

Abstract
Although proteasome inhibitors (PIs) are used as anticancer drugs to treat various cancers, their relative therapeutic efficacy on stem cells vs. bulk cancers remains unknown. Here, we show that stem cells derived from gliomas, GSCs, are up to 1,000‐fold more sensitive to PIs (IC 50, 27–70 nM) compared with their differentiated controls (IC 50, 47 to »100 μM). The stemness of GSCs correlates to increased ubiquitination, whose misregulation readily triggers apoptosis. PI‐induced apoptosis of GSCs is independent of NF‐κB but involves the phosphorylation of c‐Jun N‐terminal kinase as well as the transcriptional activation of endoplasmic reticulum (ER) stress‐associated proapoptotic mediators. In contrast to the general notion that ER stress‐associated apoptosis is signaled by prolonged unfolded protein response (UPR), GSC‐selective apoptosis is instead counteracted by the UPR. ATF3 is a key mediator in GSC‐selective apoptosis. Pharmaceutical uncoupling of the UPR from its downstream apoptosis sensitizes GSCs to PIs in vitro and during tumorigenesis in mice. Thus, a combinational treatment of a PI with an inhibitor of UPR‐coupled apoptosis may enhance targeting of stem cells in gliomas.
Keywords: apoptosis, c‐Jun N‐terminal kinase, glioma stem cells, proteasome inhibitors, ubiquitin proteasome system
Subject Categories: Autophagy & Cell Death, Cancer, Stem Cells
Introduction
Gliomas, malignancies originating from glial cells in the brain, are the most common malignant tumors occurring in the central nervous system and one of the most expensive cancers to treat 1. Despite advances in therapeutic strategies over past decades, patient outcomes are very poor and the median patient survival is still < 2 years after diagnosis, making gliomas a significant public health issue 1. To date, alkylating agents, including temozolomide (TMZ) and nitrosoureas, are the most commonly used chemotherapy treatments in the clinic 2. However, malignant gliomas are resistant to conventional chemotherapy and are highly recurrent in a local fashion after surgical removal, mainly due to the diffuse invasion of tumor cells into the brain 3. One reason for this poor responsiveness is the phenotypic heterogeneity of gliomas, which are composed of a mixture of various cell types, including more differentiated astrocyte‐like cells and a rare subpopulation that displays stem cell characteristics called glioma stem cells (GSCs) 4, 5, 6. Similar to other cancer stem cells, GSCs are defined as a highly tumorigenic cell subset responsible for in vivo tumor growth and propagation through their characteristic capacities for indefinite self‐renewal and differentiation into a non‐tumorigenic bulk tumor cell population 7. GSCs have been implicated in resistance of the tumor to chemo‐ and radiotherapy 5, 8, angiogenesis by elevated expression of VEGF 9, and aggressive invasive phenotype 10, contributing to tumor recurrence and the failure of conventional therapies. As conventional therapies targeting the rapidly proliferating bulk of tumor cells are likely to fail in targeting the GSC population, GSCs are emerging as an attractive therapeutic target to control glioma growth and progression 4.
The Ub‐proteasome system (UPS) mediates Ub‐dependent degradation of short‐lived proteins through the proteasome 11, 12. The UPS targets short‐lived regulators involved in various processes such as cell cycle control, DNA repair, apoptosis, tumor growth, and stress response to maintain cellular homeostasis 13, 14, 15, 16. The timely degradation of these substrates is essential for cancer cell growth and survival. Moreover, owing to uncontrolled proliferation, cancer cells accumulate abnormal proteins more rapidly and, thus, are more sensitive to proteasomal inhibition than normal cells 13. Many PIs such as lactacystin, MG132, bortezomib/PS341 (marketed as Velcade®), epoxomicin, and SC68896 show antiproliferative or proapoptotic activity in various hematological and solid malignancies at IC50 values of 1–10 μM 13, 17. Bortezomib is the first FDA‐approved drug for the treatment of multiple myeloma and mantle cell lymphoma 17 and demonstrates anticancer activity in vitro against various cancer cell lines, including those derived from colon, ovary, pancreas, lung, breast, bladder, and prostate cancers 18. Recent studies suggest that PIs can also induce apoptosis in human glioma cell lines and primary glioblastoma multiforme (GBM) explants 19, 20. PIs were also shown to induce apoptosis in glioma‐derived stem‐like cells 21, 22, 23, 24, 25. In a recent siRNA screening to identify genes important for GBM cell survival, 22% (12/55) of the hits were components of the 20S and 26S proteasome subunits 26. Although the action mechanisms still remain poorly understood, extensive studies have shown that inactivation of the NF‐κB pathway significantly contributes to the apoptotic death of cancer cells caused by PIs at 1–10 μM 27. Despite the demonstrated therapeutic efficacy of PIs as anticancer agents, clinical studies have reported that many solid tumors do not respond well to PI treatments, possibly because of inaccessibility to cancer stem cells within the tumors, limiting their therapeutic potential 28. It remains poorly understood whether and, if so, to what degree PIs have differential cytotoxic activities on stem cells embedded in bulk tumors.
In this study, we show that PIs selectively kill GSCs (IC50, 27–70 nM) as compared with their non‐stem differentiated controls (IC50, 47 μM to » 100 μM). GSC‐selective apoptosis is independent of NF‐κB but requires the transcription factor ATF3. The ATF3‐dependent killing of GSCs involves the transcriptional activation of so‐called endoplasmic reticulum (ER) stress‐associated apoptosis. In contrast to known properties of many non‐stem cancer cells, however, pharmaceutical uncoupling of the UPR from apoptosis renders GSCs even more sensitive to PIs. Our results suggest that a combination treatment of a PI with an inhibitor of UPR‐coupled apoptosis may be useful to target stem cells in gliomas.
Results
Validation of the in vitro culture model of GSCs
We have previously established a suspension culture model of human primary GSC lines (XO1, XO2, XO3, XO4, XO6, XO8, XO9, and XO10) from gliomas of patients 29, 30. In this study, we induced the differentiation of XO6, XO8, and XO10 GSCs by passaging them seven times on an adhesive surface in serum‐containing media. Semi‐quantitative PCR and immunoblotting analyses of GSCs and their differentiated, non‐stem glioma cancer cells (DFCs; see Materials and Methods) showed that GSC‐specific stemness markers, such as Nestin, Musashi‐1 and SOX2, were selectively expressed in GSCs and gradually downregulated in the course of differentiation (Fig 1A and B). Similar passage‐dependent downregulation was observed with general stem cell markers, including NANOG, OCT4, i‐NOS, and Olig2. The expression of GFAP and βIII‐tubulin, markers of astrocyte and neuronal differentiation, respectively, was activated at passages 2 through 4 and subsequently returned to basal levels (Fig 1B). Following differentiation, the doubling time increased from 16 h in GSCs to 32 h in DFCs (Fig 1C).
Figure 1. Validation of the in vitro culture model of GSCs.
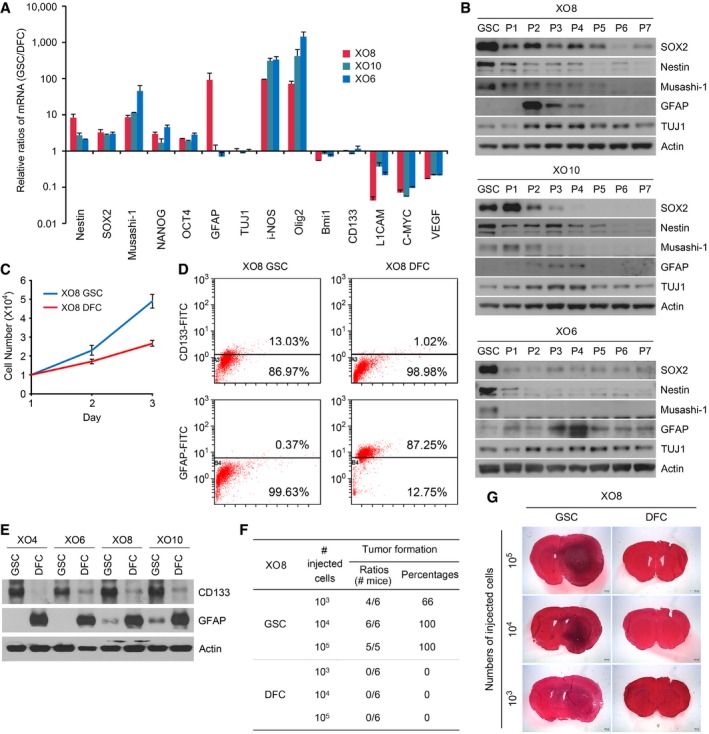
- GSCs contain the higher levels of stemness markers (Nestin, SOX2, Musashi‐1, NANOG, and OCT4) compared with their DFCs. Total RNAs from GSCs and DFCs were subjected to real‐time PCR. The average relative amounts of GSCs were normalized with those of DFCs. Shown are means + SD values of the normalized mRNA levels of GSCs obtained from the three independent experiments.
- Immunoblotting analysis of markers for stemness (SOX2, Nestin, and Musashi‐1) and differentiation [GFAP and TUJ1 (neuron‐specific class III beta‐tubulin)] in GSCs in comparison with GSC‐derived cells at different passage numbers (P1–P7).
- Measurement of the cell doubling time. Approximately 3 × 104 cells for each of XO8 GSCs and XO8 DFCs were seeded in a 60‐mm culture dish and counted every day. The cells were treated with trypsin–EDTA, and the number of viable cells was counted with a hemocytometer. The doubling time was calculated from the cell growth curve over 5 days. Shown are means ± SD values obtained from three independent experiments.
- FACS analysis of CD133 and GFAP using X08 GSCs and DFCs. Data are from representative experiments repeated at least three times.
- Immunoblotting analysis of CD133 and GFAP in GSCs in comparison with DFCs.
- Quantitation of (G).
- Equal numbers of XO8 GSCs and DFCs were injected orthotropically into mouse brains. After 6 weeks, brains were harvested and tumorigenic potential was assessed by evaluating tumor formation. Representative microphotographs of the brain sections of mice injected with XO8 GSCs and XO8 DFC controls.
Source data are available online for this figure.
As an alternative way to assess the stemness of GSCs and the differentiation status of DFCs, we examined the expression profiles of the stemness marker CD133 and the differentiation marker GFAP in GSC spheres using flow cytometry in comparison with DFCs. Consistent with our previous observations with other XO GSCs 29, 30, approximately 13% of XO8 GSCs were CD133+ as compared with 1% for DFCs (Fig 1D). Moreover, 0.37% of XO8 GSCs were GFAP+ as compared with 97.3% for DFCs (Fig 1D). These results were confirmed using Western blot analysis (Fig 1E).
We have previously validated the high tumorigenicity of XO1 GSCs in mice 31, 32. To determine the tumorigenicity of XO8 GSCs, we injected XO8 GSCs and DFCs into the brains of BALB/c nude mice and compared tumorigenesis. None of the mice receiving 103, 104, or 105 XO8 DFCs developed tumors. By contrast, only 103 XO8 GSCs was sufficient to induce tumors (Fig 1F and G). These results validate the model of GSCs and DFCs of this study.
Stem cells derived from gliomas are hypersensitive to PIs
Despite the demonstrated therapeutic efficacy of PIs on various cancers, their relative cytotoxicity to stem cells vs. bulk cancer cells has not been systematically characterized. To develop a strategy targeting stem cells, we treated GSCs and DFCs with various anticancer agents and measured their cytotoxicity using mitochondrial NADH dehydrogenase activity (see Materials and Methods). GSCs showed no significant difference to TMZ (IC50, 3 mM) and cisplatin (IC50, 22 μM), first‐ and second‐line chemotherapeutic drugs for glioma, respectively, as compared with their non‐stem controls (Fig 2A and B). Similar results were obtained with other types of anticancer agents (adriamycin, taxol, and TRAIL) as well as the autophagic inhibitor bafilomycin A1 and the ER stressor thapsigargin (Fig 2C–G). Strikingly, XO8 GSCs (IC50, 75 nM) were unusually sensitive to the proteasome inhibitor MG132 as compared with TMZ (IC50, 3 mM) and cisplatin (IC50, 22 μM) (Fig 2H). Moreover, XO8 GSCs (IC50, 75 nM) were strikingly more sensitive to MG132 than GSC‐derived non‐stem controls (DFCs) (IC50, » 100 μM), two glioma‐derived non‐stem cell lines [U87 (IC50, 47 μM) and U251 (IC50, 75 μM)], and two non‐glioma cancer cell lines [HeLa (IC50, » 100 μM) and HEK293 (IC50, » 100 μM)] (Fig 2H). The hypersensitivity of GSCs to PIs was similarly observed when cell viability was measured using trypan blue exclusion assay (Fig 2I). As an alternative measurement, we also determined the efficacy of MG132 to inhibit the self‐renewal of GSCs using in vitro colony formation assays (Fig 2J). MG132 completely inhibited the clonogenic growth of XO8 GSCs at an even lower concentration (IC50, 20 nM). To our knowledge, these IC50 values of GSCs are the lowest compared with those of any other cancer types reported (Fig 2H).
Figure 2. GSCs are hypersensitive to PIs compared with their DFCs.
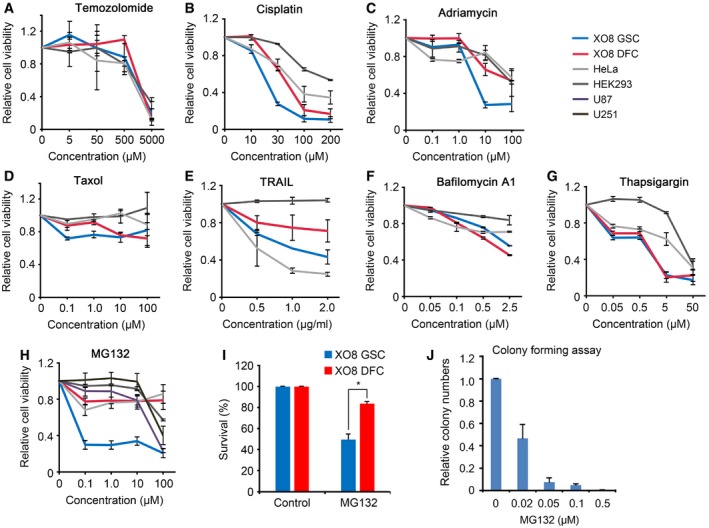
-
A–HCell viability assays were performed as described in the Materials and Methods after cells were treated with temozolomide (A), cisplatin (B), adriamycin (C), taxol (D), TRAIL (E), bafilomycin A1 (F), thapsigargin (G), and MG132 (H). Means ± SD from at least three independent experiments are shown as relative indexes after normalization to those of individual control cells treated with DMSO.
-
IXO8 GSCs and DFCs were treated with 50 nM MG132 for 24 h. Cell viability was measured using trypan blue exclusion assay. Error bars represent the mean ± SD from three independent experiments. For statistical analysis, Student's t‐test (two‐sided, paired) was used (*P < 0.05).
-
JXO8 GSCs were treated with MG132 were cultured in 0.3% soft agar as described in the Materials and Methods. The colonies bigger than 100 μm in diameter in four randomly chosen fields were subjected to blind counting. Bars, means + SD from three independent experiments.
To test whether MG132 selectively kills stem cells in a mixture, XO8 GSCs and DFCs were differentially stained with green (PKH67) and red (PKH26) fluorescent dyes, respectively, and cultured as a mixture in serum‐free media, followed by treatment with 50 nM MG132 for 17 h. Fluorescent microscopy showed that green fluorescence‐labeled XO8 GSCs were selectively eliminated, while red fluorescence‐labeled XO8 DFCs remained largely resistant (Fig 3A and B). Flow cytometry showed that the ratio of GSCs (green) decreased from 52 to 26%, whereas DFCs (red) increased from 45 to 67% (Fig 3C).
Figure 3. GSCs are hypersensitive to proteasomal inhibition.
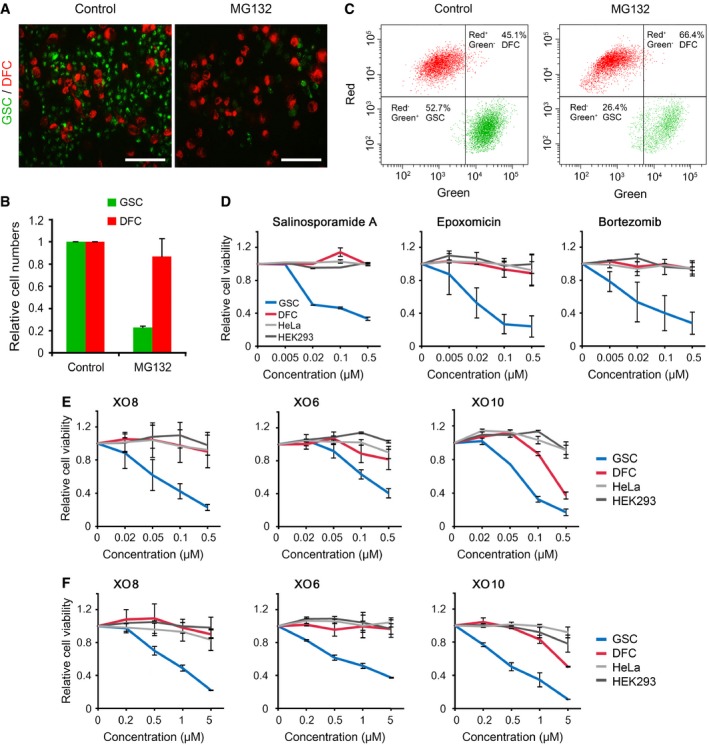
- XO8 GSCs and DFCs were differentially labeled with PKH67 (green) and PKH26 (red), respectively, and mixed before plated on laminin‐coated plates. The cells were treated with 50 nM MG132 and subjected to fluorescent microscopy. Scale bar, 100 μm.
- Quantitation of (A). Cell numbers from three independent experiments were normalized to those of control cells treated with DMSO and presented as means + SD (P = 0.0035, n = 3). Student's t‐test (two‐sided, paired) was used for statistical analysis.
- The harvested cells from (A) were subjected to FACS analysis.
- XO8 GSCs were treated with various PIs (salinosporamide A, epoxomicin, and PS341), followed by cell viability assays. Bars, means ± SD from three independent experiments.
- XO6, XO8, and XO10 GSCs were treated with MG132 and subjected to cell viability assay. Bars, means ± SD from three independent experiments.
- XO6, XO8, and XO10 GSCs were treated with MG132 and subjected to cell viability assay in serum‐containing media. Bars, means ± SD from three independent experiments.
Source data are available online for this figure.
To determine whether MG132 exerts cytotoxicity to GSCs through proteasomal inhibition, we treated XO8 GSCs and DFCs with various PIs that have differential specificities to the β5, β2, and β1 subunits of the 20S particle. XO8 GSCs showed similar sensitivity to all the tested PIs, including bortezomib (IC50, 40 nM), epoxomicin (IC50, 27 nM), and salinosporamide A (IC50, 29 nM) (Fig 3D). The results were validated using XO6 (IC50, 335 nM) and XO10 (IC50, 79 nM) GSCs independently established from patients with glioma (Fig 3E). Next, we determined whether the cytotoxicity of PIs is influenced by the presence of serum in media during treatment. Even when cultured on the surface in serum‐containing media during MG132 treatment, GSCs showed IC50 values (XO6, 1.4 μM; XO8, 560 nM; XO10, 450 nM) significantly lower than those of DFCs (XO6, » 5 μM; XO8, » 5 μM; XO10, » 5 μM) (Fig 3F). These results suggest that stem cells derived from gliomas are more sensitive to misregulation in the UPS as compared with bulk cancer mass.
The stemness of GSCs correlates to increased ubiquitination
We speculated that GSCs may be hypersensitive to misregulation in proteasomal degradation of ubiquitinated proteins. Indeed, immunoblotting analysis of Ub conjugates showed that the overall level of ubiquitination was significantly higher in XO8 GSCs as compared with in XO8 DFCs (Fig 4A). To determine whether a dynamic change occurs in ubiquitination during differentiation, we monitored ubiquitination in the course of differentiation. Cellular ubiquitination was highest in XO8 GSCs and gradually downregulated as cell passaging was repeated (Fig 4B). Decreased ubiquitination during differentiation was similarly observed with XO6 GSCs (Fig 4C). In vitro proteasome activity assays using extracts of XO6, XO8, and XO10 GSCs showed that proteasomal model substrates were cleaved faster in GSCs than in non‐stem cells (Fig 4F–I). Proteasomal activities of the stem cells were higher than those of other non‐stem, non‐glioma cancer cells, such as HeLa and HEK293 (Fig 4F–I). Immunoblotting and semi‐quantitative RT–PCR analyses revealed no significant changes between GSCs and DFCs in the numbers of the 26S proteasome and some Ub ligases (Figs 4D and E, and 5A–D) (note that part of Fig 5C is also shown as the left panel of Fig 4A). These results suggest that the stemness of glioma‐derived stem cells may depend on UPS‐dependent degradation of key regulators.
Figure 4. The stemness of GSCs correlates to increased ubiquitination, and PIs induce GSC‐specific apoptosis.
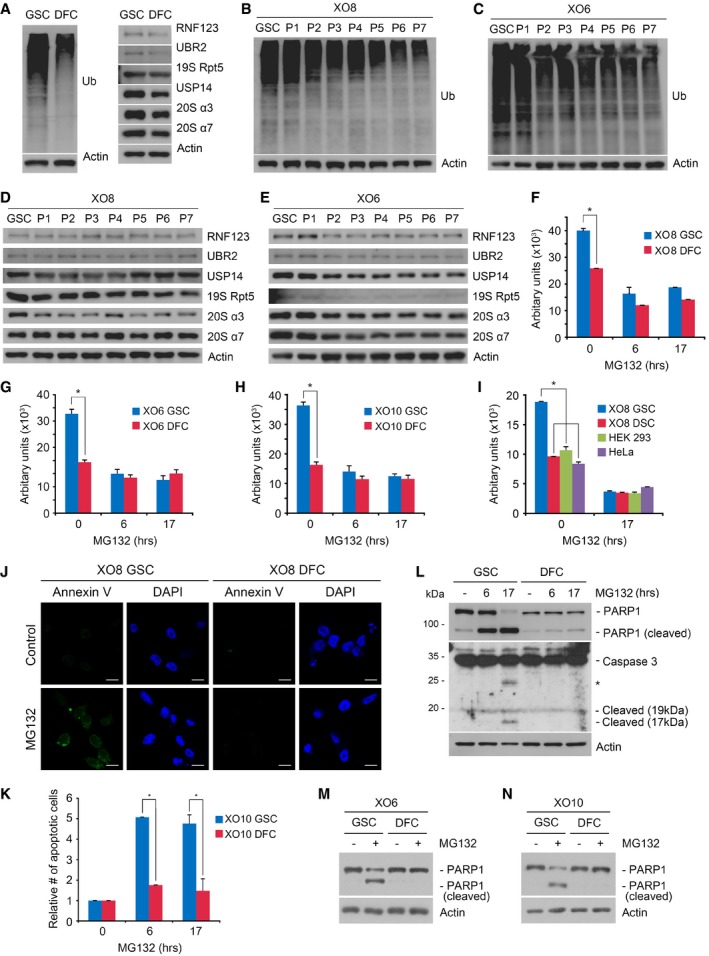
-
AImmunoblotting of XO8 GSCs and DFCs.
-
BImmunoblotting analysis of Ub in XO8 GSCs and XO8 GSC‐derived cells in the course of differentiation (P1–P7).
-
CSimilar to (B) except that XO6 GSCs were used.
-
DImmunoblotting analysis of ubiquitin ligases and proteasomal subunits in XO8 GSCs and XO8 GSC‐derived cells in the course of differentiation (P1–P7).
-
ESimilar to (C) except that XO6 GSCs were used.
-
F–IIn vitro proteasome activity assays were performed as described in the Materials and Methods. Bars represent means + SD from three independent experiments. For statistical analysis, Student's t‐test (two‐sided, paired) was used. *P‐values: (F) 0.0039; (G) 0.0052; (H) 0.0009, (I) DFC, 0.00032; HEK293, 0.0039; HeLa, 0.00051.
-
JXO8 GSCs and XO8 DFCs treated with MG132 as described in the Materials and Methods were labeled with Annexin V and visualized using fluorescent microscopy. Scale bar, 100 μm.
-
KCells from (J) were subjected to flow cytometry analysis. Annexin V‐positive cell numbers were obtained from three independent experiments and normalized to those of controls cells treated with DMSO. Bars, means + SD from three independent experiments. For statistical analysis, Student's t‐test (two‐sided, paired) was used. *P‐values: 6 h, 0.0012; 17 h, 0.0041.
-
LCells treated with 50 nM MG132 were subjected to immunoblotting analysis of apoptotic markers. Asterisk indicates non‐specific band.
-
M, NSimilar to (L) except that XO6 and XO10 cells were used.
Source data are available online for this figure.
Figure 5. Analysis of the mRNA expression of proteasomal subunits and the NF‐κB pathway in GSCs.
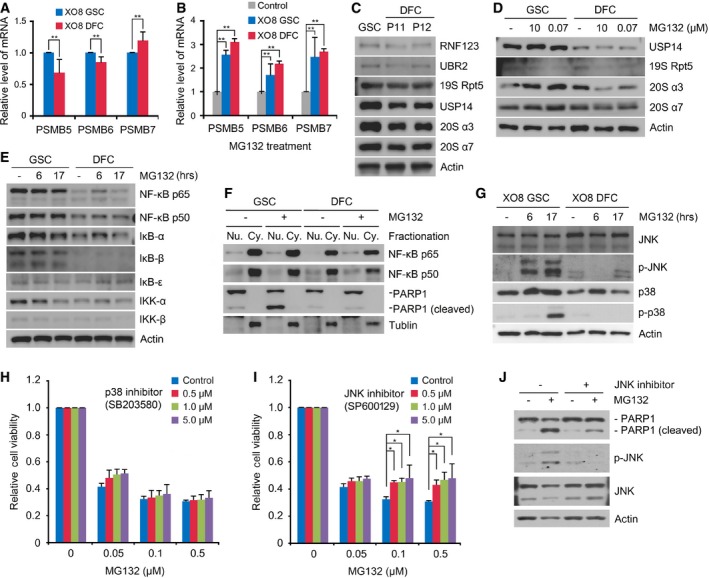
- Real‐time PCR analysis of the proteasomal subunits PSMB5, PSMB6, and PSMB7 in XO8 GSCs and XO8 DFCs. Shown are means + SD values from the three independent experiments following normalization to those of GSCs. For statistical analysis, Student's t‐test (two‐sided, paired) was used. **P‐values: PSMB5, 0.053; PSMB6, 0.034; PSMB7, 0.067.
- Similar to (A) except that the cells were treated with 50 nM MG132 for 17 h. Shown are means + SD values from the three independent experiments following normalization to those of control cells treated with DMSO. For statistical analysis, Student's t‐test (two‐sided, paired) was used. **P‐values: PSMB5, 0.022; PSMB6, 0.08; PSMB7, 0.603.
- Same as (C) except that the cells were treated with MG132 for 17 h.
- Immunoblotting analysis of the NF‐κB pathway in XO8 GSCs and XO8 DFCs.
- Fractionation analysis of NF‐κB in XO8 GSCs and XO8 DFCs treated with 50 nM MG132 for 17 h. PARP1 and tubulin were used as markers for the nucleus and cytoplasm, respectively.
- Immunoblotting analysis of JNK and p38.
- Cell viability assay was performed as in Fig 2 with cells treated with MG132 alone or in combination with SB203580. Shown are means + SD values from three independent experiments following normalization to those of control cells treated with DMSO. For statistical analysis, Student's t‐test (two‐sided, paired) was used. P‐values: 0.724, 0.502, 0.439 for 0.5, 1.0, and 5.0 μM SB203580, respectively, at 0.1 μM MG132, and 0.760, 0.683, 0.498 for 0.5, 1.0, and 5.0 μM SB203580, respectively, at 0.5 μM MG132.
- Experimental conditions were similar to (H) except that SP600129 was used. Shown are means + SD values from three independent experiments following normalization to those of control cells treated with DMSO. For statistical analysis, Student's t‐test (two‐sided, paired) was used. *P‐values: 0.009, 0.002, 0.07 for 0.5, 1.0, and 5.0 μM SP600129, respectively, at 0.1 μM MG132, and 0.018, 0.025, 0.09 for 0.5, 1.0, and 5.0 μM SP600129, respectively, at 0.5 μM MG132.
- Immunoblotting analysis of XO8 GSCs treated with 50 nM MG132 alone or in combination with the JNK inhibitor SP600129.
Proteasomal misregulation triggers an NF‐κB‐independent apoptosis selectively in glioma stem cells
To investigate the mechanism by which GSCs are selectively killed by PIs, XO8 GSCs and DFCs were treated with 50 nM MG132 and stained with Annexin V. Immunofluorescence microscopy 17 h post‐treatment revealed that the majority of GSCs, but not DFCs, became Annexin V‐positive (Fig 4J). Flow cytometry showed that the Annexin V‐positive population in MG132‐treated GSCs sharply expanded as early as 6 h (Fig 4K). Immunoblotting analysis confirmed that caspase‐3 and PARP1 were cleaved in 50 nM MG132‐treated XO8 GSCs at 6 and 17 h (Fig 4L). A similar GSC‐specific PARP1 cleavage was observed in XO6 and XO10 GSCs (Fig 4M and N). These results suggest that glioma‐derived stem cells contain an apoptotic switch hypersensitive to proteasomal misregulation as compared with glioma‐derived non‐stem cells or non‐glioma immortalized cell lines.
The inhibition of NF‐κB has been shown to be a dominating molecular mechanism underlying the therapeutic effects of PIs (typically at 1–10 μM) in many cancer types 18, 33, 34. However, we observed no significant differences in the levels of the NF‐κB subunits p65 and p50 and the IκB family members (IκB‐α, IκB‐β, and IκB‐ε) when GSCs and DFCs were treated with 50 nM MG132 (Fig 5E). This result was further validated with IKK‐α and IKK‐β and their phosphorylated forms that mediate IκB phosphorylation (Fig 5E). Subcellular fractionation retrieved comparable amounts of NF‐κB p65 and p50 subunits from the nucleus (Fig 5F). These results suggest that stem cells derived from gliomas operate an NF‐κB‐independent apoptotic circuit which is turned on under 50 nM MG132.
JNK mediates GSC‐selective apoptosis under proteasomal misregulation
Recent studies showed that p38 and c‐Jun N‐terminal kinase (JNK) of the mitogen‐activated protein kinase (MAPK) pathway underlie the therapeutic efficacy of 1–10 μM PIs in lung cancer, glioblastoma, and multiple myeloma 20, 35. Immunoblotting analyses showed that GSCs treated with 50 nM MG132 strongly induced JNK phosphorylation at 6 h post‐treatment, followed by p38 phosphorylation at 17 h (Fig 5G). By contrast, no significant phosphorylation of p38 or JNK was detected in XO8 DFCs under the same conditions (Fig 5G). We then compared the role of JNK and p38 in GSC‐specific apoptosis when treated with 50 nM MG132. Cytotoxicity assays showed that GSC‐specific apoptosis was significantly inhibited by the JNK1/2 inhibitor SP600129 (Fig 5I) but not by the p38 inhibitor SB203580 (Fig 5H). Immunoblotting assays confirmed that the JNK inhibitor abolished JNK phosphorylation as well as PARP1 cleavage (Fig 5J). These results suggest that GSC‐specific apoptosis is mediated through in part JNK phosphorylation and in part JNK‐independent pathway(s).
Proteasomal inhibition transcriptionally activates a group of UPR‐associated proapoptotic proteins selectively in GSCs
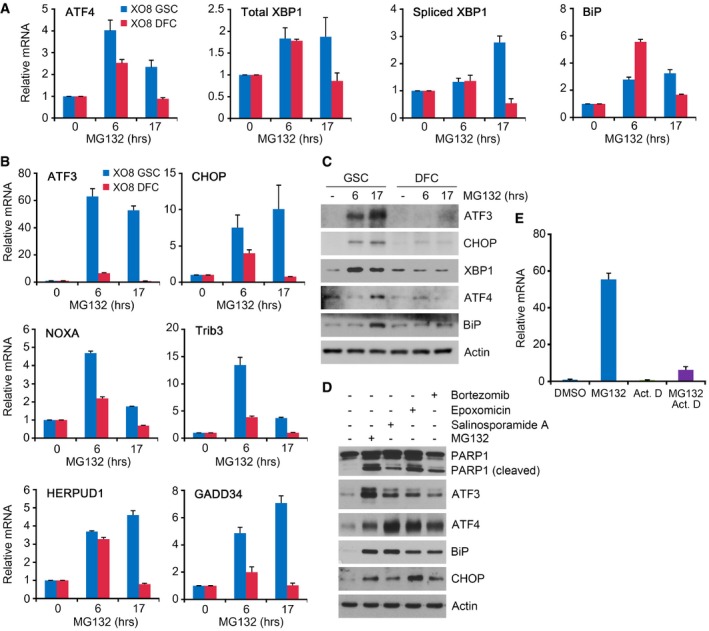
Proteasomal inhibition causes the accumulation of misfolded proteins in the ER, triggering the UPR 36. During the early phase, the UPR increases the synthesis of folding factors to enhance folding capacity and halts protein synthesis to reduce ER loading. However, if proteasomal inhibition prolongs, the UPR triggers so‐called ER stress‐associated apoptosis 36. Notably, our microarray analysis revealed a strong transcriptional induction of UPR core components (e.g., BiP, XBP1, and ATF4) as well as ER stress‐associated proapoptotic proteins specifically in GSCs treated with 50 nM MG132 for 6 and 17 h (Appendix Tables S2–S5). These proapoptotic proteins include ATF3, CHOP, DDIT4, HERPUD1, GADD34, TRIB3, NOXA, and DNAJB1, all of which are known to be induced during the late‐phase UPR. The induction of ER‐associated proapoptotic proteins was reproduced using semi‐quantitative PCR (Fig 6A and B) and immunoblotting (Fig 6C and D) analyses.
Figure 6. The expressions of ER stress‐associated proapoptotic genes are selectively induced by MG132 in GSCs.
-
A, BXO8 GSCs and XO8 DFCs were treated with 50 nM MG132 for indicated hours. The expression of genes was assessed by real‐time PCR using total RNA extracted from MG132‐treated cells. Mean + SD values from the three independent experiments are shown after normalization to those of control cells treated with DMSO.
-
CXO8 GSCs and XO8 DFCs treated with 50 nM MG132 as described in (A) were subjected to immunoblotting.
-
DXO8 GSCs were treated with MG132, salinosporamide A, epoxomicin, or bortezomib for 24 h. Cell lysates were subjected to immunoblot analysis with anti‐PARP‐1, anti‐CHOP, anti‐ATF3, anti‐ATF4, and anti‐BiP antibodies.
-
EXO8 cells were treated with 50 nM MG132 in the absence or presence of 5 μM actinomycin D (ActD) for 6 h. The level of ATF3 mRNA was measured using semi‐quantitative real‐time PCR in comparison with β‐actin. Mean + SD values from three independent experiments are shown after normalization to those of control cells treated with DMSO.
Source data are available online for this figure.
ATF3 mediates GSC‐specific apoptosis under proteasomal inhibition
It is known that the proapoptotic transcription factor ATF3 is transcriptionally induced by the PERK‐eIF2α‐ATF4 pathway during the late UPR 37, 38. When GSCs and DFCs were treated with 50 nM MG132, the mRNA level of ATF3 increased 31‐folds in GSCs (Fig 6B, and Appendix Tables S2 and S4), which correlated with the increased protein level of ATF3 (Fig 6C and D). The treatment of actinomycin D, a transcriptional inhibitor, abolished such an induction of ATF3 mRNA in PI‐treated GSCs, suggesting that this induction is largely transcriptional (Fig 6E). The induction of ATF3 is specific to GSCs as no such induction was observed in DFCs under the same conditions. To test the role of ATF3 in apoptosis of GSCs, we knocked down ATF3 using lentiviral shRNA in GSCs treated with 50 nM MG132. ATF3 shRNA #5 and #6 showed high knockdown efficiency and, thus, strongly inhibited the cleavage of PARP1 and caspase‐3 in GSCs (Fig 7A and B). By contrast, shRNA #1 showed poor knockdown and barely inhibited such cleavages (Fig 7A and B). Thus, ATF3 mediates apoptosis selectively in glioma‐derived stem cells under proteasomal inhibition.
Figure 7. ATF3 and NOXA mediate low‐dose PI‐induced apoptosis in GSCs.
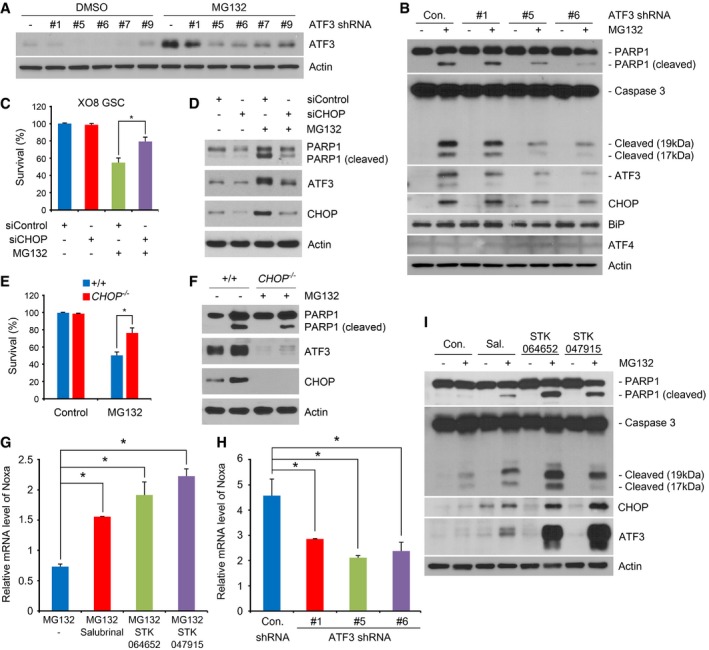
- HEK293 cells were transfected with ATF3 shRNA or vector (pLKO1), treated with 50 nM MG132, and then subjected to immunoblotting.
- Individualized XO8 GSCs were transduced with lentiviral shRNAs against ATF3 or control as described in the Materials and Methods. After 4 days, individualized GSCs were treated with 50 nM MG132 and subjected to immunoblotting.
- XO8 GSCs were transfected with control or CHOP siRNA for 48 h and then treated with 50 nM MG132 for 24 h. Cell viability was determined using the trypan blue exclusion assay. Error bars represent the mean ± SD from three independent experiments (*P < 0.05, n = 3). For statistical analysis, Student's t‐test (two‐sided, paired) was used.
- Immunoblotting analysis of (C). The cells were collected, lysed, and subjected to immunoblot analysis with indicated antibodies.
- Wild‐type and CHOP −/− MEF cells were treated with MG132 for 24 h. Cell viability was determined using trypan blue exclusion assay. Error bars represent the mean ± SD from three separate experiments (*P < 0.05, n = 3). For statistical analysis, Student t‐test (two‐sided, paired) was used.
- Cell lysates obtained in (E) were subjected to immunoblot analysis with anti‐PARP‐1, anti‐CHOP, or anti‐ATF3 antibody.
- The expression of NOXA was assessed with real‐time PCR using total RNA extracted from XO8 GSCs treated with 50 nM MG132 alone or in combination with each inhibitor of ER stress‐driven apoptosis (salubrinal; 15 μM, STK064652; 20 μM, STK047915; 20 μM). The mean + SD values from the three independent experiments are shown after normalization to those of control cells treated with DMSO. For statistical analysis, Student's t‐test (two‐sided, paired) was used. *P‐values: salubrinal, 0.0003; STK064650, 0.0036; STK047915, 0.0019.
- Similar to (C) except that XO8 GSCs transduced with lentiviral shRNAs were treated with MG132. The mean + SD values from the three independent experiments are shown after normalization to those of control cells treated with DMSO. For statistical analysis, Student's t‐test (two‐sided, paired) was used. *P‐values: #1, 0.026; #5, 0.0033; #6, 0.0482.
- XO8 GSCs were treated with 20 nM MG132 alone or in combination with each inhibitor as described in (C) and subjected to immunoblotting. Sal., salubrinal.
Source data are available online for this figure.
Transcriptionally induced ATF3 during the late UPR is known to form a dimer with CHOP to mediate apoptosis 38. Consistently, we found that the mRNA and protein levels of CHOP were commonly induced in GSCs but not DFCs (Appendix Tables S2 and S4). Moreover, knockdown of CHOP in XO8 GSCs resulted in the loss of hypersensitivity to 50 nM MG132 (Fig 7C), which correlated to decreases in the cleavage of PARP and the level of ATF3 (Fig 7D). A similar result was observed with CHOP −/− mouse embryonic fibroblasts (MEFs) relative to wild‐type MEFs (Fig 7E and F). These results suggest that ATF3 may function as a dimer with CHOP to mediate apoptosis in GSCs.
The transcriptional targets of ATF3 include the BH3‐only protein NOXA, a proapoptotic member of the Bcl‐2 family 39. NOXA has been shown to inhibit Bcl‐2, activating Bax/Bak‐mediated mitochondrial release of cytochrome C 39. Consistently, we found that NOXA is co‐induced with ATF3 and CHOP (Appendix Table S5). Moreover, RNA interference assay of ATF3 showed that the levels of ATF3 and NOXA correlate with each other in 50 nM MG132‐treated GSCs (Fig 7G). These results suggest that ATF3 is a key regulator that mediates apoptosis in glioma‐derived stem cells under proteasomal inhibition.
ER‐associated apoptosis in PI‐treated GSCs is induced independently of the UPR
To determine whether the UPR does indeed trigger apoptosis in PI‐treated GSCs, we compared which of the two events first occurs at 6 and 17 h post‐50 nM MG132 treatment. It is expected that UPR‐associated apoptosis occurs during the late UPR. Surprisingly, a strong induction of proapoptotic proteins was obvious as early as 6 h, that is, earlier than that of the UPR whose induction became obvious at 17 h as compared with 6 h (Fig 6A and B). Immunoblotting analyses after 6 h confirmed marked induction of proapoptotic proteins (e.g., ATF3 and CHOP) but not UPR markers (e.g., XBP1, ATF4, and BiP) (Fig 6C). These results suggest that the apoptosis in PI‐treated GSCs is non‐canonical in that it occurs earlier than the UPR.
The UPR plays a protective role in PI‐induced apoptosis of GSCs
The UPR induces apoptosis through the PERK‐eIF2α‐ATF4‐CHOP pathway 40, 41, 42, 43. To confirm that apoptosis observed in PI‐treated GSCs is not caused by ER stress, we employed chemical inhibitors known to uncouple the UPR from apoptosis. Salubrinal selectively inhibits UPR‐driven apoptosis by constitutively phosphorylating eIF2α 44. If apoptosis in GSCs requires the UPR, salubrinal would block apoptosis in GSCs. Unexpectedly, the result showed that salubrinal did not inhibit apoptosis in 20 nM MG132‐treated GSCs. Moreover, salubrinal exerted an opposite effect, that is, rendered GSCs more sensitive to 20 nM MG132 as determined by the cleavage of PARP1 and caspase‐3 as well as the levels of ATF3 and CHOP (Fig 7I). To confirm this result, we disconnected the UPR from apoptosis using STK047915 which selectively inhibits the IRE1‐ASK1‐JNK pathway. Similar to salubrinal, STK047915 also exhibited a powerful synergy with 20 nM MG132 in inducing apoptosis in GSCs (Fig 7H). Yet another inhibitor of UPR‐driven apoptosis, STK064652, also exerted a similar synergy with 20 nM MG132 (Fig 7H). Semi‐quantitative PCR analyses confirmed that these inhibitors of ER‐associated apoptosis increased the mRNA level of NOXA in GSCs treated with 50 nM MG132 (Fig 7C). These results suggest that the early UPR protects GSCs from the cytotoxicity of PIs and that low‐dosage PIs kill GSCS through a previously unknown, non‐canonical ER stress‐associated apoptosis, which does not require the UPR. Based on these results, we propose that PIs and UPR blockers may be used for combination treatments for gliomas.
A combinational treatment of the proteasome inhibitor MG132 and UPR blocker STK047915 synergistically inhibits the tumor formation of GSCs
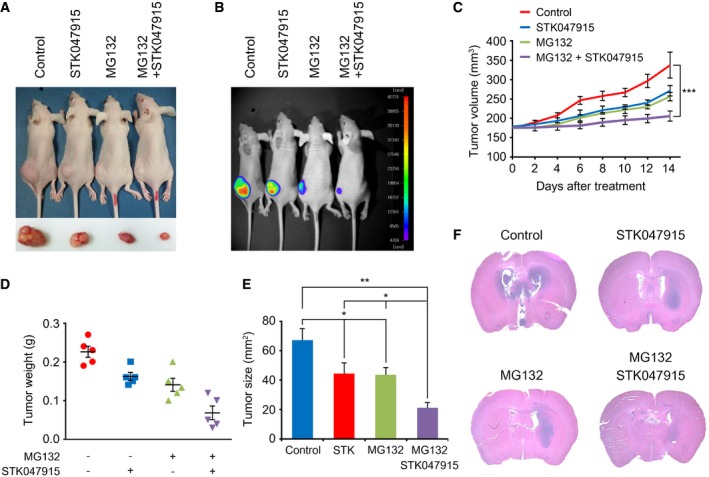
To assess the therapeutic efficacy of MG132 and STK047915 on gliomas in mice, we injected XO8 GSCs subcutaneously (s.c.) in the flank of mice. When the tumor volume reached approximately 180 mm3, GSC‐bearing mice were administered intraperitoneally (i.p.) with 1 mg/kg MG132 and 10 mg/kg STK alone or in combination on every day for 2 weeks. Consistently with our in vitro data, the single treatment of MG132 (1 mg/kg) or STK047915 (10 mg/kg) significantly decreased the tumor growth (Fig 8A–E). Importantly, a combination of STK047915 and MG132 almost completely blocked tumor formation (Fig 8A–E), indicating a synergistic effect between the two compounds.
Figure 8. A combinational treatment of the proteasome inhibitor MG132 and UPR blocker STK047915 synergistically inhibits the tumor formation of GSCs.
-
A, BXO8 GSCs were subcutaneously (s.c.) injected in the flank of mice (n = 5). When the tumor volume reached approximately 180 mm3, GSC‐bearing mice were administered intraperitoneally (i.p.) with 1 mg/kg MG132 and 10 mg/kg STK alone or in combination on every day for 2 weeks. Tumors were dissected from the subcutaneous regions of nude mice. Inset picture shows the tumor in respective group. Therapeutic effects were monitored using bioluminescence imaging.
-
COn each day of treatment as outlined in (A, B), tumor volume was calculated. Error bars represent the mean ± SD from five calculations (***P < 0.0037). For statistical analysis, Student's t‐test (two‐sided, paired) was used.
-
DOn each day of treatment as outlined in (A, B), total tumor weight was measured. Error bars represent the mean ± SD from five measurements.
-
EOn each day of treatment as outlined in (A, B), tumor size was calculated. Error bars represent the mean ± SD from five calculations. For statistical analysis, Student's t‐test (two‐sided, paired) was used. P‐values: *, 0.0388; **, 0.0153.
-
FXO10 GSC were orthotopically injected into the brains of BALB/c nude mice. STK047915, MG132, or both were injected intraperitoneally to GSC‐bearing mice at day 14 after XO10 (5 × 105 cells) inoculation. The PBS was used as a control. Representative photographs of H&E staining from each group show tumor growth. Magnification, ×1.
Next, to validate these results using an orthotopic model, we injected XO10 GSCs into the brains of BALB/c nude mice. When tumor sizes were assessed using histological staining, the treatment of MG132 or STK047915 resulted in a significant reduction in tumor sizes. Moreover, a synergistic effect was obtained with a combination treatment of MG132 or STK047915 (Fig 8F). These results suggest that a combinational treatment of a proteasome inhibitor and a UPR blocker synergistically inhibits the tumor formation of GSCs in mice (see Fig 9).
Figure 9. A model illustrating the mechanism by which proteasome inhibitors (PIs) selectively kill GSCs.
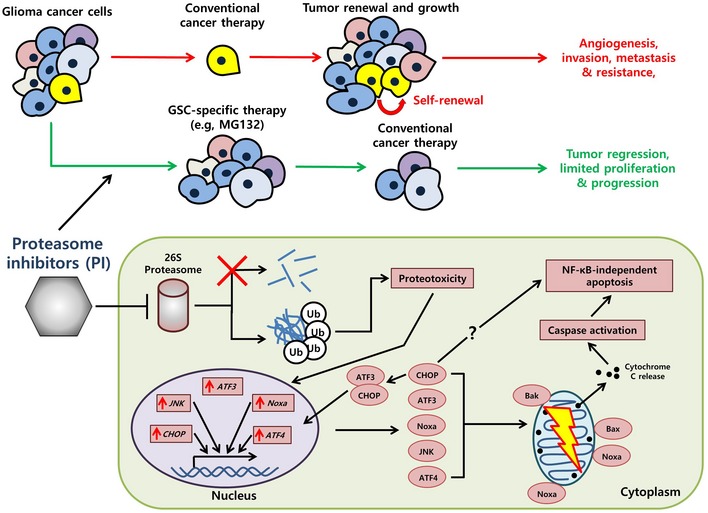
A key factor in low‐dosage proteasome inhibition and its selective efficacy against GSCs lies in the sustained accumulation of ubiquitinated conjugates due to the failure of the proteasome (red cross). The resulting proteotoxicity then can activate the apoptotic cascade, among which genes such as ATF3, CHOP, and JNK are upregulated and translated into their respective protein counterparts. ATF3 and CHOP also form a heterodimer which serves to maintain the apoptotic cascade. The ATF3‐CHOP axis‐induced cell death is independent of classical ER stress‐associated apoptosis and may instead directly play a role via NOXA to induce mitochondrial dysfunction (yellow lightning) and eventually cell death.
Discussion
In this study, we demonstrated that PIs selectively kill GSCs (IC50, 27–70 nM) relative to their DFCs (IC50, » 100 μM). Based on its therapeutic efficacy on cultured cells, bortezomib has been exploited to treat glioblastoma in a phase I clinical trial as a single agent and in a phase II trial in combination with a HDAC inhibitor 45, 46. Unfortunately, these clinical trials failed because of low response rates on recurrent glioblastoma. Given our results indicating the hypersensitivity of GSCs to PIs, one possible explanation for this failure is that bortezomib conferred cytotoxicity only to GSCs but not to the tumor bulk, which would not apparently affect total tumor masses. In fact, our results (Fig 2) suggest that more differentiated cells, which comprise of the bulk mass in tumors, are approximately 1,000‐fold more resistant to PIs. As already suggested by others 47, some differentiated cells that survive PI treatment may acquire stem cell properties through de‐differentiation. Another explanation for the failure in these clinical trials is that bortezomib may not efficiently cross the blood–brain barrier (BBB). Thus, maximal therapeutic efficacy may be achieved once a BBB‐penetrating PI is developed to kill GSCs in combination with other antitumor reagents targeting differentiated tumor cells.
Our results show that the levels of cellular ubiquitination and proteasomal activities in GSCs are markedly higher compared with their DFCs and are gradually downregulated through the course of differentiation (Fig 4B, C and F), indicating that the genomewide program in the UPS is subjected to dynamic changes during the maintenance of stemness in GSCs and their differentiation process. Also, recent genomic analyses to describe cancer‐associated genetic abnormalities using 72 cancer cell lines showed that genes associated with UPS are a point of vulnerability in many cancer cells 48. Relevant to this speculation, a previous quantitative mass spectrometric study on the role of the UPS in pluripotent embryonic stem cells (ESCs) showed that a significant number of proteins involved in the maintenance of stemness are ubiquitinated and actively degraded in ESCs compared with their DFCs 49. In another study, a screening of 312 ligases ended up with 17 Ub ligases whose knockdown resulted in apoptotic death of cancer cell lines 23. Notably, knockdown of these 17 Ub ligases in glioblastoma stem cells induced differentiation associated with death or hypersensitivity to stressors 23. These results together suggest that the apoptotic machinery in GSCs is highly sensitive to subtle misregulation in the timely degradation of key molecules, readily triggering programmed cell death in GSCs (Fig 9).
Studies have shown that the cytotoxicity caused by PIs is mainly associated with enhanced apoptosis 50, 51. The apoptotic effects of PIs at 1–10 μM have been attributed to the failure to degrade IκB and the consequential inhibition of NF‐κB activation. 18, 33, 34. Our results show that PIs at 20–70 nM do not significantly affect the functions of NF‐κB and IκB as well as IKK, predicting the presence of an NF‐κB‐independent apoptotic pathway. One proapoptotic mediator that we found links PI‐generated death signals to apoptotic core machinery is JNK, but not p38, of the MAPK pathway 20, 35, 52. Intriguingly, we found that the JNK pathway is responsible for only part of the apoptotic death of GSCs because a small‐molecule inhibitor of JNK significantly, but only partially, protects GSCs from the cytotoxicity of PIs. This leakiness of JNK inhibitors can be explained by our finding that 50 nM MG132 strongly induces an ER‐associated apoptotic pathway (Appendix Tables S4 and S5, and Fig 6). Among these, the proapoptotic transcription factor ATF3 is likely to function as a key signaling mediator underlying the hypersensitivity of GSCs to proteasomal inhibition. This proposition is supported by the following results. First, our microarray analyses identified ATF3 to be the most prominent hit, with an mRNA level ~30.9‐fold induced in 50 nM MG132‐treated GSCs (Appendix Tables S2 and S4). Second, knockdown of ATF3 using RNA interference assay markedly inhibited the cleavage of PI‐induced apoptosis in GSCs, but not in DFCs (Fig 7A and B). Third, the induction of ATF3 and CHOP temporally correlated with each other during PI treatment of GSCs. Consistent with these results are the findings by others that ATF3 is activated by PERK‐eIF2α‐ATF4 during prolonged ER stress and transfers death signals from stressed ER to the mitochondrial apoptotic machinery 37, 38.
Studies have shown that ATF3 forms a dimer with CHOP to induce the expression of genes involved in the stress responses and apoptosis 53. Consistently, our results show that ATF3 is co‐induced with CHOP during apoptosis in GSCs treated with 50 nM MG132 (Fig 6C) and that CHOP is required for PI‐induced selective apoptosis of GSCs (Fig 7C–F). Although it remains to be determined whether ATF3 directly interacts with CHOP in PI‐treated GSCs, ATF3 may work with CHOP to mediate PI‐induced apoptosis in GSCs. Another question concerning the identity of the downstream signaling molecule of ATF3 remains to be addressed. Our results suggest that ATF3 is transcriptionally co‐induced with NOXA during apoptosis in 50 nM MG132‐treated GSCs (Appendix Tables S2 and S4, and Fig 6B) and that ATF3‐knockdown downregulates the level of NOXA mRNA in MG132‐treated GSCs (Fig 7G). We suggest that the transcriptional activation of NOXA by ATF3 may be a mechanism underlying the hypersensitivity of GSCs to PIs.
The UPR is triggered by accumulating misfolded proteins in the ER lumen and attempts to restore homeostasis. There is a general notion that ER stress‐induced apoptosis is triggered by terminal UPR when early UPR fails to restore ER functions 36, 54. Recent study showed that ER stress‐induced UPR in colon cancer stem cells induces differentiation which enhances the sensitivity to chemotherapy 55. Our microarray analyses also show the transcriptional induction of the UPR and proapoptotic genes associated with ER stress in GSCs treated with 50 nM MG132. Surprisingly, we found that so‐called ER stress‐associated apoptosis in 50 nM MG132‐treated GSCs is not a consequence of failed UPR but is directly triggered, possibly on the cytosolic surface of the ER membrane, by unknown signals from the troubled cytosol in which proteasomal degradation of key molecules is impaired. Thus, apoptosis observed in 50 nM MG132‐treated GSCs is mechanistically different from ER stress‐associated apoptosis in other cancer cell types. Is the ER‐associated apoptosis in PI‐treated GSCs then functionally independent from the UPR? If the apoptosis is dependent on the UPR, the inhibition of UPR‐mediated apoptosis will rescue GSCs from the cytotoxicity of PIs. Surprisingly, we found that pharmacological inhibition of UPR‐mediated apoptosis is synergistic with 20 nM MG132 in killing GSCs, suggesting that the UPR, at least its early phase, plays a protective role in GSCs treated with PIs. Given the synergistic cytotoxic activity observed in this study, PIs and inhibitors of ER stress‐induced apoptosis may provide a means for a combination treatment for gliomas (see Fig 9). Indeed, we confirmed that a combinational treatment of the proteasome inhibitor MG132 and UPR blocker STK047915 synergistically inhibited the tumor formation of GSCs in mice (Fig 8).
Materials and Methods
In vitro culture of GSCs
We previously established XO6, XO8, and XO10 GSCs 29, 30, 56, 57. The GSCs were cultured in suspension using serum‐free media containing Dulbecco's modified Eagle's medium (DMEM)/F12 media (Life Technologies), 20 ng/ml epidermal growth factor (EGF; R&D Systems), 20 ng/ml basic fibroblast growth factor (bFGF; R&D Systems), and 0.4 μl/ml B‐27 (Life Technologies). B‐27 is a serum‐free supplement for neural cell culture. EGF and bFGF were replenished every 2 days. The cells were cultured at 37°C under 5% CO2 and 20% O2, with relative humidity higher than 95%. Cells were passaged every 5–6 days when spheres became 150–200 μm in diameters. For passaging, GSC spheres were collected and dispersed into individual cells by mixing with Accutase (Sigma), an enzymatic mixture with proteolytic and collagenolytic activities. Following incubation at 37°C for 2 min and gentle agitation, the dispersed cells were reseeded into fresh media at about 100,000 cells/ml. When necessary, individualized cells were allowed to attach on coverslips or culture plates in media supplemented with 20 μg/ml laminin (Sigma).
Differentiation of GSCs
To induce differentiation of GSCs into non‐stem cells, GSC spheres were collected and dispersed into individual cells by treating with Accutase (Sigma) and gentle agitation and then allowed to grow in DMEM/F12 media containing 10% fetal bovine serum (FBS: Thermo Fisher Scientific Inc.) in adherent conditions. After seven passages of adherent culture in serum‐containing media, the cells were determined to be fully differentiated. HEK293, HeLa, U87, and U251 cells were cultured in DMEM supplemented with 10% FBS.
Other cell lines
CHOP −/− and corresponding +/+ MEF cell lines were kindly provided by Dr. Randal J. Kaufman (Sanford Burnham Medical Research Institute, CA, USA). MEF cells were maintained in DMEM supplemented with β‐mercaptoethanol (55 μM; Invitrogen, Carlsbad, CA, USA) and cultured to 60–80% confluency.
Cytotoxicity assay of anticancer reagents and stressors
To compare the cytotoxicity of anticancer reagents to GSCs and DFCs under similar conditions, we dispersed GSC spheres in serum‐free media and DFCs in serum‐containing media into individual cells. Both individualized GSCs and DFCs were plated on 96‐well plates and subsequently cultured adherently in serum‐free media supplemented with laminin (20 μg/ml) for 8 h. Alternatively, individualized GSCs and DFCs were both cultured in serum‐containing media for 8 h. GSCs and DFCs, cultured in the same serum‐free or serum‐containing media, were treated with various stressors in the same media for 17 h prior to cell viability assay. Cell viability was assessed using EZ‐Cytox cell viability assay kit (Dojindo Laboratory) that uses water‐soluble tetrazolium salt (WST), according to the manufacturer's guidelines. Assay reagent solution (10 μl) provided by the manufacturer was added to DMSO‐treated or anticancer reagents and/or stressor‐treated cells and incubated for 4–6 h at 37°C in CO2 incubator; optical density was then measured at 450 nm wavelength.
Alternatively, cell viability was also measured using trypan blue exclusion assay as previously described (Lee et al, 2008). GSCs plated on 6‐well plate were treated with MG132 for 24 h and subsequently harvested, mixed with equal volume of 0.4% trypan blue (Amresco Inc.), and loaded over a counting chamber. The total number of cells in each well was counted under a microscope. To determine the doubling time (DT), cells were collected every day for 5 days counted with the hemocytometer stained with trypan blue. DT was calculated using the following formula: DT = (t−t 0) × log2/(logN−logN 0), where t, t 0 indicate time points at counting and initial plating, respectively, and N, N 0 indicate numbers of cells at aforementioned time points. Results are presented as mean DT ± SD of five consecutive passages.
Soft agar colony formation assay
GSCs and DFCs were individualized using Accutase (Sigma) and both cultured adherently in serum‐free media supplemented with laminin (20 μg/ml) for 8 h. The cells were treated with MG132 for 17 h and recovered for 24 h in the absence of MG132. Following harvesting, GSCs and DFCs were resuspended in serum‐free media containing 0.3% agarose, seeded on the top of 0.8% agarose (5,000 cells/35‐mm dish), and incubated for 2–3 weeks. Colonies bigger than 100 μm in diameter were blind‐counted from four microscopic fields. Three independent experiments were performed.
Live cell staining and flow cytometry
The staining of live GSCs and DFCs was performed using PKH67 Green and PKH26 Red Fluorescent Cell Linker kits (Sigma), respectively, according to the manufacturer's instruction. Briefly, individualized GSCs and DFCs were washed with serum‐free media and resuspended in a manufacturer‐provided solution (Diluent C). The cells in Diluent C were mixed with an equal volume of Diluent C containing 4 μM of each fluorescent dye, followed by incubation for 5 min at room temperature. After the labeling reaction was stopped by adding serum‐containing growth media, the cells were washed twice and resuspended in serum‐free media. Equal volumes of green‐labeled GSCs and red‐labeled DFCs were mixed with each other. The mixture of GSCs and DFCs was cultured for 8 h in serum‐free media supplemented with laminin (20 μg/ml) on plates or coverslips, followed by incubation with DMSO or 50 nM MG132 for 17 h.
For flow cytometry, harvested cells were washed with phosphate‐buffered saline (PBS) and fixed in 0.1% paraformaldehyde for 15 min. The fixed cells were filtered using a cell strainer with a pore size of 40 μm and analyzed using the BD LSR II or BD CellQuest Pro flow cytometer (BD Biosciences). For fluorescent microscopy, the cells plated on coverslips were fixed with 4% paraformaldehyde for 15 min and analyzed using microscopy. The ratio between GSCs labeled in green and DFCs labeled in red was determined by blind counting. Three independent experiments were performed.
The expression profiles of CD133 and GFAP in cultured cells were also analyzed using flow cytometry. Briefly, 1 × 106 cells were incubated in 100 μl of 1% BSA in PBS containing 1 μg of CD16/CD32 (eBioscience) for 30 min on ice to block unspecific Fc interaction. The cells were labeled with FITC‐conjugated anti‐CD133, FITC‐conjugated anti‐GFAP, and eFluor 660‐conjugated anti‐ESA antibodies for 1 h. Labeled cells were resuspended in PBS with 1% FBS and analyzed by flow cytometry using a Beckman Coulter Expo (Brea, CA). Isotypic IgG and unstained cells served as negative controls.
Annexin V staining
To measure the apoptotic status, DMSO‐ or drug‐treated cells were stained with Annexin V using the EzWay™ Annexin V‐FITC Apoptosis Detection kit (Komabiotech) according to the manufacturer's instructions. Cells were harvested by centrifugation at 1,000 g for 5 min at room temperature, washed with PBS once, and stained with Ab‐Annexin V‐FITC for 15 min at room temperature in the dark. The harvested cells were washed with PBS and immediately analyzed using flow cytometry. Adherent cells were also similarly stained with Ab‐Annexin V‐FITC, followed by examination with fluorescence microscopy.
Tumorigenesis assay in mice
The tumorigenic potential of XO8 GSCs was compared to that of DFCs by in vivo tumorigenicity titration. Animal studies were conducted according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85‐23, revised in 1996) and the protocols (12‐0304) approved by the Institutional Animal Care and Use Committee at Seoul National University. XO8 GSCs or DFCs were resuspended in 3 μl of Dulbecco's PBS and injected stereotactically into the striatum of BALB/c nude mice via stereotactic device (coordinates: 1 mm anterior, 2 mm lateral to the bregma, 3 mm depth). The injection procedure caused no lethality in the animals. After 6 weeks, the mice were sacrificed at the onset of neurological symptoms for the analysis of tumor histology and immunohistochemistry. Mice were anesthetized with an intraperitoneal injection of 20 mg/kg Zoletil (Virbac) and 10 mg/kg xylazine (Bayer). Brains were harvested by cardiac perfusion with 4% paraformaldehyde and further fixed at 4°C overnight. After fixation, the brains were dehydrated using sucrose and embedded in optimum cutting temperature compound (Tissue‐Tek) and stored at −80°C. The brains were sectioned using a cryostat into 10‐μm slices and stained with hematoxylin and eosin using standard protocols. Bright‐field images were acquired using a microscope (BX21; Olympus).
In vivo xenograft and orthotopic studies in mice
All animal procedures were carried out in accordance with animal care guidelines approved by the Korea University Institutional Animal Care and Use Committee (IACUC). Five‐week‐old female BALB/c nude mice were obtained from the Shizuoka Laboratory Animal Center (Shizuoka, Japan) and housed in a specific pathogen‐free environment. All surgery was performed under Zoletil® 50/xylazine anesthesia, and animal suffering was minimized. For the brain orthotopic model, XO8 GSCs were resuspended in sterile PBS at a density of 3 × 106 cells/ml, and 30,000 cells were stereotactically implanted in the right cerebral cortices of nude mice. For the xenograft model, XO10 GSCs stably infected with a luciferase expression vector were likewise resuspended in sterile PBS at a density of 3 × 106 cells/ml, among which 30,000 cells were injected subcutaneously in the right flank of mice. One week after implantation, animals received 50 mg/kg d‐luciferin to verify tumor implantation via luciferase bioluminescence (NightOwl LB 981 Molecular Imaging System; Berthold Technologies) in vivo imaging system. After a few weeks when tumor volume had reached approximately 180 mm3, mice were randomized into four drug treatment groups: control (n = 5), STK047915 (n = 5), MG132 (n = 5), and STK047915 + MG132 (n = 5). The mice were administered intraperitoneally (i.p.) with 10 mg/kg STK047915, 1 mg/kg MG132, or in combination daily for 14 days. Tumor volume was calculated every 2 days for 21 days according to the following equation: tumor volume (mm3) = π/6 × length × (width)2. Maximum tumor area and its corresponding section were calculated via MetaMorph software (Molecular Devices).
Antibodies and other reagents
We used antibodies against the following proteins: PARP1 (1:5,000), ubiquitin (1:2,500), NF‐κBp65 (1:2,500), NF‐κBp50 (1:2,500), IκB‐α (1:2,500), IκB‐β (1:2,500), IκB‐ε (1:2,500), actin (1:10,000) from Sigma, Nestin (1:2,500), Musashi‐1 (1:2,500) from Merck Millipore, SOX2 (1:3,000) from R&D Systems, glial fibrillary acidic protein (GFAP; 1: 2,500) from DakoCytomation, βIII‐tubulin (1:2,500) from Covance, caspase‐3 (1:2,500), IKK‐α (1:2,500), IKK‐β (1:2,500), phospho‐IKKαβ (1:2,500), JNK (1:2,500), phospho‐JNK (1:2,500), p38 (1:2,500), phospho‐p38 (1:2,500) from Cell Signaling Technology, USP14 (1:2,000) from Bethyl Laboratory, polyubiquitinated proteins (FK1; 1:2,500), 19S Rpt5 (1:2,000), 20S α3 (1:2,000), 20S α7 (1:2,000) from ENZO Life, RNF123 (1:2,500) from Abcam, and UBR2 (1:2,500) from Novus Biologicals. We purchased adriamycin, taxol, and thapsigargin from Sigma; salubrinal and TMZ from Santa Cruz Biotechnology; MG132, cisplatin, bafilomycin A1, SB203580, and SP600125 from Merck Millipore; and epoxomicin from ENZO Life Science. PS341, salinosporamide A, and TRAIL were generous gifts from Drs. D. Finley, J. Clardy (Harvard University), and Jae J. Song (Yonsei University), respectively.
Real‐time PCR
Total RNAs were extracted from cultured cells using the RNeasy Plus kit (Qiagen) following the manufacturer's instructions. One microgram of total RNA was used for reverse transcription to synthesize cDNA using Superscript RT III cDNA synthesis kit (Life Technologies), followed by real‐time PCR using SYBR FAST ABI Prism qPCR kit (Kapa Biosystems). Real‐time PCR was performed on ABI Prism 7900 sequence detection system (Life Technologies) using the comparative cycle threshold (CT) method as described in the manufacturer's instructions. CT was determined in the exponential phase of the amplification curve. All samples were assayed in triplicate. The relative CT value of target transcripts was normalized to that of human GAPDH transcripts. Primers used for real‐time PCR are listed in Appendix Table S1.
Immunoblot assay
Subconfluent cells were harvested and washed with ice‐cold PBS twice. The cells were lysed on ice for 15 min in 1% Nonidet P‐40 lysis buffer (20 mM Tris–HCl, pH 8.0, 137 mM NaCl, 1% Nonidet P‐40, 10% glycerol, and 1 mM sodium vanadate) containing a protease inhibitor cocktail (Sigma). The cell extracts were cleared by centrifugation at 15,000 g for 15 min at 4°C, and protein concentration was determined using the bicinchoninic acid (BCA) assay (ThermoFisher Scientific). Proteins were separated using SDS–PAGE and electronically transferred to polyvinylidene difluoride (PVDF) membrane. The membrane was blocked for 1 h with 1% non‐fat dry milk in PBS containing 0.1% Tween‐20 and incubated with primary antibodies for 1 h at room temperature or overnight at 4°C. Immunoreactive bands were detected using horseradish peroxidase‐conjugated IgG as a secondary antibody and visualized using enhanced chemiluminescence (ThermoFisher Scientific) according to the manufacturer's protocol.
Proteasome activity assay
Cells were plated and incubated for 8 h on laminin‐coated plates in serum‐free GSC growth media to allow cell attachment and treated with 50 nM MG132 for the times indicated. Following MG132 treatment in the cytotoxicity assay section, cells were harvested, washed with cold PBS, and resuspended in cold buffer I (50 mM Tris–HCl, pH 7.5, 20 μM ATP, 5 mM MgCl2, 1 mM DTT, 20% glycerol). Cell extracts were prepared by at least seven cycles of freezing and thawing and cleared by centrifugation at 15,000 g for 10 min at 4°C. Protein concentration was determined using the BCA assay kit (ThermoFisher Scientific). Total 10 μl of cell lysates (1 μg/μl) was mixed with 10 μl of 300 μM succinyl‐LLVY‐7‐amino‐4‐methylcoumarin (AMC; Merck Millipore) and 85 μl of assay buffer (20 mM Tris–HCl, pH 7.5, and 20% glycerol), followed by incubation at 37°C for 30 min. The amount of released fluorescent AMC was measured using the spectrofluorometer Infinite M200 (TECAN) at 440 nm emission with 380 nm excitation wavelength.
Cell fractionation
Cells were harvested, washed with cold PBS twice, and lysed by incubating in a lysis buffer (20 mM HEPES, pH 7.4, 10 mM KCl, 2 mM MgCl2, and 0.5% Nonidet P‐40) containing a protease inhibitor cocktail (Sigma) for 10 min. The cell lysates were centrifuged at 1,500 g for 5 min to sediment the nuclei. The resulting supernatant was centrifuged again at 15,000 g for 10 min to obtain the cytoplasmic fraction. The nuclear pellets were washed three times with lysis buffer, resuspended in the same lysis buffer containing 0.5 M NaCl, and centrifuged at 15,000 g for 10 min. The resulting supernatant is the nuclear fraction.
Microarray
Total RNAs were extracted from cells treated with 50 nM MG132 for 17 h in serum‐free media by RNeasy® Plus kit (Qiagen). Microarray analysis was performed using the Illumina HumanHT‐12 v4 Expression BeadChip (Illumina Inc.). Isolated RNAs were labeled with biotin and the biotinylated cRNAs were prepared from 0.55 g total RNA using the Illumina TotalPrep RNA Amplification Kit (Ambion). After fragmentation, 0.75 g of cRNAs was hybridized to the Illumina HumanHT‐12 Expression Beadchip according to the manufacturer's guidelines. Illumina GenomeStudio v2009.2 (Gene Expression Module v1.5.4) was used to array data export processing and analysis. The data in GEOArchive files were deposited in the Gene Expression Omnibus (GEO) of NCBI (http://www.ncbi.nlm.nih.gov/geo/) under the accession number of GSE62356. Only the probes that fulfill the criteria in which the detection probability value (pval) is lower than 0.05 were analyzed. Quantile normalization was applied for normalization of gene expression value. The correlation coefficient between the log fold change of raw data and that after quantile normalization was 0.996 for GSCs and 0.980 for differentiated cells, suggesting the value was adjusted without significant loss from raw data. All probe intensities conformed to the same distribution for all sample arrays through the quantile normalization. Fold change (FC) was calculated by comparing the expression in the DMSO‐treated cells with that in the MG132‐treated cells.
Viral transduction
ATF3‐specific shRNAs in lentiviral pLKO.1 vector were obtained from MISSION shRNA of Sigma (TRCN0000013571; #1, TRCN0000013572; #5, TRCN0000329689; #6, TRCN0000329690; #7, TRCN0000013568; #9), and pLKO.1 vector was used as a control. Lentiviruses were produced using the BLOCK‐iT Lentiviral RNAi expression system (Invitrogen) according to the manufacturer's protocol. Briefly, 293 FT cells were co‐transfected with ATF3 shRNA‐expressing viral vectors and manufacturer‐provided viral packaging plasmid mixture (pLP1, pLP2, and pLP/VSVG). The next day, the media was changed with serum‐free GSC growth media and incubated for 2–3 days. The virus‐containing media was collected and cleared by centrifugation (2,500 g for 10 min) and filtration (0.45 μm). Virus‐containing media was diluted four times with fresh serum‐free GSC growth media and used for GSCs transduction. For viral transduction of GSCs, GSC spheres were individualized by Accutase (Sigma) treatment and cultured in virus‐containing media supplemented with 20 ng/ml EGF and bFGF (R&D Systems) for 4–5 days. The viral‐transduced GSCs spheres were collected, individualized, and attached on the plates in the serum‐free GSC growth media containing laminin for 8 h and then treated with MG132 for 17 h.
Statistical analysis
In cell viability assay, each point value represents the mean ± or + SD from at least three independent experiments unless otherwise specified and depending on the nature of the experimental settings. P‐values were determined using the Student's t‐test. P < 0.05 was considered statistically significant. In Fig 3B, cells stained with red or green colors were counted from four randomly chosen fields. In Fig 3D, colonies bigger than 100 μm diameter in four randomly chosen fields were subjected to blind counting. The data were analyzed using GraphPad Prism 7.0 statistical software (San Diego, CA, USA). Other information for statistical analysis is described in the figure legends.
Author contributions
YDY participated in conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing; D‐HL, HK, SRM, CJ, SHP, KSS, JH, SKK, K‐JP, YJL and BYK participated in conception and design, and collection and assembly of data; HC‐M participated in collection and assembly of data, data analysis, and interpretation; SAC participated in collection and assembly of data, data analysis and interpretation, and manuscript writing; DMP participated in provision of study materials; S‐HK, SCO and AC participated in conception and design, data analysis and interpretation, and financial support; YTK participated in conception and design, financial support, collection and assembly of data, data analysis and interpretation, manuscript writing, and final approval of manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Drs. Daniel Finley, Jon Clardy (Harvard University), and Jae J. Song (Yonsei University) for providing us with PS341, salinosporamide A, and TRAIL, respectively; Kyoung Tae Seo (Soongsil University) and Su Jin Yoo (University of Wisconsin‐Madison) for technical assistance; and Edward Kwak (Viagen Biotech Inc., USA) for editorial assistance. This work was supported by the SNU Nobel Laureates Invitation Program (to A.C.), the Basic Science Research Programs of the NRF funded by the Ministry of Science, ICT and Future Planning (MSIP) (NRF‐2016R1A2B3011389 to Y.T.K. and 2015R1D1A1A01058303 to D.H.L.) and the Ministry of Education (NRF‐2013R1A1A2058983 to Y.D.Y), Seoul National University Hospital (to Y.T.K.), the National Cancer Institute grant CA140554 (to Y.J.L), the World Class Institute (WCI) Program of the NRF funded by the MSIP (WCI 2009‐002 to B.Y.K.), the R&D Convergence Program (CAP‐16‐03‐KRIBB to B.Y.K.) of NST of Korea, the Bio and Medical Technology Development Program (NRF‐2014M39Ab5073938 to B.Y.K.) of MSIP and KRIBB Research Initiative Program, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF) (to A.C.), and the Israel Science Foundation (ISF) (to A.C.). A.C. is an Israel Cancer Research Fund (ICRF), USA Professor.
EMBO Reports (2017) 18: 150–168
Contributor Information
Yong J Lee, Email: leeyj@upmc.edu.
Bo Yeon Kim, Email: bykim@kribb.re.kr.
Yong Tae Kwon, Email: yok5@snu.ac.kr.
References
- 1. Iacob G, Dinca EB (2009) Current data and strategy in glioblastoma multiforme. J Med Life 2: 386–393 [PMC free article] [PubMed] [Google Scholar]
- 2. Lim SK, Llaguno SR, McKay RM, Parada LF (2011) Glioblastoma multiforme: a perspective on recent findings in human cancer and mouse models. BMB Rep 44: 158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van den Bent MJ, Hegi ME, Stupp R (2006) Recent developments in the use of chemotherapy in brain tumours. Eur J Cancer 42: 582–588 [DOI] [PubMed] [Google Scholar]
- 4. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432: 396–401 [DOI] [PubMed] [Google Scholar]
- 5. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444: 756–760 [DOI] [PubMed] [Google Scholar]
- 6. Kang MJ, Kim HP, Lee KS, Yoo YD, Kwon YT, Kim KM, Kim TY, Yi EC (2013) Proteomic analysis reveals that CD147/EMMPRIN confers chemoresistance in cancer stem cell‐like cells. Proteomics 13: 1714–1725 [DOI] [PubMed] [Google Scholar]
- 7. Huang Z, Cheng L, Guryanova OA, Wu Q, Bao S (2010) Cancer stem cells in glioblastoma–molecular signaling and therapeutic targeting. Protein Cell 1: 638–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eramo A, Ricci‐Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R (2006) Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 13: 1238–1241 [DOI] [PubMed] [Google Scholar]
- 9. Folkins C, Shaked Y, Man S, Tang T, Lee CR, Zhu Z, Hoffman RM, Kerbel RS (2009) Glioma tumor stem‐like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal‐derived factor 1. Cancer Res 69: 7243–7251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Molina JR, Hayashi Y, Stephens C, Georgescu MM (2010) Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 12: 453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tasaki T, Sriram SM, Park KS, Kwon YT (2012) The N‐end rule pathway. Annu Rev Phys Chem 81: 261–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sriram SM, Kwon YT (2010) The molecular principles of N‐end rule recognition. Nat Struct Mol Biol 17: 1164–1165 [DOI] [PubMed] [Google Scholar]
- 13. Adams J (2004) The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5: 417–421 [DOI] [PubMed] [Google Scholar]
- 14. Lee MJ, Kim DE, Zakrzewska A, Yoo YD, Kim SH, Kim ST, Seo JW, Lee YS, Dorn GW II, Oh U et al (2012) Characterization of arginylation branch of N‐end rule pathway in G‐protein‐mediated proliferation and signaling of cardiomyocytes. J Biol Chem 287: 24043–24052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sriram SM, Banerjee R, Kane RS, Kwon YT (2009) Multivalency‐assisted control of intracellular signaling pathways: application for ubiquitin‐ dependent N‐end rule pathway. Chem Biol 16: 121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee MJ, Pal K, Tasaki T, Roy S, Jiang Y, An JY, Banerjee R, Kwon YT (2008) Synthetic heterovalent inhibitors targeting recognition E3 components of the N‐end rule pathway. Proc Natl Acad Sci USA 105: 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu WK, Cho CH, Lee CW, Wu K, Fan D, Yu J, Sung JJ (2010) Proteasome inhibition: a new therapeutic strategy to cancer treatment. Cancer Lett 293: 15–22 [DOI] [PubMed] [Google Scholar]
- 18. Russo A, Bronte G, Fulfaro F, Cicero G, Adamo V, Gebbia N, Rizzo S (2010) Bortezomib: a new pro‐apoptotic agent in cancer treatment. Curr Cancer Drug Targets 10: 55–67 [DOI] [PubMed] [Google Scholar]
- 19. Vlashi E, Mattes M, Lagadec C, Donna LD, Phillips TM, Nikolay P, McBride WH, Pajonk F (2010) Differential effects of the proteasome inhibitor NPI‐0052 against glioma cells. Transl Oncol 3: 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP (2005) Proteasome inhibitor PS‐341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM). Oncogene 24: 344–354 [DOI] [PubMed] [Google Scholar]
- 21. Bota DA, Alexandru D, Keir ST, Bigner D, Vredenburgh J, Friedman HS (2013) Proteasome inhibition with bortezomib induces cell death in GBM stem‐like cells and temozolomide‐resistant glioma cell lines, but stimulates GBM stem‐like cells' VEGF production and angiogenesis. J Neurosurg 119: 1415–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asklund T, Kvarnbrink S, Holmlund C, Wibom C, Bergenheim T, Henriksson R, Hedman H (2012) Synergistic killing of glioblastoma stem‐like cells by bortezomib and HDAC inhibitors. Anticancer Res 32: 2407–2413 [PubMed] [Google Scholar]
- 23. Low J, Blosser W, Dowless M, Ricci‐Vitiani L, Pallini R, de Maria R, Stancato L (2012) Knockdown of ubiquitin ligases in glioblastoma cancer stem cells leads to cell death and differentiation. J Biomol Screen 17: 152–162 [DOI] [PubMed] [Google Scholar]
- 24. Unterkircher T, Cristofanon S, Vellanki SH, Nonnenmacher L, Karpel‐Massler G, Wirtz CR, Debatin KM, Fulda S (2011) Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin Cancer Res 17: 4019–4030 [DOI] [PubMed] [Google Scholar]
- 25. Kahana S, Finniss S, Cazacu S, Xiang C, Lee HK, Brodie S, Goldstein RS, Roitman V, Slavin S, Mikkelsen T et al (2011) Proteasome inhibitors sensitize glioma cells and glioma stem cells to TRAIL‐induced apoptosis by PKCepsilon‐dependent downregulation of AKT and XIAP expressions. Cell Signal 23: 1348–1357 [DOI] [PubMed] [Google Scholar]
- 26. Thaker NG, Zhang F, McDonald PR, Shun TY, Lewen MD, Pollack IF, Lazo JS (2009) Identification of survival genes in human glioblastoma cells by small interfering RNA screening. Mol Pharmacol 76: 1246–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fribley A, Wang CY (2006) Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol Ther 5: 745–748 [DOI] [PubMed] [Google Scholar]
- 28. Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R et al (2003) A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348: 2609–2617 [DOI] [PubMed] [Google Scholar]
- 29. Soeda AON, Lee D, Iwama T, Park DM (2014) Surface protein dynamics in glioma stem cells. Austin J Neurosurg 1: 7–13 [Google Scholar]
- 30. Park DM, Li J, Okamoto H, Akeju O, Kim SH, Lubensky I, Vortmeyer A, Dambrosia J, Weil RJ, Oldfield EH et al (2007) N‐CoR pathway targeting induces glioblastoma derived cancer stem cell differentiation. Cell Cycle 6: 467–470 [DOI] [PubMed] [Google Scholar]
- 31. Inagaki A, Soeda A, Oka N, Kitajima H, Nakagawa J, Motohashi T, Kunisada T, Iwama T (2007) Long‐term maintenance of brain tumor stem cell properties under at non‐adherent and adherent culture conditions. Biochem Biophys Res Commun 361: 586–592 [DOI] [PubMed] [Google Scholar]
- 32. Oka N, Soeda A, Inagaki A, Onodera M, Maruyama H, Hara A, Kunisada T, Mori H, Iwama T (2007) VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem Biophys Res Commun 360: 553–559 [DOI] [PubMed] [Google Scholar]
- 33. Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl Bancroft C, Sausville E, Adams J, Elliott P, Van Waes C (2001) Novel proteasome inhibitor PS‐341 inhibits activation of nuclear factor‐kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res 7: 1419–1428 [PubMed] [Google Scholar]
- 34. McConkey DJ, Zhu K (2008) Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat 11: 164–179 [DOI] [PubMed] [Google Scholar]
- 35. Zanotto‐Filho A, Braganhol E, Battastini AM, Moreira JC (2012) Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/Akt and NFkappaB pathways, mitochondrial dysfunction, and activation of p38‐JNK1/2 signaling. Invest New Drugs 30: 2252–2262 [DOI] [PubMed] [Google Scholar]
- 36. Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13: 89–102 [DOI] [PubMed] [Google Scholar]
- 37. Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24: 1365–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sok J, Wang XZ, Batchvarova N, Kuroda M, Harding H, Ron D (1999) CHOP‐dependent stress‐inducible expression of a novel form of carbonic anhydrase VI. Mol Cell Biol 19: 495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000) Noxa, a BH3‐only member of the Bcl‐2 family and candidate mediator of p53‐induced apoptosis. Science 288: 1053–1058 [DOI] [PubMed] [Google Scholar]
- 40. Boyce M, Yuan J (2006) Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ 13: 363–373 [DOI] [PubMed] [Google Scholar]
- 41. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666 [DOI] [PubMed] [Google Scholar]
- 42. Cha‐Molstad H, Sung KS, Hwang J, Kim KA, Yu JE, Yoo YD, Jang JM, Han DH, Molstad M, Kim JG et al (2015) Amino‐terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol 17: 917–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. He L, Jang JH, Choi HG, Lee SM, Nan MH, Jeong SJ, Dong Z, Kwon YT, Lee KS, Lee KW et al (2013) Oligomycin A enhances apoptotic effect of TRAIL through CHOP‐mediated death receptor 5 expression. Mol Carcinog 52: 85–93 [DOI] [PubMed] [Google Scholar]
- 44. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D et al (2005) A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307: 935–939 [DOI] [PubMed] [Google Scholar]
- 45. Phuphanich S, Supko JG, Carson KA, Grossman SA, Burt Nabors L, Mikkelsen T, Lesser G, Rosenfeld S, Desideri S, Olson JJ (2010) Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J Neurooncol 100: 95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, Schwerkoske J, Mazurczak M, Gross H, Pajon E et al (2012) Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol 14: 215–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES (2011) Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 146: 633–644 [DOI] [PubMed] [Google Scholar]
- 48. Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JL et al (2012) Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov 2: 172–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Buckley SM, Aranda‐Orgilles B, Strikoudis A, Apostolou E, Loizou E, Moran‐Crusio K, Farnsworth CL, Koller AA, Dasgupta R, Silva JC et al (2012) Regulation of pluripotency and cellular reprogramming by the ubiquitin‐proteasome system. Cell Stem Cell 11: 783–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu X, Yue P, Chen S, Hu L, Lonial S, Khuri FR, Sun SY (2007) The proteasome inhibitor PS‐341 (bortezomib) up‐regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL‐induced apoptosis despite up‐regulation of c‐FLIP and survivin expression in human NSCLC cells. Cancer Res 67: 4981–4988 [DOI] [PubMed] [Google Scholar]
- 51. Song JJ, Szczepanski MJ, Kim SY, Kim JH, An JY, Kwon YT, Alcala MA Jr, Bartlett DL, Lee YJ (2010) c‐Cbl‐mediated degradation of TRAIL receptors is responsible for the development of the early phase of TRAIL resistance. Cell Signal 22: 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang Y, Ikezoe T, Saito T, Kobayashi M, Koeffler HP, Taguchi H (2004) Proteasome inhibitor PS‐341 induces growth arrest and apoptosis of non‐small cell lung cancer cells via the JNK/c‐Jun/AP‐1 signaling. Cancer Sci 95: 176–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xu L, Su L, Liu X (2012) PKCdelta regulates death receptor 5 expression induced by PS‐341 through ATF4‐ATF3/CHOP axis in human lung cancer cells. Mol Cancer Ther 11: 2174–2182 [DOI] [PubMed] [Google Scholar]
- 54. Sano R, Reed JC (2013) ER stress‐induced cell death mechanisms. Biochim Biophys Acta 1833: 3460–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wielenga MC, Colak S, Heijmans J, van Lidth de Jeude JF, Rodermond HM, Paton JC, Paton AW, Vermeulen L, Medema JP, van den Brink GR (2015) ER‐stress‐induced differentiation sensitizes colon cancer stem cells to chemotherapy. Cell Rep 13: 490–494 [DOI] [PubMed] [Google Scholar]
- 56. Park DM, Jung J, Masjkur J, Makrogkikas S, Ebermann D, Saha S, Rogliano R, Paolillo N, Pacioni S, McKay RD et al (2013) Hes3 regulates cell number in cultures from glioblastoma multiforme with stem cell characteristics. Sci Rep 3: 1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee DH, Szczepanski M, Lee YJ (2008) Role of Bax in quercetin‐induced apoptosis in human prostate cancer cells. Biochem Pharmacol 75: 2345–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7