

Abstract Abstract
Mechanical ventilation, a lifesaving intervention for patients with acute respiratory distress syndrome (ARDS), also unfortunately contributes to excessive mechanical stress and impaired lung physiological and structural integrity. We have elsewhere established the pivotal role of increased nicotinamide phosphoribosyltransferase (NAMPT) transcription and secretion as well as its direct binding to the toll-like receptor 4 (TLR4) in the progression of this devastating syndrome; however, regulation of this critical gene in ventilator-induced lung injury (VILI) is not well characterized. On the basis of an emerging role for epigenetics in enrichment of VILI and CpG sites within the NAMPT promoter and 5′UTR, we hypothesized that NAMPT expression and downstream transcriptional events are influenced by epigenetic mechanisms. Concomitantly, excessive mechanical stress of human pulmonary artery endothelial cells or lipopolysaccharide (LPS) treatment led to both reduced DNA methylation levels in the NAMPT promoter and increased gene transcription. Histone deacetylase inhibition by trichostatin A or Sirt-1–silencing RNA attenuates LPS-induced NAMPT expression. Furthermore, recombinant NAMPT administration induced TLR4-dependent global H3K9 hypoacetylation. These studies suggest a complex epigenetic regulatory network of NAMPT in VILI and ARDS and open novel strategies for combating VILI and ARDS.
Keywords: lung endothelium, epigenetics, DNA methylation, epigenetic modifiers, histone acetylation
Acute respiratory distress syndrome (ARDS) is a devastating inflammatory syndrome affecting over 200,000 people a year that is associated with significantly high morbidity and mortality rates.1 Mechanical ventilation, a lifesaving intervention, paradoxically contributes directly to an inflammatory syndrome effectuated by excessive mechanical stress known as ventilator-induced lung injury (VILI).2 VILI is indistinguishable in pathobiology from ARDS and includes the most common features of ARDS-like hypoxemia, inflammation, and pulmonary edema and aggravates earlier insult.2 However, the mechanisms underlying the pathobiology are very poorly understood and need further evaluation.
Our genomic-intensive approaches using preclinical models of ARDS and VILI identified nicotinamide phosphoribosyltransferase (NAMPT) as a novel mediator of VILI.3 NAMPT, as an intracellular molecule, is a rate-limiting enzyme in nicotinamide adenine dinucleotide (NAD) biosynthesis, influencing the metabolism of the cell and fueling sirtuin activity.4 NAMPT also exists as an extracellular molecule where circulating plasma levels represent a biomarker of disease as well as contribute to the development and severity of VILI as an inflammatory stimulus.5,6 Multiple preclinical models using murine and canine models have exhibited NAMPT localization to lung leukocytes, epithelium, and the endothelium.5 Reduced NAMPT activity through silencing, moderating the catalytic activity (neutralizing antibodies), or utilizing mice with partial NAMPT genetic deletion (Nampt+/−) confer significant protection against VILI.6 Conversely, intratracheal administration of recombinant NAMPT increases neutrophilic alveolitis and contributes to increased inflammation in the lungs via induction of NF-ĸB.6 Additional biochemical assays exploring the activation of NFkB pathways in mouse models of VILI and human lung endothelium identified toll-like receptor 4 (TLR4) as a direct binding partner for NAMPT independent of sepsis-related stimuli via NAMPT’s unique similarities to MD-2 protein sequence.7 Although our earlier studies have also elucidated specific transcription factors that bind and augment NAMPT promoter, no studies to date have evaluated the epigenetic regulation of NAMPT expression with excessive mechanical stress.8
Epigenetics has emerged as a critical novel regulator of gene transcription. The major epigenetic mechanisms, including DNA methylation, histone modifications, and noncoding RNA–mediated posttranscriptional regulation of messenger RNA (mRNA), have been implicated in a multitude of physiological processes, such as cancer, cellular differentiation, inflammation,9-11 and VILI. In this study, we confirmed an epigenetic regulation of NAMPT promoter and expression as well as the epigenetic role of NAMPT/TLR4 pathway in SIRT1–induced H3K9 acetylation. These studies reveal the complex interplay of NAMPT in VILI and the critical role of epigenetics in NAMPT regulation and function.
Material and methods
Cell culture and reagents
Human pulmonary artery endothelial cells (ECs) obtained from Lonza (Walkersville, MD) were grown under standard conditions in EBM-2 media supplemented with growth kit provided by the manufacturer.12 ECs between passages 6 and 8 were used in all experiments. Lipopolysaccharide (LPS) was obtained from Sigma-Aldrich (St. Louis, MO) and used at 100 ng/mL at various time points as indicated in the experiments. Mechanical stress was imparted by coating cells on collagen-coated flexible membrane plates and stretching them at regular cyclic intervals at 5% or 18% magnitude using a FlexCell FX5000 apparatus (Greensboro, NC).
DNA methylation analysis
Genomic DNA and RNA from EC were isolated simultaneously using the Zymo Duet kit (Irvine, CA), and 1 μg of isolated genomic DNA was bisulfite converted as recommended using the EZ DNA lightning kit from Zymo Research (Irvine, CA). An aliquot of bisulfite-converted DNA was used to amplify the NAMPT promoter region using primers specifically designed to discriminate bisulfite-converted and unconverted DNA. The polymerase chain reaction (PCR) products were cloned into PGL4 sequencing vector from Invitrogen (Carlsbad, CA), plated on ampicillin selection LB agar plates, and left overnight at 37°C for colony growth. Several colonies were picked from each plate and sequenced directly at Eton Biosciences (San Diego, CA). The sequenced clones were then used for methylation analysis using the BISMA software.
Real-time PCR and semiquantitative PCR
Complementary DNA (cDNA) was generated using high-capacity cDNA reverse-transcription kit from Life Technologies (Carlsbad, CA) according to the manufacturer’s recommendation. Changes in NAMPT transcripts were quantified by real-time PCR in a Bio-Rad CFX96 apparatus using SYBR green reagents obtained from Life Technologies, primers specific for NAMPT, and an aliquot of cDNA. Primers specific for glyceraldehyde 3-phosphate dehydrogenase were used for normalization. All reactions were performed in triplicate, and the changes were evaluated by the −ΔΔCt method. All primers designed for quantification were designed on exon boundaries to eliminate genomic DNA detection.
Silencing Sirt-1
Small interfering RNA (siRNA) specific for Sirt-1 or nontargeting siRNA were obtained from IDT (Coralville, IA) and transfected into ECs (5 nM, 72 hours), and RNA from these cells were either used in quantitative PCR (qPCR) to assay for NAMPT expression or lysed in sample buffer and electrophoresed to validate NAMPT protein expression.
Western blot analysis
Treated cells were lysed as described in protein lysis buffer,12 which was subsequently separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, blocked with 5% nonfat dry milk in tris-buffered saline and Tween 20 for 1 hour, and incubated with the primary antibody in 1% nonfat dry milk overnight. Primary antibody was detected by incubation with horseradish peroxidase-coupled secondary antibody diluted in 1% nonfat dry milk for 1 hour at room temperature. The protein blots were detected using ECL (Thermoscientific), and the bands obtained were quantified using ImageJ.
Results
Changes in DNA methylation in response to mechanical stress and LPS
Excessive mechanical stress and endotoxin challenges to the lung endothelium are known to increase NAMPT gene expression.5,6,8,13 To assess the changes in DNA methylation occurring in the NAMPT promoter, we performed PCR using primers specific for bisulfite-converted DNA and targeting the NAMPT promoter. The PCR products were cloned into PCR4.1 topo vectors, and multiple clones were selected for sequencing. Our data demonstrated significant changes occurring in both the CpG islands in response to excessive mechanical stress or endotoxin challenges compared with untreated static cells (Fig. 1). These data suggest that excessive mechanical stress and endotoxin challenges might induce active demethylation in the NAMPT promoter that could be linked to subsequent increases in mRNA transcripts.
Figure 1.

Endotoxin and mechanical stress induces decreases in DNA methylation in the nicotinamide phosphoribosyltransferase (NAMPT) promoter. Bisulfite sequencing of DNA from human lung endothelial cells (ECs) demonstrated decreases in DNA methylation occurring in both of the CpG islands of the NAMPT promoter. Compared with unstretched or unstimulated cells, 18% cyclic stretch (CS), and endotoxin challenges (100 ng/mL), DNA methylation was significantly decreased. Blue bars depict unmethylated alleles, and red bars depict methylated alleles. Each row is representative of a unique allele, and each column depicts individual CpG sites.
Histone deacetylase (HDAC) activity is critical for increased NAMPT gene transcription
LPS is a known trigger for increased HDAC activity in the lung endothelium.14 In our studies, qPCR analysis of mRNA validated our earlier findings and showed significantly increased gene expression of NAMPT in response to LPS. However, pretreatment of EC with the class I and II HDAC inhibitor trichostatin A (TSA; 400 nM and 1,000 nM) for 20 hours followed by vehicle or LPS for 4 hours reduced basal-level NAMPT gene transcript levels (∼40%) and inhibited endotoxin-mediated increases in NAMPT mRNA similar to basal levels (Fig. 2A). Not only were the gene transcripts reduced by TSA, but also analysis of NAMPT protein expression by Western blots revealed that, although endotoxin increased NAMPT protein expression significantly at 24 hours, pretreatment with TSA (1,000 nM) for 1 hour followed by 24 hours of LPS induction attenuated the induced increases in NAMPT protein expression. It is important to note that, although pretreatment with 1,000 nM TSA was effective at preventing the upregulation of NAMPT protein expression, treatment with a lower dose of TSA (400 nM) under similar conditions did not alter the induced increases in NAMPT protein expression (Fig. 2B). In addition, in our experiments with mechanical stress, ECs were treated with either 5% cyclic stretch (CS) to mimic physiological stretch or 18% CS to mimic pathological stretch with and without TSA. While NAMPT protein expression in response to mechanical stress was significantly increased, pretreatment with TSA attenuated mechanical stress–induced NAMPT protein expression (Fig. 2C). These observations demonstrated the importance of HDAC activity in the induced increases in gene expression and that pharmacological inhibition by HDAC inhibitors, such as TSA, can be effective in reducing the endotoxin- and mechanical stress–induced NAMPT protein expression.
Figure 2.

Trichostatin A (TSA)–attenuated lipopolysaccharide (LPS) induced nicotinamide phosphoribosyltransferase (NAMPT) expression in human lung endothelial cells (ECs). A, Human lung ECs were pretreated with either vehicle (Veh) or histone deacetylase (HDAC) class I and II–specific inhibitor TSA (400 nM and 1,000 nM) for 20 hours and were further unstimulated or stimulated with LPS (100 ng/mL) for 4 hours. TSA showed marginal statistically insignificant decreases for NAMPT transcripts at both doses in unstimulated cells as assayed by quantitative polymerase chain reaction with primers specific for NAMPT and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Pretreatment with TSA-attenuated LPS induced increases in NAMPT messenger RNA. All samples are represented as fold-change of gene expression over Veh; n = 3 independent experiments. Two asterisks indicate P < 0.01 for LPS + TSA − 1,000 nM versus LPS, and one asterisk indicates P < 0.01 for LPS versus Veh. B, TSA pretreatment (1,000 nM) for 1 hour followed by LPS (100 ng/mL) for 24 hours inhibited the induced NAMPT protein analyzed by Western blot (∼2-fold inhibition). Pretreatment with TSA at 1,000 nM, but not 400 nM, significantly attenuated LPS-induced NAMPT protein expression. C, 18% cyclic stretch (CS) significantly increased NAMPT protein expression over 5% CS. These induced increases were significantly decreased with pretreatment of endothelial cells with TSA (1,000 nM).
Effects of silencing Sirt-1 on NAMPT expression
NAD-dependent class III HDACs, known as sirtuins, are distinct from HDAC classes I and II. To understand the regulation of NAMPT by Sirt-1, siRNA specific for Sirt-1 or nontargeting negative control were transfected into ECs, and NAMPT mRNA levels were assayed by qPCR. The results demonstrated significant increases in NAMPT gene transcripts (∼3-fold) in Sirt-1–silenced cells compared with negative control transfected cells (Fig. 3A). In addition, ECs were transfected with siRNA specific for Sirt-1 or negative control and were treated with either vehicle or LPS (100 ng/mL). The results showed that silencing of Sirt-1 significantly upregulated NAMPT protein expression and paralleled increases in negative control–transfected, LPS-induced cells (Fig. 3B). These data essentially showed that Sirt-1 negatively regulates NAMPT gene and protein transcripts in human lung endothelium.
Figure 3.
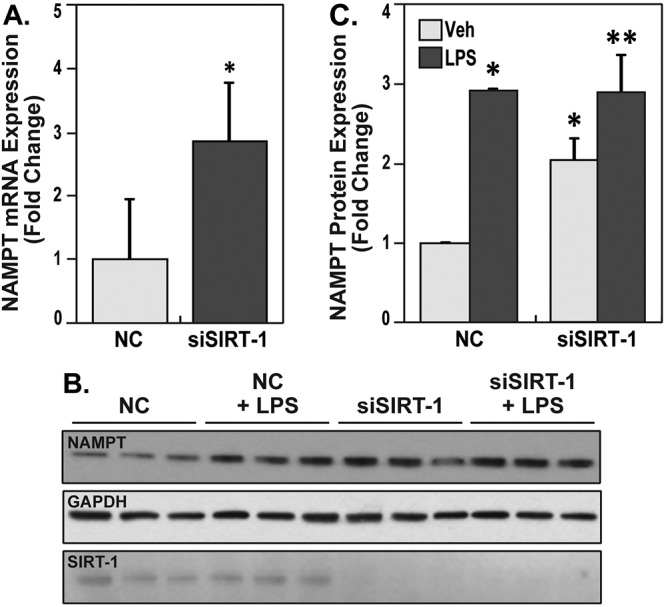
Nicotinamide phosphoribosyltransferase (NAMPT) gene and protein expression is upregulated by silencing Sirt-1. A, Human lung endothelial cells (ECs) were transfected with negative control (NC) or small interfering RNA (siRNA) specific for Sirt-1 (siSIRT-1) for 72 hours, and NAMPT messenger RNA (mRNA) transcripts were assayed by quantitative polymerase chain reaction (qPCR). qPCR assays show that silencing of Sirt-1 significantly increased NAMPT transcripts; single asterisk indicates P < 0.01. B, endothelial cells (ECs) were transfected with NC or siSIRT-1 for 72 hours and were either unstimulated or stimulated with lipopolysaccharides (LPS) 24 hours and 48 hours after siRNA transfection. Cell lysates were electrophoresed, and antibodies specific for NAMPT, Sirt-1, or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were used to assay for protein levels. Three independent experiments were run on the same blot. C, Densitometric summary of the increases in NAMPT protein expression upon silencing of Sirt-1, LPS treatment, and simultaneous treatment of LPS and silencing Sirt-1. The bar graphs represent integrated density data of NAMPT normalized to GAPDH and expressed as a fold-change of each treatment over negative control–treated human ECs. The results show silencing Sirt-1 significantly increased NAMPT levels, but LPS and LPS + siSIRT-1 stimulated NAMPT levels beyond silencing alone. Furthermore, LPS-induced increases of NAMPT subsequent to Sirt-1 silencing were not synergistic compared with LPS alone (n = 3). One asterisk indicates P < 0.05 compared with NC. Two asterisks indicate P < 0.01 compared with siSIRT-1 vehicle (Veh) group.
Sirt-1 expression is increased in ECs in response to LPS
Because NAMPT transcripts were upregulated in endotoxin-treated cells, and several reports have demonstrated decreases in Sirt-1 protein or Sirt-1 activity in concomitantly increasing gene transcription, we hypothesized that decreases in Sirt-1 activity would regulate the sustained increases in NAMPT gene transcripts. However, surprisingly, investigation of Sirt-1 transcripts in ECs by qPCR analysis showed significant increases in SIRT1 gene transcripts compared with vehicle-treated cells even at 2 hours and showed significant upregulation at 4 hours (∼2-fold upregulation; Fig. S1).
Global acetylation is downregulated in response to LPS and recombinant human NAMPT (rhNAMPT) in ECs in a TLR4-dependent manner
Because Sirt-1 expression is significantly increased after LPS treatment, we investigated the modulation of Sirt-1’s most common downstream histone substrate, H3K9. Consistent with increased Sirt-1, we observed severe depletion of global H3K9 acetylation levels. The depletion of H3K9 acetylation levels was effectively reversed by treatment with the TLR4 inhibitor CLI-095 (Fig. 4). These data suggested that TLR4-mediated signaling in lung ECs downregulates global H3K9 acetylation, and this may be facilitated by increases in Sirt-1 in human lung endothelium.
Figure 4.
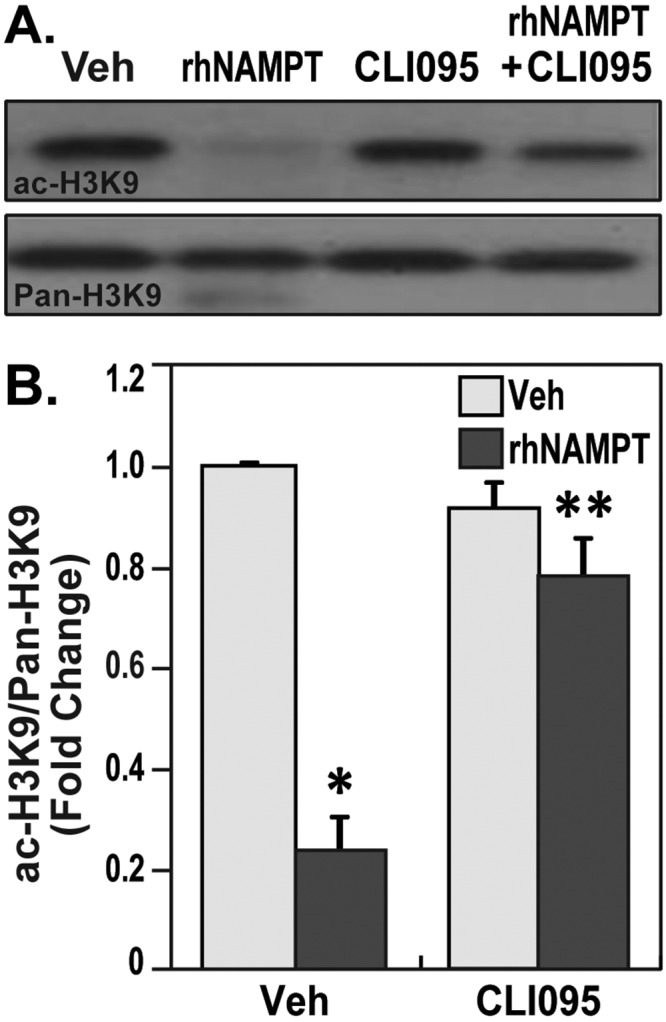
Nicotinamide phosphoribosyltransferase (NAMPT) reduces the acetylation of H3K9 in a toll-like receptor 4 (TLR4)–dependent manner. A, Human lung endothelial cells (ECs) were pretreated with vehicle (Veh) or CLI-095 (5 μM) for 1 hour and stimulated with recombinant human NAMPT (rhNAMPT; 1 μg/mL) for 4 hours. The cells were lysed, and Western blot was performed with antibodies specific for acetyl H3K9. Total H3K9 was used as loading control. Results demonstrate the extracellular NAMPT reduced the H3K9 acetylation, which is reversed by TLR4 inhibitor. B, Densitometric summary of the attenuation of rhNAMPT-induced H3K9 acetylation by TLR4 inhibitors. rhNAMPT-induced EC demonstrated reduced H3K9 acetylation (∼65% reduction). Pretreatment with TLR4 inhibitor alone did not significantly alter H3K9 acetylation levels but prevented the rhNAMPT-induced reduction of H3K9 acetylation. Bar graphs represent data as fold-change of integrated density of ac-H3K9 normalized to pan-H3K9 for each sample. Two independent experiments were performed per condition; one asterisk indicates P < 0.05 comparing the rhNAMPT-stimulated cells versus the TLR4 inhibitor–pretreated and rhNAMPT-stimulated cells.
Discussion
In VILI, NAMPT is an established candidate gene that is highly upregulated. The current data, for the first time, suggest differential DNA methylation profiles in two CpG islands in the NAMPT promoter in response to LPS and excessive mechanical stress. Furthermore, the data demonstrate other modes of epigenetic regulation, including HDAC-mediated reductions in NAMPT expression.
Preliminary analysis showed dose-dependent increases in NAMPT expression upon pharmacological inhibition of DNA methyltransferases.8 As a complementary approach, in vitro methylation of the NAMPT promoter luciferase constructs by CpG methyltransferase also showed decreased expression (data not shown). Using bisulfite sequencing of DNA from human lung endothelium, we extended our studies to investigate changes in methylation occurring in the NAMPT promoter at single-base resolution and demonstrated decreases in DNA methylation occurring in response to excessive mechanical stress and endotoxin challenges. Decreases in DNA methylation in the promoter are correlated with increases in gene transcription.15 Though our data lack experimental evidence to support binding of transcription factors to demethylated sites, our observation of decreased methylation concomitant with increased gene transcription suggests a direct link between changes in DNA methylation and transcriptional upregulation.
In addition to DNA methylation, chromatin modifications contribute to gene transcription. Chromatin modifications are mediated by histone acetyl transferases, which add acetyl groups to histones, thereby activating transcription, and deacetylases, which remove acetyl groups on histones to repress transcription.16,17 Recent studies have shown that HDAC activity is required for gene transcription, and LPS is known to induce HDAC activity.18 We observed that inhibition of class I and II HDACs in vitro using TSA significantly reduced basal-level expression of NAMPT and attenuated endotoxin-mediated NAMPT upregulation. Our earlier publication has described the intricate involvement of signal transducer and activator of transcription 5 (STAT5) in the regulation of NAMPT in response to mechanical stress.8 Because HDAC activity is required for transcription of STAT5-responsive genes,19 inhibition of HDAC activity may lead to decreased binding of STAT5 transcription factors to the NAMPT promoter, thus leading to the attenuated responses. In contrast to the observations of HDAC inhibition attenuating NAMPT gene and protein expression, silencing of Sirt-1, a class III HDAC, resulted in significantly increased NAMPT gene and protein expression, suggesting distinct mechanisms of action between class I and II HDACs versus class III HDACs. Relevant to our findings for STAT5-mediated transcription, Sirt-1 can also directly interact with STAT5 and alters its acetylation to modulate STAT5-mediated transcriptional responses.20
In our earlier published studies, we have conclusively demonstrated that secreted NAMPT binds to and activates the TLR4 receptor, and this interaction is a primary cause for the induction of proinflammatory cytokine production in mouse lungs and in human lung ECs, akin to LPS induction of TLR4.7 Although the role of LPS in inflammatory gene transcription is well known, the mechanistic role of rhNAMPT in inflammatory cytokine induction is not completely understood. Thus, we utilized recombinant NAMPT as an agonist for TLR4 activation and demonstrated significantly decreased H3K9 acetylation in human lung ECs. This result is consistent with other studies that have demonstrated LPS-mediated downregulation of H3K9 acetylation occurring during inflammation of the brain21 and also in LPS-challenged HLMVECs mimicking lung infection.18 To address the mechanisms driving downregulation of H3K9 acetylation, we utilized pharmacological inhibition of TLR4 receptor to establish the essential role of TLR4 pathway. The results demonstrated that CLI-095–mediated pharmacological inhibition of TLR4 receptor attenuated the rhNAMPT-induced decreases in H3K9 acetylation, which suggests that the changes in acetylation on the histone tail occurred in a TLR4-dependent manner. Furthermore, the transcript levels of Sirt-1, the upstream deacetylase for H3K9, are increased during TLR4 activation (through LPS) in the human lung endothelium. In contrast, LPS treatment of other cell types, including macrophages, decreased Sirt-1 expression at both the mRNA and protein levels, leading to increased H3K9 acetylation. Because H3K9 acetylation is a downstream target of Sirt-1, increases in Sirt-1 in the human lung endothelium during infection are a potential mechanism driving downregulation of H3K9 acetylation.22 H3K9 acetylation is an important regulatory mark for gene transcription, and modulations in H3K9 acetylation may potentially influence the epigenetic landscape of the endothelium and thus the transcription of inflammatory cytokines. It is interesting to note that increases in H3K9 acetylation in gene promoters are positively correlated with increased transcription; however, we observed global decreases in H3K9 acetylation. To understand the mechanisms in depth, use of ChIP-Seq to study the localization of acetyl H3K9 marks in the genome may increase our understanding of proinflammatory and antiinflammatory gene expression patterns in the lung endothelium. To our knowledge, this is the first report to suggest a global epigenetic modifying role for rhNAMPT.
ARDS and VILI are morbid syndromes with a poor mechanistic insight into the transcriptional activation of important inflammatory genes. In our studies, we have shown that NAMPT, an important gene upregulated in VILI, is influenced by epigenetic mechanisms in the human lung endothelium. Specifically, using bisulfite sequencing, we have demonstrated decreases in methylation occurring in the NAMPT promoter and 5ʹUTR that are positively correlated with increased gene transcription. To our knowledge, this is the first instance of changes in the NAMPT promoter that has been reported to occur in the setting of increased gene transcription. In silico analysis of the NAMPT gene promoter for promoter-binding elements has also identified several transcription factors, including PAX5 and E2F1, at sites of demethylation. This becomes increasingly important, because our earlier report has demonstrated increased STAT5 binding to the mechanical stress–inducible region of NAMPT 5ʹUTR upon pharmacological inhibition of methylation; however, the consensus binding motif for STAT transcription factors does not contain a CpG site, suggesting that a synergistic function with transcription factors that bind the demethylated sites is possible. In addition to demonstrating changes in DNA methylation, we also demonstrate that HDAC inhibitors specific for class I and II HDACs can attenuate the LPS-induced increases in NAMPT gene transcripts. In contrast to class I and II HDAC inhibition, class III HDAC inhibition significantly increased NAMPT gene transcripts. However, the increases in Sirt-1 mRNA, simultaneous with decreases in global H3K9 acetylation and increases in NAMPT transcription, suggest complicated mechanisms that need further evaluation. Finally, we also intricately linked the decreases in H3K9 acetylation occurring in ECs during inflammation to the direct binding of rhNAMPT to the TLR4 receptor. Thus we have identified and demonstrated that NAMPT, a key ARDS and VILI candidate gene, is epigenetically regulated during transcription and potentially contributes to the epigenetic landscape during excessive mechanical stress and endotoxin challenges.
Appendix.
Figure S1.

Sirt-1 expression is increased by toll-like receptor 4 (TLR4) activation. Quantitative PCR (qPCR) assays demonstrated increased expression of Sirt-1 messenger RNA transcripts in lipopolysaccharide (LPS)–mediated TLR4 activation in human lung endothelial cells. RNA from cells activated with LPS was reverse transcribed and probed with primers specific for Sirt-1. The results showed that Sirt-1 was increased as early as 2 hours and stayed elevated at 4 hours. The data are for n = 3. Asterisk indicates P < 0.05 compared with 0 hours. mRNA: messenger RNA.
Source of Support: This study is supported by National Institutes of Health grants R01HL94394, P01HL126609, and R01HL91889.
Conflict of Interest: None declared.
Supplements
Appendix (519.8KB, pdf)
References
- 1.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, Gattinoni L, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016;315(8):788–800. [DOI] [PubMed]
- 2.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med 2013;369(22):2126–2136. [DOI] [PubMed]
- 3.Kamp R, Sun X, Garcia JG. Making genomics functional: deciphering the genetics of acute lung injury. Proc Am Thorac Soc 2008;5(3):348–353. [DOI] [PMC free article] [PubMed]
- 4.Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol 2007;23(2):164–170. [DOI] [PubMed]
- 5.Ye SQ, Simon BA, Maloney JP, Zambelli-Weiner A, Gao L, Grant A, Easley RB, et al. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med 2005;171(4):361–370. [DOI] [PubMed]
- 6.Hong SB, Huang Y, Moreno-Vinasco L, Sammani S, Moitra J, Barnard JW, Ma SF, et al. Essential role of pre-B-cell colony enhancing factor in ventilator-induced lung injury. Am J Respir Crit Care Med 2008;178(6):605–617. [DOI] [PMC free article] [PubMed]
- 7.Camp SM, Ceco E, Evenoski CL, Danilov SM, Zhou T, Chiang ET, Moreno-Vinasco L, et al. Unique toll-like receptor 4 activation by NAMPT/PBEF induces NFκB signaling and inflammatory lung injury. Sci Rep 2015;5:13135. [DOI] [PMC free article] [PubMed]
- 8.Sun X, Elangovan VR, Mapes B, Camp SM, Sammani S, Saadat L, Ceco E, et al. The NAMPT promoter is regulated by mechanical stress, signal transducer and activator of transcription 5, and acute respiratory distress syndrome–associated genetic variants. Am J Respir Cell Mol Biol 2014;51(5):660–667. [DOI] [PMC free article] [PubMed]
- 9.Joss-Moore LA, Albertine KH, Lane RH. Epigenetics and the developmental origins of lung disease. Mol Genet Metab 2011;104(1–2):61–66. [DOI] [PMC free article] [PubMed]
- 10.Weinhold B. Epigenetics: the science of change. Environ Health Perspect 2006;114(3):A160–A167. [DOI] [PMC free article] [PubMed]
- 11.Wierda RJ, Geutskens SB, Jukema JW, Quax PH, van den Elsen PJ. Epigenetics in atherosclerosis and inflammation. J Cell Mol Med 2010;14(6A):1225–1240. [DOI] [PMC free article] [PubMed]
- 12.Wang T, Chiang ET, Moreno-Vinasco L, Lang GD, Pendyala S, Samet JM, Geyh AS, et al. Particulate matter disrupts human lung endothelial barrier integrity via ROS- and p38 MAPK-dependent pathways. Am J Respir Cell Mol Biol 2010;42(4):442–449. [DOI] [PMC free article] [PubMed]
- 13.Adyshev DM, Elangovan VR, Moldobaeva N, Mapes B, Sun X, Garcia JG. Mechanical stress induces pre-B-cell colony-enhancing factor/NAMPT expression via epigenetic regulation by miR-374a and miR-568 in human lung endothelium. Am J Respir Cell Mol Biol 2014;50(2):409–418. [DOI] [PMC free article] [PubMed]
- 14.Xing S, Nie F, Xu Q, Deng Y, Li W, Yang Z, Zhao X, et al. HDAC is essential for epigenetic regulation of Thy-1 gene expression during LPS/TLR4-mediated proliferation of lung fibroblasts. Lab Invest 2015;95(10):1105–1116. [DOI] [PubMed]
- 15.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011;25(10):1010–1022. [DOI] [PMC free article] [PubMed]
- 16.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011;21(3):381–395. [DOI] [PMC free article] [PubMed]
- 17.Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J 2005;25(3):552–563. [DOI] [PubMed]
- 18.Thangjam GS, Dimitropoulou C, Joshi AD, et al. Novel mechanism of attenuation of LPS-induced NF-kappaB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am J Respir Cell Mol Biol 2014;50(5):942–952. [DOI] [PMC free article] [PubMed]
- 19.Rascle A, Johnston JA, Amati B. Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol Cell Biol 2003;23(12):4162–4173. [DOI] [PMC free article] [PubMed]
- 20.Yamamoto M, Iguchi G, Fukuoka H, et al. SIRT1 regulates adaptive response of the growth hormone–insulin-like growth factor-I axis under fasting conditions in liver. Proc Natl Acad Sci U S A 2013;110(37):14948–14953. [DOI] [PMC free article] [PubMed]
- 21.Soliman ML, Rosenberger TA. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol Cell Biochem 2011;352(1–2):173–180. [DOI] [PubMed]
- 22.Shen Z, Ajmo JM, Rogers CQ, Liang X, Le L, Murr MM, Peng Y, You M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am J Physiol Gastrointest Liver Physiol 2009;296(5):G1047–G1053. [DOI] [PMC free article] [PubMed]