −7.140379375 | KCNJ2 | −11.17813985 |
| ACTC1 | 95.9064689 | LAMA1 | −6.744129897 |
| ADA | 4.141622168 | LGR4 | 2.714392814 |
| ADAMTS12 | −3.258729977 | LIFR | −3.810556211 |
| ADGRA3 | −3.822489543 | LRP4 | 2.558093918 |
| ADGRG6 | −12.13024322 | LURAP1L | −3.804082107 |
| ADRA2C | 3.255290428 | MCAM | 6.967111342 |
| AKAP6 | 10.28542192 | MSX1 | 5.22209982 |
| BCHE | −5.487451706 | NCALD | 5.594113689 |
| C5orf30 | 6.225149585 | NCAM1 | 6.92291223 |
| CADPS | 3.087142808 | NCKAP5 | −3.099513479 |
| CCL2 | −20.1807767 | NRN1 | 72.7478546 |
| CDH10 | 4.813123244 | NTF4 | 3.876836386 |
| CDH18 | 5.362029696 | OLFML2B | 3.951511144 |
| CFH | −29.66566966 | PDE5A | −16.36720262 |
| COMP | 12.8007186 | PERP | −3.618870672 |
| CTSC | −8.547382511 | PLAC8 | 5.083368648 |
| DKK1 | 5.50202578 | PODXL | 22.77536965 |
| DUSP2 | 5.766688863 | PRRX2 | 3.70223168 |
| EMCN | 7.616999049 | RASD2 | 7.94451159 |
| EMX2 | 5.806593483 | RCAN2 | 3.082643063 |
| ERG | 6.475541649 | RIMS1 | 3.079968681 |
| EXOC6 | −5.861872829 | RTN4RL1 | 3.90925993 |
| FRAS1 | 4.465130742 | SCN9A | −25.38799166 |
| GCLC | −5.092460846 | SDPR | −4.349046474 |
| GDF15 | −9.364839758 | SFRP1 | −9.129036858 |
| GK | −9.841969507 | SHC3 | −5.652915738 |
| GPC4 | 15.38302986 | SIM1 | 4.173372549 |
| HGF | −38.06457212 | SLC7A8 | 18.1691909 |
| HOXA9 | 7.436936653 | SMAD6 | 3.728750848 |
| HOXD10 | 11.51907568 | SVIP | −3.662563103 |
| HOXD11 | 2.998537142 | TBX15 | 9.499693723 |
| HSPA4L | −6.417888004 | TBX18 | 5.023753737 |
| HTR1F | 5.230995929 | TGFB2 | −8.750447068 |
| HTR2A | 3.224492221 | TIAM2 | 3.641838934 |
| IFIT2 | −3.09684411 | TNFSF15 | −3.208561644 |
| IFITM10 | 3.017262095 | TOM1L1 | −3.559699575 |
| IGFBP5 | −83.6131559 | TSPAN18 | 6.53260725 |
| IL11RA | 2.752604431 | ZIC1 | 8.165245944 |
| ITGA8 | 4.471799319 | ZNF385D | 5.732989384 |
Source of Support: This work was funded in part by grants to SMM and EDA from Vanderbilt Clinical and Translational Science Award UL1TR000445 from the National Center for Advancing Translational Sciences. Additional funding was also provided by NIH grant PPG-5P01HL108800-04 (JEL) and by the Research Centers in Minority Institutions Program grant G12 MD007586 and the Meharry Clinical and Translational Research Center grant U54 MD007593 from the National Institute on Minority Health and Health Disparities of the NIH (SP).
Supplements
Appendix (443.5KB, pdf)
References
- 1.Austin ED, Kawut SM, Gladwin MT, Abman SH. Pulmonary hypertension: NHLBI workshop on the primary prevention of chronic lung diseases. Ann Am Thorac Soc 2014;11(suppl 3):S178–S185. [DOI] [PMC free article] [PubMed]
- 2.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62(25 suppl):D34–D41. [DOI] [PubMed]
- 3.Stevens D, Sharma K, Szidon P, Rich S, McLaughlin V, Kesten S. Severe pulmonary hypertension associated with COPD. Ann Transplant 2000;5(3):8–12. [PubMed]
- 4.Mukerjee D, St George D, Coleiro B, Knight C, Denton CP, Davar J, Black CM, Coghlan JG. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis 2003;62(11):1088–1093. [DOI] [PMC free article] [PubMed]
- 5.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009;53(17):1573–1619. [DOI] [PubMed]
- 6.Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet 2003;361(9368):1533–1544. [DOI] [PubMed]
- 7.Runo JR, Vnencak-Jones CL, Prince M, Loyd JE, Wheeler L, Robbins IM, Lane KB, et al. Pulmonary veno-occlusive disease caused by an inherited mutation in bone morphogenetic protein receptor II. Am J Respir Crit Care Med 2003;167(6):889–894. [DOI] [PubMed]
- 8.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res 2014;115(1):189–202. [DOI] [PMC free article] [PubMed]
- 9.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002;105(14):1672–1678. [DOI] [PubMed]
- 10.West JD, Austin ED, Gaskill C, Marriott S, Baskir R, Bilousova G, Jean JC, et al. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. Am J Physiol Cell Physiol 2014;307(5):C415–C430. [DOI] [PMC free article] [PubMed]
- 11.Austin ED, Ma L, LeDuc C, Rosenzweig EB, Borczuk A, Phillips JA III, Palomero T, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012;5(3):336–343; clinical perspective 343. [DOI] [PMC free article] [PubMed]
- 12.Maarman G, Lecour S, Butrous G, Thienemann F, Sliwa K. A comprehensive review: the evolution of animal models in pulmonary hypertension research; are we there yet? Pulm Circ 2013;3(4):739–756. [DOI] [PMC free article] [PubMed]
- 13.Chow K, Fessel JP, Ihida-Stansbury K, Schmidt EP, Gaskill C, Alvarez D, Graham B, et al. Dysfunctional resident lung mesenchymal stem cells contribute to pulmonary microvascular remodeling. Pulm Circ 2013;3(1):31–49. [DOI] [PMC free article] [PubMed]
- 14.Jun D, Garat C, West J, Thorn N, Chow K, Cleaver T, Sullivan T, et al. The pathology of bleomycin-induced fibrosis is associated with loss of resident lung mesenchymal stem cells that regulate effector T-cell proliferation. Stem Cells 2011;29(4):725–735. [DOI] [PMC free article] [PubMed]
- 15.Marriott S, Baskir RS, Gaskill C, Menon S, Carrier EJ, Williams J, Talati M, et al. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am J Physiol Cell Physiol 2014;307(8):C684–C698. [DOI] [PMC free article] [PubMed]
- 16.Fatima S, Zhou S, Sorrentino BP. Abcg2 expression marks tissue-specific stem cells in multiple organs in a mouse progeny tracking model. Stem Cells 2012;30(2):210–221. [DOI] [PMC free article] [PubMed]
- 17.Newman JH, Fanburg BL, Archer SL, Badesch DB, Barst RJ, Garcia JG, Kao PN, et al. Pulmonary arterial hypertension: future directions: report of a National Heart, Lung and Blood Institute/Office of Rare Diseases workshop. Circulation 2004;109(24):2947–2952. [DOI] [PubMed]
- 18.Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: clinical patterns. Am Rev Respir Dis 1984;129(1):194–197.
- 19.Simonneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004;43(12 suppl):S5–S12. [DOI] [PubMed]
- 20.Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM, Hedges LK, Stanton KC, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174(5):590–598. [DOI] [PMC free article] [PubMed]
- 21.Kim K-A, Wagle M, Tran K, Zhan X, Dixon MA, Liu S, Gros D, et al. R-spondin family members regulate the Wnt pathway by a common mechanism. Mol Biol Cell 2008;19(6):2588–2596. [DOI] [PMC free article] [PubMed]
- 22.Königshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, Jahn A, Rose F, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Investig 2009;119(4):772–787. [DOI] [PMC free article] [PubMed]
- 23.Faraday N, Yanek LR, Yang XP, Mathias R, Herrera-Galeano JE, Suktitipat B, Qayyum R, et al. Identification of a specific intronic PEAR1 gene variant associated with greater platelet aggregability and protein expression. Blood 2011;118(12):3367–3375. [DOI] [PMC free article] [PubMed]
- 24.Cohen ED, Ihida-Stansbury K, Lu MM, Panettieri RA, Jones PL, Morrisey EE. Wnt signaling regulates smooth muscle precursor development in the mouse lung via a tenascin C/PDGFR pathway. J Clin Investig 2009;119(9):2538–2549. [DOI] [PMC free article] [PubMed]
- 25.Severyn CJ, Shinde U, Rotwein P. Molecular biology, genetics and biochemistry of the repulsive guidance molecule family. Biochem J 2009;422(3):393–403. [DOI] [PMC free article] [PubMed]
- 26.Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 2001;411(6835):321–325. [DOI] [PubMed]
- 27.Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab 2014;11(1):1–9. [DOI] [PMC free article] [PubMed]
- 28.Pate KT, Stringari C, Sprowl-Tanio S, TeSlaa T, Hoverter NP, McQuade MM, Garner C, et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J 2014;33(13):1454–1473. [DOI] [PMC free article] [PubMed]
- 29.Cho H, Park C, Hwang I-Y, Han SB, Schimel D, Despres D, Kehrl JH. Rgs5 targeting leads to chronic low blood pressure and a lean body habitus. Mol Cell Biol 2008;28(8):2590–2597. [DOI] [PMC free article] [PubMed]
- 30.Sellers RS, Radi ZA, Khan NK. Pathophysiology of cyclooxygenases in cardiovascular homeostasis. Vet Pathol 2010;47(4):601–613. [DOI] [PubMed]
- 31.Ono M, Inkson CA, Kilts TM, Young MF. WISP-1/CCN4 regulates osteogenesis by enhancing BMP-2 activity. J Bone Miner Res 2011;26(1):193–208. [DOI] [PMC free article] [PubMed]
- 32.Iozzi S, Remelli R, Lelli B, Diamanti D, Pileri S, Bracci L, Roncarati R, Caricasole A, Bernocco S. Functional characterization of a small-molecule inhibitor of the DKK1-LRP6 interaction. ISRN Mol Biol 2012;2012:9. doi:10.5402/2012/823875. [DOI] [PMC free article] [PubMed]
- 33.Wipff J, Kahan A, Hachulla E, Sibilia J, Cabane J, Meyer O, Mouthon L, et al. Association between an endoglin gene polymorphism and systemic sclerosis-related pulmonary arterial hypertension. Rheumatology 2007;46(4):622–625. [DOI] [PubMed]
- 34.Coghlan JG, Mukerjee D. The heart and pulmonary vasculature in scleroderma: clinical features and pathobiology. Curr Opin Rheumatol 2001;13(6):495–499. [DOI] [PubMed]
- 35.Fleming J, Nash RA, Mahoney WM Jr., Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009;11(2):103–110. [DOI] [PMC free article] [PubMed]
- 36.Fleming JN, Nash RA, McLeod DO, Fiorentino DF, Shulman HM, Connolly MK, Molitor JA, et al. Capillary regeneration in scleroderma: stem cell therapy reverses phenotype? PLoS ONE 2008;3(1):e1452. doi:10.1371/journal.pone.0001452. [DOI] [PMC free article] [PubMed]
- 37.Königshoff M, Eickelberg O. WNT signaling in lung disease: a failure or a regeneration signal? Am J Respir Cell Mol Biol 2010;42(1):21–31. [DOI] [PubMed]
- 38.Foronjy RF, Majka SM. The potential for resident lung mesenchymal stem cells to promote functional tissue regeneration: understanding microenvironmental cues. Cells 2012;1(4):874–885. [DOI] [PMC free article] [PubMed]
- 39.Wang R, Ahmed J, Wang G, Hassan I, Strulovici-Barel Y, Hackett NR, Crystal RG. Down-regulation of the canonical Wnt β-catenin pathway in the airway epithelium of healthy smokers and smokers with COPD. PLoS ONE 2011;6(4):e14793. doi:10.1371/journal.pone.0014793. [DOI] [PMC free article] [PubMed]
- 40.Baarsma HA, Spanjer AIR, Haitsma G, Engelbertink LH, Meurs H, Jonker MR, Timens W, Postma DS, Kerstjens HA, Gosens R. Activation of WNT/β-catenin signaling in pulmonary fibroblasts by TGF-β1 is increased in chronic obstructive pulmonary disease. PLoS ONE 2011;6(9):e25450. doi:10.1371/journal.pone.0025450. [DOI] [PMC free article] [PubMed]
- 41.Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, Sugano S, Akiyama T. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 2004;23(52):8520–8526. [DOI] [PubMed]
- 42.Kamiya N, Kobayashi T, Mochida Y, Yu PB, Yamauchi M, Kronenberg HM, Mishina Y. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J Bone Miner Res 2009;25(2):200–210. [DOI] [PMC free article] [PubMed]
- 43.Miura S, Singh AP, Mishina Y. Bmpr1a is required for proper migration of the AVE through regulation of Dkk1 expression in the pre-streak mouse embryo. Dev Biol 2010;341(1):246–254. [DOI] [PMC free article] [PubMed]
- 44.Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 2006;25(57):7469–7481. [DOI] [PubMed]
- 45.Dees C, Schlottmann I, Funke R, Distler A, Palumbo-Zerr K, Zerr P, Lin NY, et al. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann Rheum Dis 2014;73(6):1232–1239. [DOI] [PubMed]
- 46.Valencia A, Román-Gómez J, Cervera J, Such E, Barragán E, Bolufer P, Moscardó F, Sanz GF, Sanz MA. Wnt signaling pathway is epigenetically regulated by methylation of Wnt antagonists in acute myeloid leukemia. Leukemia 2009;23(9):1658–1666. [DOI] [PubMed]
- 47.Lee SJ, Jeong JY, Oh CJ, Park S, Kim JY, Kim HJ, Kim NM, et al. Pyruvate dehydrogenase kinase 4 promotes vascular calcification via SMAD1/5/8 phosphorylation. Sci Rep 2015;5:16577. doi:10.1038/srep16577. [DOI] [PMC free article] [PubMed]
- 48.Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, Diebold I, et al. Elafin reverses pulmonary hypertension via caveolin-1-dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med 2015;191(11):1273–1286. [DOI] [PMC free article] [PubMed]
- 49.Semenov MV, Zhang X, He X. DKK1 antagonizes Wnt signaling without promotion of LRP6 internalization and degradation. J Biol Chem 2008;283(31):21427–21432. [DOI] [PMC free article] [PubMed]
- 50.Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc Natl Acad Sci USA 2011;108(32):13253–13257. [DOI] [PMC free article] [PubMed]
- 51.Yamamoto H, Komekado H, Kikuchi A. Caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of β-catenin. Dev Cell 2006;11(2):213–223. [DOI] [PubMed]
- 52.Tahir SA, Yang G, Goltsov A, Song KD, Ren C, Wang J, Chang W, Thompson TC. Caveolin-1-LRP6 signaling module stimulates aerobic glycolysis in prostate cancer. Cancer Res 2013;73(6):1900–1911. [DOI] [PMC free article] [PubMed]
- 53.El Wakil A, Bandulik S, Guy N, Bendahhou S, Zennaro MC, Niehrs C, Mari B, Warth R, Barhanin J, Lalli E. Dkk3 is a component of the genetic circuitry regulating aldosterone biosynthesis in the adrenal cortex. Hum Mol Genet 2012;21(22):4922–4929. [DOI] [PubMed]
- 54.Wang F-S, Ko J-Y, Lin C-L, Wu H-L, Ke H-J, Tai P-J. Knocking down dickkopf-1 alleviates estrogen deficiency induction of bone loss. A histomorphological study in ovariectomized rats. Bone 40(2):485–492. [DOI] [PubMed]
- 55.Zhang Q-G, Wang R, Khan M, Mahesh V, Brann DW. Role of dickkopf-1, an antagonist of the Wnt/β-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci 2008;28(34):8430–8441. [DOI] [PMC free article] [PubMed]
- 56.Kouzmenko AP, Takeyama K-i, Ito S, Furutani T, Sawatsubashi S, Maki A, Suzuki E, et al. Wnt/β-catenin and estrogen signaling converge in vivo. J Biol Chem 2004;279(39):40255–40258. [DOI] [PubMed]
- 57.Park H, Jung HY, Choi H-J, Kim DY, Yoo J-Y, Yun C-O, Min J-K, Kim Y-M, Kwon Y-G. Distinct roles of DKK1 and DKK2 in tumor angiogenesis. Angiogenesis 2014;17(1):221–234. [DOI] [PMC free article] [PubMed]
- 58.Min J-K, Park H, Choi H-J, Kim Y, Pyun B-J, Agrawal V, Song B-W, et al. The WNT antagonist Dickkopf2 promotes angiogenesis in rodent and human endothelial cells. J Clin Investig 121(5):1882–1893. [DOI] [PMC free article] [PubMed]
- 59.De Langhe SP, Sala FG, Del Moral P-M, Fairbanks TJ, Yamada KM, Warburton D, Burns RC, Bellusci S. Dickkopf-1 (DKK1) reveals that fibronectin is a major target of Wnt signaling in branching morphogenesis of the mouse embryonic lung. Dev Biol 2005;277(2):316–331. [DOI] [PubMed]
- 60.Christodoulides C, Laudes M, Cawthorn WP, Schinner S, Soos M, O’Rahilly S, Sethi JK, Vidal-Puig A. The Wnt antagonist Dickkopf-1 and its receptors are coordinately regulated during early human adipogenesis. J Cell Sci 2006;119(12):2613–2620. [DOI] [PMC free article] [PubMed]
- 61.Jeoung NH, Harris RA. Pyruvate dehydrogenase kinase-4 deficiency lowers blood glucose and improves glucose tolerance in diet-induced obese mice. Am J Physiol Endocrinol Metab 2008;295(1):E46–E54. [DOI] [PMC free article] [PubMed]
- 62.Pugh ME, Robbins IM, Rice TW, West J, Newman JH, Hemnes AR. Unrecognized glucose intolerance is common in pulmonary arterial hypertension. J Heart Lung Transplant 2011;30(8):904–911. [DOI] [PMC free article] [PubMed]
- 63.Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, Gupta M, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009;323(5922):1747–1750. [DOI] [PMC free article] [PubMed]
- 64.Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol 2012;2012:95068. doi:10.1155/2012/950658. [DOI] [PMC free article] [PubMed]
- 65.Kestler HA, Kuhl M. Generating a Wnt switch: it’s all about the right dosage. J Cell Biol 2011;193(3):431–433. [DOI] [PMC free article] [PubMed]
- 66.Ring A, Kim Y-M, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev 2014;10(4):512–525. [DOI] [PMC free article] [PubMed]
- 67.Caneparo L, Huang Y-L, Staudt N, Tada M, Ahrendt R, Kazanskaya O, Niehrs C, Houart C. Dickkopf-1 regulates gastrulation movements by coordinated modulation of Wnt/βcatenin and Wnt/PCP activities, through interaction with the Dally-like homolog Knypek. Genes Dev 2007;21(4):465–480. [DOI] [PMC free article] [PubMed]
- 68.Gray JD, Kholmanskikh S, Castaldo BS, Hansler A, Chung H, Klotz B, Singh S, Brown AM, Ross ME. LRP6 exerts non-canonical effects on Wnt signaling during neural tube closure. Hum Mol Genet 2013;22(21):4267–4281. [DOI] [PMC free article] [PubMed]
- 69.de Jesus Perez VA, Alastalo TP, Wu JC, Axelrod JD, Cooke JP, Amieva M, Rabinovitch M. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt–β-catenin and Wnt–RhoA–Rac1 pathways. J Cell Biol 2009;184(1):83–99. [DOI] [PMC free article] [PubMed]
- 70.Yuan K, Orcholski ME, Panaroni C, Shuffle EM, Huang NF, Jiang X, Tian W, et al. Activation of the Wnt/planar cell polarity pathway is required for pericyte recruitment during pulmonary angiogenesis. Am J Pathol 2015;185(1):69–84. [DOI] [PMC free article] [PubMed]
- 71.Shen Q, Fan J, Yang X-R, Tan Y, Zhao W, Xu Y, Wang N, et al. Serum DKK1 as a protein biomarker for the diagnosis of hepatocellular carcinoma: a large-scale, multicentre study. Lancet Oncol 2012;13(8):817–826. [DOI] [PubMed]