

Abstract
Hippo signaling controls organ size by regulating cell proliferation and apoptosis. Yes‐associated protein (YAP) is a key downstream effector of Hippo signaling, and LATS‐mediated phosphorylation of YAP at Ser127 inhibits its nuclear localization and transcriptional activity. Here, we report that Nemo‐like kinase (NLK) phosphorylates YAP at Ser128 both in vitro and in vivo, which blocks interaction with 14‐3‐3 and enhances its nuclear localization. Depletion of NLK increases YAP phosphorylation at Ser127 and reduces YAP‐mediated reporter activity. These results suggest that YAP phosphorylation at Ser128 and at Ser127 may be mutually exclusive. We also find that with the increase in cell density, nuclear localization and the level of NLK are reduced, resulting in reduction in YAP phosphorylation at Ser128. Furthermore, knockdown of Nemo (the Drosophila NLK) in fruit fly wing imaginal discs results in reduced expression of the Yorkie (the Drosophila YAP) target genes expanded and DIAP1, while Nemo overexpression reciprocally increased the expression. Overall, our data suggest that NLK/Nemo acts as an endogenous regulator of Hippo signaling by controlling nuclear localization and activity of YAP/Yorkie.
Keywords: 14‐3‐3, cell density, Hippo signaling, NLK, YAP
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
Precise control of the organ size is crucial during animal development and tissue regeneration. Therefore, the size‐control mechanisms of tissue homeostasis have become a long‐standing topic of interest. As loss of the organ size control is linked to many human diseases, including cancer and degenerative diseases, regulation of the organ size could be an attractive therapeutic strategy. Recently, Hippo signaling has been identified as a major signaling pathway to control the organ size; dysregulation of this pathway results in aberrant growth 1.
The Hippo pathway is evolutionarily conserved from nematodes to humans and controls a variety of cellular processes, such as cell proliferation and apoptosis, culminating in the organ size regulation 2. This signaling pathway was originally discovered via genetic screening in Drosophila to identify genes involved in the regulation of the organ size. Hippo, Warts, Salvador, and Yorkie are core components of the pathway which, when mutated, result in dramatic overgrowth of mutant tissues 3, 4, 5, 6, 7. In addition, a number of novel regulatory components in the Hippo pathway have emerged 3, 8, 9. In mammals, the Hippo pathway consists of the serine/threonine kinases MST1/2 (mammalian Ste20‐like kinase, Hippo orthologs) and LATS1/2 (large tumor suppressor kinase, Warts orthologs), the scaffolding protein WW45 (Salvador ortholog), a transcriptional co‐activator YAP (Yes‐associated protein, Yorkie ortholog) and its paralogue TAZ (known as WWTR1) 10. Activation of the Hippo signaling by extracellular stimuli, mechanical sensation, or cell density promotes cytoplasmic retention of YAP/TAZ via interaction with 14‐3‐3, followed by β‐TrCP‐mediated proteasomal degradation of YAP/TAZ 11, 12, 13. These processes depend on LATS1/2‐mediated direct phosphorylation at different sites of YAP 11, 14. YAP phosphorylation on S127 is essential to associate with 14‐3‐3, while phosphorylation on S397 by LATS1/2 creates a phospho‐degron motif for β‐TrCP binding 12. Conversely, dephosphorylated YAP/TAZ functions through the TEAD family transcriptional factors in the nucleus to activate genes involved in cell proliferation and tissue overgrowth 15. Therefore, YAP/TAZ phosphorylation by LATS1/2 has been accepted as a critical process that regulates YAP/TAZ transcriptional activity.
Nemo‐like kinase (NLK) belongs to the atypical MAP‐kinase family and participates in a variety of biological responses, including cell motility and embryogenesis 16, 17. NLK is thought to act as a proline (P)‐directed serine/threonine kinase 16. As several transcriptional regulators have been identified as NLK substrates, NLK is known to play crucial regulatory roles in diverse signaling pathways, including Wnt/β‐catenin, Notch, FOXO, and mTOR signaling 18, 19, 20, 21. However, the functional interaction between NLK and Hippo‐YAP signaling has been unknown.
Here, we show that NLK phosphorylates YAP at Ser128, which leads to reduction in LATS‐mediated phosphorylation at Ser127. NLK thus promotes YAP nuclear localization and transcriptional activity via dissociation from 14‐3‐3. The level of YAP phosphorylation at Ser128, as well as the nuclear localization and overall NLK levels, becomes decreased upon augmentation of the cell density. Consistently, we further show that knockdown of NLK in Drosophila reduces expression of YAP target genes in imaginal discs. Overall, our data suggest that NLK‐mediated YAP phosphorylation is a bona fide mechanism for regulation of YAP activity in vivo.
Results and Discussion
NLK interacts with and phosphorylates YAP
Previous reports have shown that NLK is involved in a variety of signaling pathways, including Wnt, Notch, Foxo, and mTOR signaling. This prompted us to examine whether NLK is able to regulate Hippo signaling. To this end, we overexpressed core components of the Hippo pathway with or without NLK and examined mobility shifts, which might be induced by NLK‐mediated phosphorylation. As shown in Fig EV1A, NLK led to a strong mobility shift of YAP but not other components such as MST1, WW45, and TEAD1. Meanwhile, overexpression of two forms of kinase‐negative NLK constructs (K155M, later on referred as “KM”, and T286V, later on referred as “TV”) 16 did not induce a mobility shift for YAP, indicating the kinase activity of NLK was required for the mobility shift of YAP (Figs 1A and EV1B). Elimination of the mobility shift by treatment with λ‐phosphatase as well as an in vitro kinase assay further confirmed that YAP was phosphorylated directly by NLK (Figs EV1C and 1B). In line with these findings, we observed that both endogenous and exogenous NLK interacted with YAP, and the two kinase‐negative forms of NLK interacted with YAP much more strongly than the wild type did (Figs 1C and EV1D). Taken together, these results suggest that NLK interacts with and directly phosphorylates YAP, and this phosphorylation in turn may regulate binding of NLK and its substrate.
Figure EV1. NLK induces mobility shift of YAP.
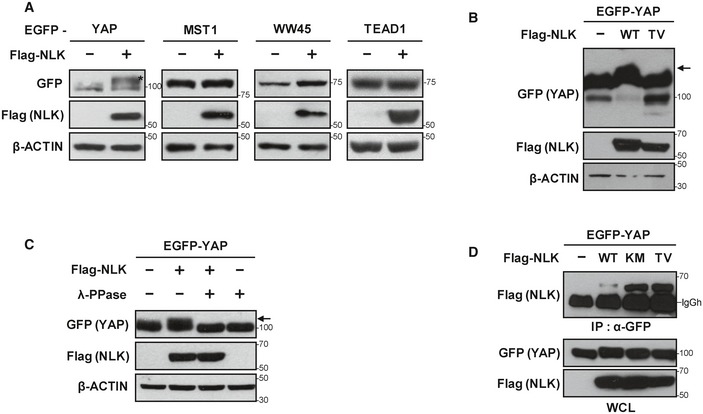
- NLK induces mobility shift of YAP but not other Hippo signaling components. HEK293T cells were transfected with plasmids indicated in the figure, and cell lysates were analyzed by immunoblotting (IB) with antibodies shown on the left.
- Mobility shift of EGFP‐YAP by NLK in Phos‐tag‐PAGE gel. Cell lysates from HEK293T cells transfected with indicated plasmids shown in the figure were separated in Phos‐tag‐PAGE gel, followed by immunoblotting with indicated antibodies. Arrows indicates phosphorylated form of GFP‐YAP.
- Removal of mobility shift by treatment with λ‐phosphatase. Mobility shift of EGFP‐YAP by co‐expression of Flag‐NLK was eliminated by treatment with λ‐phosphatase. Cell lysates were incubated with or without λ‐phosphatase and separated by SDS–PAGE, followed by immunoblotting with the indicated antibodies. Arrow indicates phosphorylated form of GFP‐YAP.
- NLK interacted with YAP. EGFP‐YAP was transfected with empty vector (−), Flag‐NLK‐WT, Flag‐NLK‐KM, or Flag‐NLK‐TV (T286V) into HEK293T cells. EGFP‐YAP was immunoprecipitated with anti‐GFP antibody, followed by immunoblotting with anti‐Flag antibody.
Figure 1. NLK phosphorylates YAP.
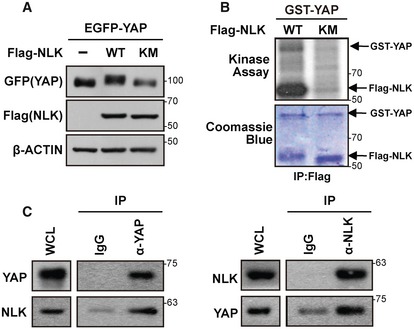
- NLK but not kinase‐negative NLK (Flag‐NLK‐KM; K155M) caused mobility shift of EGFP‐YAP. EGFP‐YAP was transfected with empty vector, Flag‐NLK‐WT, or Flag‐NLK‐KM into HEK293T cells. Mobility shift was shown by immunoblotting.
- NLK phosphorylates YAP in vitro. Flag‐NLK‐WT or Flag‐NLK‐KM was immunoprecipitated with anti‐Flag antibody from cell lysates of HEK293T cells transfected with each plasmid and subjected to in vitro kinase assay with bacterially purified GST‐YAP in the presence of [32P‐ATP]. Phosphorylation of GST‐YAP and autophosphorylation of NLK were examined by autoradiography (top panel), and Coomassie blue staining showed similar levels of GST‐YAP and Flag‐NLK (bottom panel).
- NLK interacts with YAP at the endogenous level. Immunoprecipitates from HEK293T cell lysates with indicated antibody were blotted with anti‐YAP or anti‐NLK antibody.
YAP phosphorylation at Ser128 by NLK attenuates phosphorylation at Ser127
YAP is evolutionarily conserved from nematodes to flies and to mammals, and phosphorylation of YAP at Ser127 (corresponding to human YAP) by serine/threonine kinase LATS1/2 is an important step for regulation of nuclear/cytoplasmic localization and stability (Fig 2A). Interestingly, YAP contains a highly conserved SP motif, a putative NLK phosphorylation site 22, immediately adjacent to Ser127 (Fig 2A). To examine whether NLK is involved in YAP phosphorylation at Ser128, HA‐YAP was transiently transfected into HEK293T cells along with either Flag‐NLK‐WT or Flag‐NLK‐KM and then HA‐YAP was immunoprecipitated with the anti‐HA antibody. LC–MS/MS analysis revealed phospho‐peaks of YAP at Ser128 when Flag‐NLK‐WT was transfected and phospho‐peaks of YAP at Ser127 when Flag‐NLK‐KM was transfected (Fig EV2). This finding prompted us to explore whether or not NLK affects YAP phosphorylation at Ser127. As shown in Fig 2B, endogenous phosphorylation of YAP at Ser127 was reduced by NLK‐WT but not by the NLK‐KM. Interestingly, YAP phosphorylation at Ser397, another LATS1/2 consensus site in YAP that creates a phospho‐degron motif for β‐TrCP binding, was also reduced when NLK was overexpressed (Fig 2B). Next, we used lithium chloride (LiCl), previously used as an inhibitor of NLK and GSK3β activities 23, 24, to further test whether NLK has a negative effect on YAP phosphorylation at Ser127. LiCl augmented YAP phosphorylation at Ser127 when compared to KCl (Fig EV3A). On the other hand, both the GSK3β‐specific inhibitor BIO (Fig EV3A) and knockdown of GSK3β had no effect on YAP phosphorylation at Ser127 (Fig EV3B). These data suggest that LiCl‐mediated induction of YAP phosphorylation at Ser127 was due to inhibition of NLK. To further show that NLK regulates YAP phosphorylation at Ser127 and Ser397, we used the CRSPR/Cas9 system to assess loss‐of‐function effects of NLK on YAP phosphorylation. Transient transfection of two different gRNA sequences targeting NLK with Cas9 resulted in an efficient ablation of NLK (Fig 2C). Consistent with above results, the levels of phosphorylation at Ser127/397 were significantly increased in the NLK KO cell pool (Fig 2C).
Figure 2. YAP phosphorylation at Ser128 by NLK attenuates phosphorylation at Ser127.
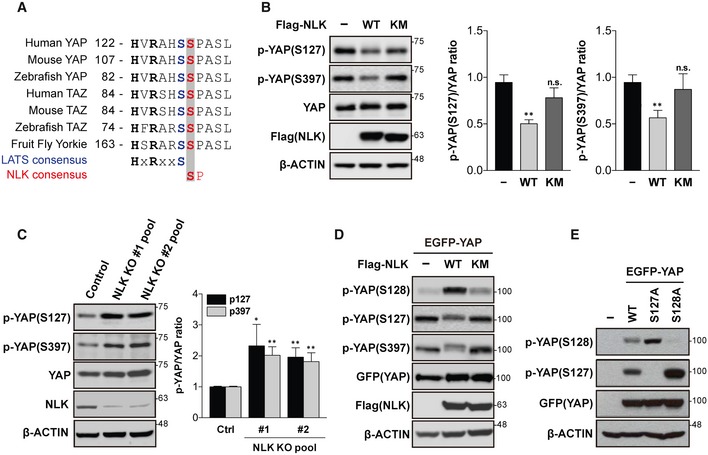
- Conservation of phosphorylation sites for LATS1/2 and NLK. Amino acid sequences of different species of YAP, Yorkie (in Drosophila), and TAZ (YAP paralog) were aligned. NLK is a MAPK (mitogen‐activated protein kinase)‐type kinase, which prefers proline‐directed motif (S/TP).
- NLK‐WT but not NLK‐KM reduces Ser127 phosphorylation of endogenous YAP. Cell lysates from HEK293 cells transfected with empty vector (−), Flag‐NLK‐WT, or Flag‐NLK‐KM were immunoblotted with antibodies shown in the figure (left panel). The ratio of p‐YAP(S127) or p‐YAP(S397)/total‐YAP of three independent Western blots was quantified (right panel). Data are presented as mean ± SEM. **P < 0.01. Student's t‐test was used for statistical analysis.
- Deficiency of NLK leads to increased YAP phosphorylation at Ser127. Lysates from wild‐type and two independent NLK knockout HEK293 cell lines were immunoblotted with antibodies indicated in the figure (left panel). The ratio of p‐YAP(S127) or p‐YAP(S397)/total‐YAP of three independent Western blots was quantified (right panel). Data are presented as mean ± SEM. *P < 0.05 and **P < 0.01. Student's t‐test was used for statistical analysis.
- Ectopic expression of NLK attenuates YAP phosphorylation at Ser127 while increases phosphorylation at Ser128. Cell lysates from HEK293 cells transfected with empty vector (−), Flag‐NLK‐WT, or Flag‐NLK‐KM were immunoblotted with antibodies shown in the figure.
- Substitution of Ser127 with alanine enhances phosphorylation at Ser128, and vice versa. Cell lysates from HEK293T cells transfected with empty vector (−), EGFP‐YAP, EGFP‐YAP‐S127A, or EGFP‐YAP‐S128A were immunoblotted with antibodies shown in the figure.
Figure EV2. Ser128 of YAP is a target residue of NLK.
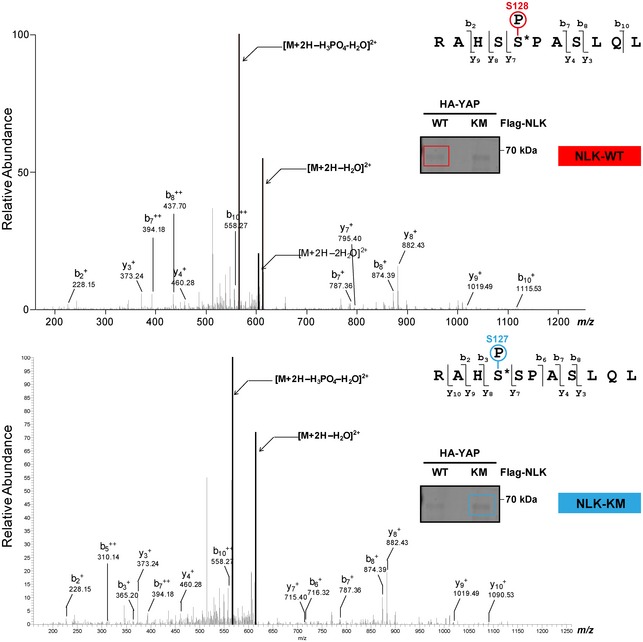
LC‐MS/MS analysis revealed phospho‐peaks of YAP‐S128 by the expression of NLK‐WT (upper panel) and phospho‐peaks of YAP‐S127 by NLK‐KM (lower panel). HA‐YAP was transiently transfected into HEK293T cells along with either Flag‐NLK‐WT or Flag‐NLK‐KM, and then, HA‐YAP was immunoprecipitated with anti‐HA antibody. Immunoprecipitates were separated on SDS–PAGE and stained with Coomassie blue. HA‐YAP bands were cut out, and LC‐MS/MS analysis was performed. Figures show the LC‐MS/MS spectra for the phosphorylation of Ser128(RAHSS*PASLQL) or Ser127(RAHS*SPASLQL), respectively (indicated by asterisks). Red and blue boxes indicate cutting areas for LC‐MS/MS analysis.
Figure EV3. Phosphorylations of Ser127 and Ser128 of YAP may be mutually exclusive.
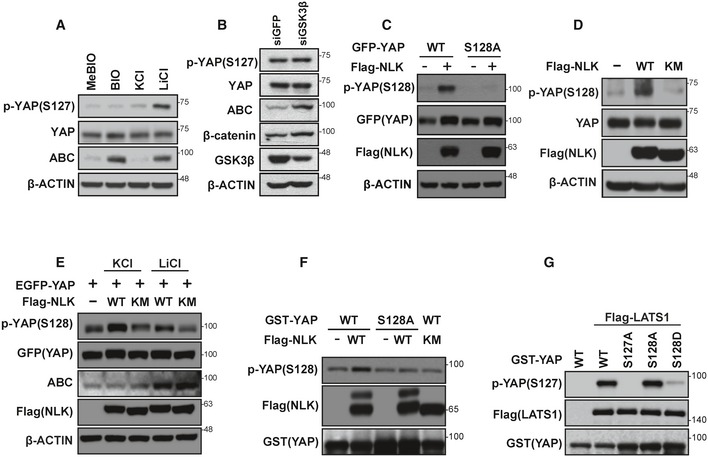
- Inhibition of NLK induces phosphorylation at Ser127 of YAP. HEK293T cells were treated with either 6‐bromoindirubin‐3′‐oxime (BIO, 4 μM; inhibitor of GSK3β), or lithium chloride (50 mM; inhibitor of GSK3β and NLK). MeBIO (4 μM) or KCl (50 mM) were used as negative controls for BIO or LiCl, respectively.
- Knockdown of GSK3β has no effect on YAP phosphorylation at Ser127. Lysates from HEK293T cells transfected with either siControl or siGSK3β were immunoblotted with antibodies indicated in the figure.
- YAP phosphorylation at Ser128 by NLK. Cell lysates from HEK293 cells transfected with empty vector (−), EGFP‐YAP, EGFP‐YAP‐S128A, and Flag‐NLK‐WT were immunoblotted with antibodies shown in the figure.
- Ectopic expression of wild‐type NLK, but not kinase‐negative NLK, enhanced the level of endogenous phospho‐YAP(Ser128). Cell lysates from HEK293T cells transfected with empty vector (−), Flag‐NLK‐WT, or Flag‐NLK‐KM were immunoblotted with antibodies indicated in the figure.
- Treatment with LiCl attenuated NLK‐mediated phosphorylation of YAP Ser128. Cell lysates from HEK293 cells transfected with empty vector (−), Flag‐NLK‐WT, or Flag‐NLK‐KM and treated KCl or LiCl. Lysates were immunoblotted with antibodies shown in the figure.
- NLK specifically phosphorylates YAP‐S128 in vitro. GST‐YAP or GST‐YAP(S128A) was incubated with immunoprecipitated Flag‐NLK‐WT or Flag‐NLK‐KM from lysates of HEK293T cells transfected with Flag‐NLK‐WT or Flag‐NLK‐KM as indicated in the figure. The level of phosphorylation at S128 was detected with pYAP‐S128 antibody.
- Phosphorylation of S128 by NLK inhibits phosphorylation of YAP‐S127 by LATS1. Immunoprecipitated Flag‐LATS1 from lysates of HEK293T cells transfected with Flag‐LATS1 was incubated with various mutant forms of GST‐YAP as shown in the figure. The level of phosphorylation at S127 was detected with pYAP‐S127 antibody.
To further confirm phosphorylation of YAP at Ser128 by NLK, we generated antibody against the phosphorylated YAP‐S128 peptide and confirmed its specificity as determined by no positive signal detected upon expression of YAP‐S128A unlike wild‐type YAP (Fig EV3C). Marked increase in endogenous YAP phosphorylation at Ser128 by NLK‐WT, but not NLK‐KM, was observed (Fig EV3D). Also, increased YAP phosphorylation at Ser128 by ectopic expression of NLK was significantly blocked by the treatment of LiCl but not KCl (Fig EV3E). To further test whether NLK phosphorylates YAP‐S128, in vitro kinase assay was performed. Phosphorylation at S128 of GST‐YAP, but not GST‐YAP‐S128A, was induced by immunoprecipitated Flag‐NLK, but not NLK‐KM (Fig EV3F). Overall, these data suggest that our antibody is specific to pYAP‐Ser128 that is phosphorylated by NLK.
Interestingly, increased phosphorylation of Ser128 by NLK showed a reverse correlation with the phosphorylation at Ser127 (Fig 2D). Phosphorylation at Ser128 was elevated in the YAP‐S127A mutant, whereas the YAP‐S128A mutant exhibited increased phosphorylation at Ser127 when compared to wild‐type YAP (Fig 2E). To further test whether the phosphorylation of S128 by NLK inhibits phosphorylation of YAP‐S127 by LATS1, in vitro kinase assay was performed. As shown in Fig EV3G, the phosphorylation at Ser127 of GST‐YAP and GST‐YAP‐S128A by immunoprecipitated Flag‐LATS1 was highly efficient, while phosphorylation at Ser127 of GST‐YAP‐S128D was strongly suppressed (Fig EV3G). Together with LC–MS analysis, which did not reveal a concomitant YAP phosphorylation on S127 and S128 (Fig EV2), these results suggest that phosphorylations of Ser127 and Ser128 of YAP may be mutually exclusive.
NLK promotes nuclear localization of YAP by inhibiting its interaction with 14‐3‐3
It has been well known that phosphorylation of YAP at Ser127 by LATS1/2 induces cytoplasmic retention of YAP through its interaction with 14‐3‐3 11. We showed that NLK increased YAP phosphorylation at Ser128 along with reduced phosphorylation at Ser127 (Fig 2D). Thus, we examined whether or not YAP phosphorylation at Ser128 by NLK affects the interaction between YAP and 14‐3‐3. Overexpression of NLK‐WT, but not NLK‐KM, completely blocked the interaction between YAP and 14‐3‐3 (Fig 3A). Consistently, NLK KO cells displayed stronger interaction between YAP and 14‐3‐3 compared with normal cells (Fig 3B). To further confirm this, we utilized the YAP mutants S127A, S128A, and S128D (a phosphorylation mimetic form) (Fig 3C). Consistent with the above findings (Figs 2E and 3A and B), YAP‐S128A showed a much stronger interaction with 14‐3‐3 than YAP‐WT did, whereas neither YAP‐S127A nor YAP‐S128D showed binding to 14‐3‐3 (Fig 3C).
Figure 3. NLK promotes nuclear localization of YAP by inhibiting interaction between YAP and 14‐3‐3.
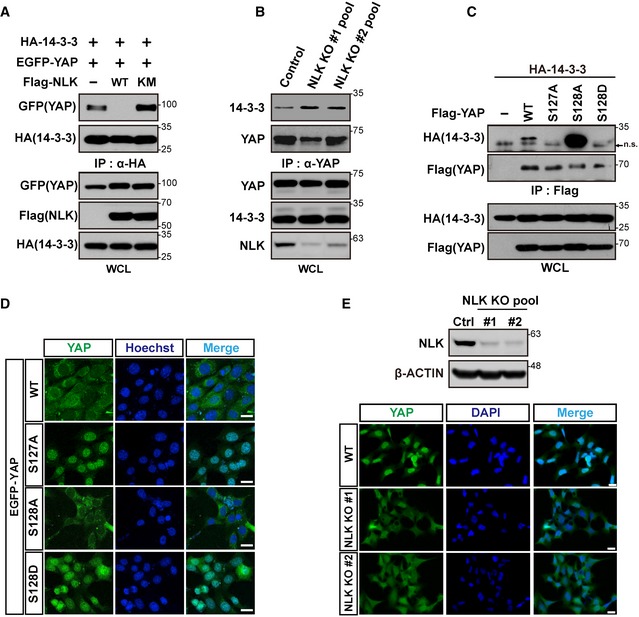
- Ectopic expression of NLK attenuates interaction between YAP and 14‐3‐3. Lysates from HEK293T cells transfected with plasmids indicated in the figure were immunoprecipitated with anti‐HA antibody and immunoblotted with anti‐GFP antibody (top two panels). Whole‐cell lysates (WCL) were immunoblotted with antibodies shown in the figure (bottom three panels).
- Deficiency of NLK leads to increased interaction between YAP and 14‐3‐3. Lysates from NLK knockout HEK293 cells were immunoprecipitated with anti‐YAP antibody and immunoblotted with anti‐14‐3‐3 antibody (top two panels). WCL were immunoblotted with antibodies indicated in the figure (bottom three panels).
- Phosphorylation status of Ser127 or Ser128 determines interaction between 14‐3‐3 and YAP. Lysates from HEK293T cells transfected with plasmids indicated in the figure were immunoprecipitated with anti‐Flag antibody and immunoblotted with anti‐HA antibodies (top two panels). Lysates were immunoblotted with anti‐HA or anti‐Flag antibodies (bottom two panels). “n.s.” arrow indicates a non‐specific band also visible in the non‐transfected lane.
- Phosphorylation status of Ser127 or Ser128 determines subcellular localization of YAP. Immunofluorescence analysis was performed in HeLa cells transfected with EGFP‐YAP, EGFP‐YAP‐S127A, EGFP‐YAP‐S128A, or EGFP‐YAP‐S128D at high cell density. Figure shows a representative of multiple areas. Nuclei were stained with Hoechst 33342. Scale bars: 20 μm.
- NLK knockout leads to increased cytoplasmic localization of endogenous YAP. To determine subcellular localization of YAP, indirect immunofluorescence analysis was performed in NLK knockout HEK293 cell pools (bottom panel). Figure shows a representative of multiple areas. Nuclei were stained with DAPI. Scale bars: 20 μm. Immunoblot analysis shows significant reduction in NLK level in NLK knockout HEK293 cell pools (top panel).
Since interaction with 14‐3‐3 leads to cytoplasmic sequestration of YAP, we next explored whether or not NLK induces nuclear localization of YAP. Overexpression of NLK‐WT, but not NLK‐KM, strongly induced nuclear localization of EGFP‐YAP (Fig EV4A). Consistently, the results from immunofluorescence and subcellular fractionation analysis clearly showed that YAP‐S128A mutant localized to the cytoplasm while the phospho‐mimetic YAP‐S128D mutant was mainly present in the nucleus, which was similar to YAP‐S127A (Figs 3D and EV4B and C). Also, normal cells exhibited strong nuclear localization of YAP at low density, but the NLK KO cell pool exhibited reduced nuclear YAP level (Fig EV4D) along with enhanced cytoplasmic localization of YAP (Fig 3E). Overall, these results indicate that NLK‐mediated phosphorylation at Ser128 leads to nuclear localization of YAP by disrupting the interaction between YAP and 14‐3‐3.
Figure EV4. NLK‐mediated phosphorylation at Ser128 leads to nuclear localization of YAP.
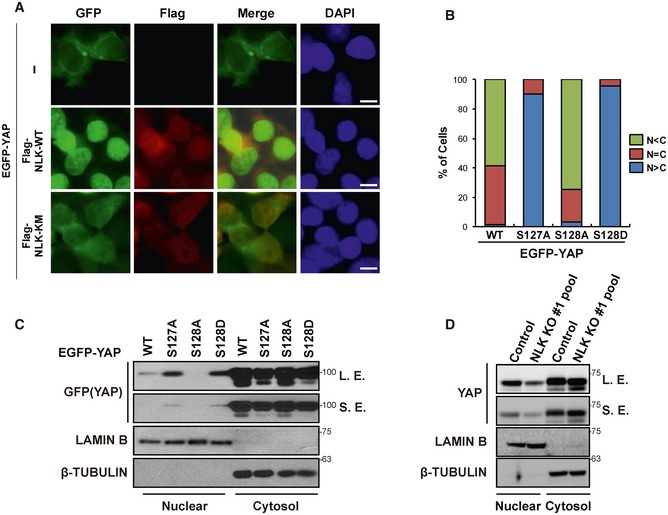
- Overexpression of NLK‐WT, but not NLK‐KM, induces nuclear localization of EGFP‐YAP. Immunofluorescence analysis was performed in HEK293T cells transfected with EGFP‐YAP along with empty vector (−), Flag‐NLK‐WT, or Flag‐NLK‐KM. Scale bars: 10 μm.
- Summary of subcellular localization of EGFP‐YAP, EGFP‐YAP‐S127A, EGFP‐YAP‐S128A, or EGFP‐YAP‐S128D, as shown in Fig 3D.
- Phosphorylation status of Ser127 or Ser128 determines subcellular localization of YAP. Nuclear and cytosolic fractions of lysates were prepared and immunoblotted with antibodies indicated in the figure. Lamin B and β‐tubulin were used as markers for nuclear and cytosolic fractions, respectively. S.E., short exposure; L.E., long exposure.
- Depletion of NLK leads to reduction in YAP levels in the nuclear fraction. Lysates from NLK knockout HEK293 cells were immunoblotted with antibodies indicated in the figure. Lamin B and β‐tubulin were used as markers for nuclear and cytosolic fractions, respectively. S.E., short exposure; L.E., long exposure.
NLK localization and protein level are regulated in a cell density‐dependent manner
Recent studies have suggested that NLK is present in the nucleus or cytoplasm, and dimerization of NLK induced by growth factors is essential for its nuclear localization and functional activation 25. Because NLK regulates YAP localization (Figs 3 and EV4) and nuclear localization of YAP is influenced by cell confluency 11, we investigated whether or not nuclear localization of NLK is changed by cell density. NLK mainly localized to nuclei of cells at low density, whereas it was redistributed in the cytoplasm at high cell density of MCF10A and NIH3T3 cells (Fig 4A). Interestingly, the protein level of NLK, but not its mRNA expression, decreased along with increasing cell density (Fig 4B and C). Consistently, the phosphorylation of endogenous YAP at Ser128 was severely reduced in cells at high cell density, concomitant with increased YAP phosphorylation at Ser127 and Ser397 (Fig 4B). These data indicate that localization as well as protein level of NLK is controlled in a cell density‐dependent manner, which affects the status of phosphorylation and cytoplasmic localization of YAP.
Figure 4. Subcellular localization and protein level of NLK are regulated by cell density.
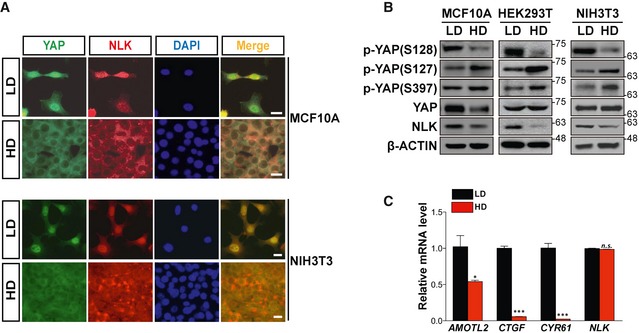
- Localization of endogenous YAP and NLK in low‐density (LD) or high‐density (HD) MCF10A (upper panel) or NIH3T3 cells (bottom panel) was examined by indirect immunofluorescence. Nuclei were stained with DAPI. Scale bars: 20 μm.
- Level of NLK decreases in a cell density‐dependent manner. Lysates from low‐density (LD) and high‐density (HD) MCF10A, HEK293T, and NIH3T3 cells were immunoblotted with antibodies indicated in the figure.
- Level of endogenous NLK mRNA is unaffected, whereas the expression of YAP target genes was reduced, at high cell density. Quantitative real‐time PCR analyses for expression of AMOTL2, CTGF, CYR61, and NLK in low‐density (LD) and high‐density (HD) NIH3T3 cells were performed. Quantification of AMOTL2, CTGF, CYR61, and NLK mRNA was normalized with the level of Gapdh. Data represent average values from a representative of multiple experiments performed in triplicate. Error bars indicate standard deviations of triplicate measurements. Data are presented as mean ± SD. *P < 0.05 and ***P < 0.005. Student's t‐test was used for statistical analysis.
NLK regulates YAP transcriptional activity and cell migration
YAP interacts with TEAD and induces expression of genes involved in the regulation of cell proliferation, anti‐apoptosis, and cell migration 15. Therefore, we next investigated whether or not NLK controls transcriptional activity of YAP. Knockout of NLK severely reduced the interaction between YAP and TEAD (Fig 5A). Consistently, reporter activities using a luciferase construct driven by 8x‐GTIIC promoter 13 were significantly reduced when NLK was knocked down (Fig EV5A). Real‐time PCR analysis revealed that the levels of endogenous CTGF and ANKRD1 mRNA, well‐known YAP/TEAD target genes 13, 15, were significantly reduced by knockdown of NLK (Fig EV5B). To investigate the role of NLK in regulation of the transcriptional activity of YAP in vivo, we tested the effects of manipulation of Nemo (Drosophila NLK) levels during fruit fly wing development on Yorkie (Drosophila YAP) target gene expression. Normal expression of expanded, as can be seen in the anterior, anti‐Ci marked control regions of the Drosophila wing imaginal discs (Figs EV5C and 5D), is low within the wing pouch and high at the wing pouch periphery 26. Nemo overproduction leads to dramatic upregulation of expanded within the wing pouch, quantified in Fig EV5C. In contrast, Nemo downregulation reduces the peripheral expression of this target (high magnifications in Fig EV5D). To test whether Nemo downregulation could suppress another Yorkie target gene, whose expression would be more uniform across the wing pouch area, we performed immunostaining against the Yorkie target DIAP1 7, which reveals a salt‐and‐pepper expression pattern within the wing pouch area 27. This analysis reveals that Nemo downregulation strongly suppressed DIAP1 throughout the RNAi‐expressed posterior domain (Fig 5C). Finally, we wished to test the epistasis between Nemo and Yorkie. For this aim, we co‐expressed the nemo‐targeting RNAi and a Hippo insensitive mutant form of Yorkie (yki[S168A]) in the posterior compartment. The resulting discs display typical yki[S168A] phenotype 28 of massive posterior overgrowth and increase in the Yorkie target gene expression (Fig 5D), indistinguishable from discs expressing yki[S168A] alone. This observation indicates that Yorkie is epistatic to Nemo in Drosophila.
Figure 5. NLK regulates transcriptional activity of YAP.
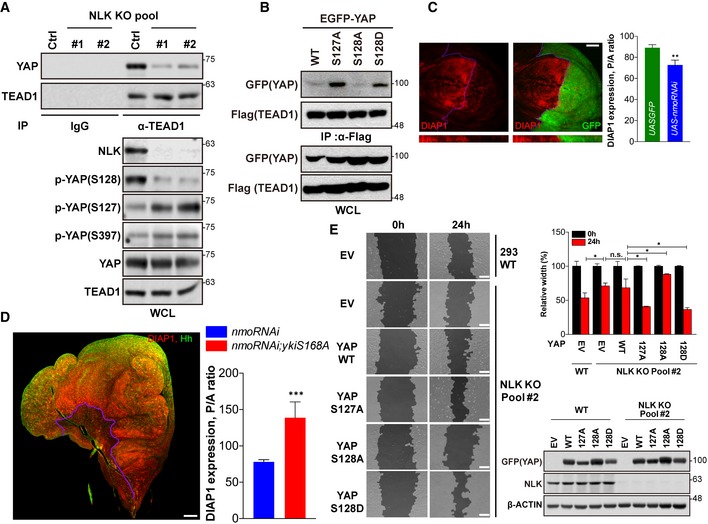
- Knockout of NLK reduces the interaction between endogenous YAP and TEAD1. Lysates from NLK knockout HEK293 cells were immunoprecipitated with anti‐TEAD1 antibody and immunoblotted with anti‐TEAD1 antibody or anti‐YAP antibody (top panel). Whole‐cell lysates (WCL) were immunoblotted with antibodies indicated in the figure.
- Phosphorylation status of Ser127 or Ser128 determines interaction between TEAD1 and YAP. Lysates from HEK293T cells transfected with plasmids indicated in the figure were immunoprecipitated with anti‐Flag antibody and immunoblotted with indicated antibodies in the figure.
- Downregulation of nemo with RNAi achieved in the posterior compartment of wing imaginal discs (co‐expression of GFP serves to mark the posterior compartment) using the hh‐Gal4 driver results in decreased expression of the Yorkie target gene DIAP1. Two different RNAi constructs produce similar effects. Panels below show Z‐stacks of magnified areas around the A/P border (left). Quantification of DIAP1 levels achieved upon posterior downregulation of nemo, measured as normalized posterior‐to‐anterior levels and compared to similar expression of GFP. Data are presented as mean ± SEM., n = 8 discs. **P < 0.01. Student's t‐test was used for statistical analysis (right). Scale bar: 50 μm.
- Yorkie is epistatic to Nemo: Co‐overexpression of the Hippo insensitive mutant yki[S168A] together with nmo‐RNAi produces typical yki[S168A] phenotypes: severe overgrowth of the affected area with concomitant increase in yki target gene expression (DIAP1). Due to lethality of the hh‐Gal4‐driven co‐overexpression, another posterior driver en‐Gal4 was used instead; anti‐Hh staining (green) was used to mark the posterior compartment (left panel). Quantification of posterior DIAP1 levels in UAS‐RNAi‐nmo/en‐Gal4 and UAS‐RNAi‐nmo/en‐Gal4; UAS‐yki[S168A], presented as in (C); n = 3–6 discs. Posterior DIAP1 levels in UAS‐RNAi‐nmo; en‐Gal4 is also significantly different from control UAS‐GFP; en‐Gal4 discs (right panel). Data are presented as mean ± SEM. ***P < 0.005. Student's t‐test was used for statistical analysis (right). Scale bar: 50 μm.
- Mutant forms of YAP which have different phosphorylation status of Ser127 or S128 differentially rescue retarded wound healing caused by NLK knockout. HEK293 wild‐type or HEK293 NLK knockout cells were transfected with indicated plasmids (“EV” stands for “empty vector”), and the wounded areas after 0 and 24 h were measured by using the TScratch program (left panel). Graphs show percentage of unfilled areas at 24 h compared to 0 h, and average values from a representative of multiple experiments performed in triplicate (right‐top panel). Lysates were immunoblotted with antibodies indicated in the figure (right‐bottom panel). Error bars indicate standard deviations of triplicate measurements. Data are presented as mean ± SD. *P < 0.05. Student's t‐test was used for statistical analysis (right). Scale bars: 200 μm.
Figure EV5. NLK positively regulates YAP activity in physiological settings.
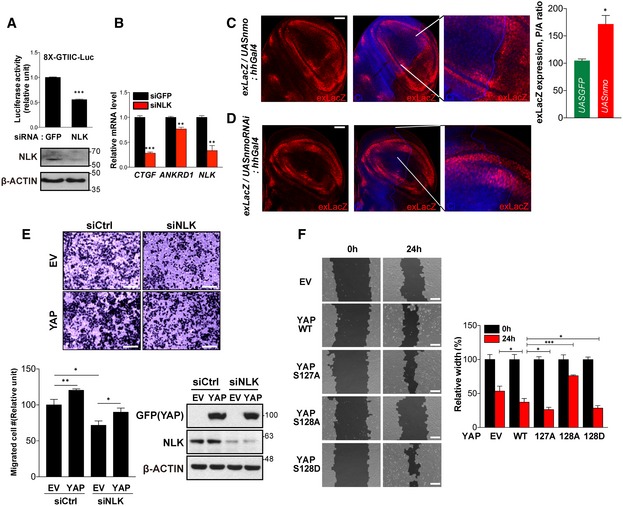
- Knockdown of NLK attenuates YAP‐dependent reporter activity. HEK293T cells were transfected with siRNA for GFP or NLK, and 8xGTIIC promoter‐driven luciferase activity was measured. Luciferase activities represent average values from a representative of multiple experiments performed in triplicate. Error bars indicate standard deviations of triplicate measurements. Lysates were immunoblotted with indicated antibodies. ***P < 0.005. Student's t‐test was used for statistical analysis.
- Knockdown of NLK reduces endogenous YAP/TEAD target gene expression. Quantitative real‐time PCR analyses were performed with RNAs from HEK293T cells transfected with siRNA for GFP or NLK. The expression of ANKRD1, CTGF, and NLK mRNA was normalized with the level of β‐actin. Data represent average values from a representative of multiple experiments performed in triplicate. Error bars indicate standard deviations of triplicate measurements. Data are presented as mean ± SD. **P < 0.01 and ***P < 0.005. Student's t‐test was used for statistical analysis.
- Overexpression of nemo achieved in the posterior compartment of wing imaginal discs (marked by the absence of anti‐Ci staining, blue) using the hh‐Gal4 driver results in expression of the yki target gene expanded (exLacZ, red) within the wing pouch, where it is normally expressed only at low levels. The third panel shows a higher magnification of a relevant area. The unaffected anterior domain serves as an internal control (left panel). Quantification of exLacZ levels achieved upon posterior overexpression of nemo, measured as normalized posterior‐to‐anterior levels and compared to similar expression of GFP (right panel). Data are presented as mean ± SEM, n = 5 discs. *P < 0.05. Student's t‐test was used for statistical analysis. Scale bar: 50 μm.
- Downregulation of nemo with RNAi achieved in the posterior compartment of wing imaginal discs (marked by the absence of Ci) results in decreased expression of the exLacZ in the periphery of the wing pouch, where it is normally expressed at high levels. The third panel shows a higher magnification of a relevant area. Two different RNAi constructs produce identical effect. Scale bar: 50 μm.
- Ectopic expression of YAP rescues retarded cell migration ability induced by knockdown of NLK. HeLa cells were transfected with control or NLK‐targeting siRNA and empty vector (EV) or EGFP‐YAP as shown in the figure. Migratory cells on lower face membrane were stained by crystal violet (upper panel). Scale bars: 200 μm. Graphs show relative migrated cell number from a representative of multiple experiments performed in triplicate (left bottom panel). Error bars indicate standard deviations of triplicate measurements. Data are presented as mean ± SD. *P < 0.05 and **P < 0.01. Student's t‐test was used for statistical analysis. Lysates from the cells transfected as above were immunoblotted as shown on the bottom right panel.
- Mutant forms of YAP which have different phosphorylation status of Ser127 or S128 differentially regulate wound healing ability. HEK293 cells were transfected with indicated plasmids in the figure, and the wounded areas after 0 and 24 h were measured by using the TScratch program (left panel). EV, empty vector. Graphs show relative values of unfilled areas and average values from a representative of multiple experiments performed in triplicate (right panel). Error bars indicate standard deviations of triplicate measurements. Data are presented as mean ± SD. *P < 0.01 and ***P < 0.005. Student's t‐test was used for statistical analysis. Scale bars: 200 μm.
Since YAP enhances cell migration and proliferation 29, we tested whether NLK regulates cell migration. Knockdown of NLK significantly reduced cell migration, which depends on YAP because its expression rescued this defect (Fig EV5E). We further tested the effect of NLK on YAP‐dependent wound healing as a measure of cell proliferation and migration. Wound healing ability was affected by the phosphorylation status of Ser127 or Ser128. Ectopic expression of YAP‐S127A and the phosphorylated mimetic form of YAP at Ser128 (YAP‐S128D) in wild‐type cells or NLK KO pool cells showed enhanced wound healing ability compared to cells expressing YAP‐WT, whereas the S128A mutation produced the opposite effect (Figs 5E and EV5F). Overall, these findings demonstrate that NLK positively regulates YAP activity in physiological settings.
YAP has oncogenic properties and mainly functions as a downstream effector of the Hippo pathway 30. LATS1/2 phosphorylates YAP at five serine/threonine residues, and phosphorylation of these sites acts as a fundamental input for YAP regulation 11. Of these phosphorylation sites, Ser127 and S397 are the most functional residues regulating YAP activity. Phosphorylation at Ser127 by LATS1/2 creates a binding consensus site for 14‐3‐3, resulting in cytoplasmic retention, while phosphorylation on S397 induces β‐TrCP‐mediated proteasomal degradation of YAP. In addition to LATS1/2 kinases, Akt is also reported to phosphorylate YAP at serine 127, thereby leading to association 14‐3‐3 and regulating apoptosis following the cellular damage 31. More recently, NDR1/2 kinases were identified as YAP‐S127 kinases and might function as tumor suppressors upstream of YAP in colon cancer 32. Conversely, YAP phosphorylation can be reversed by phosphatase, leading to YAP reactivation. Regarding the physiological importance of YAP‐S127 phosphorylation, one interesting study has addressed it by introducing a Yap‐S112A (corresponding site for the human homologue in mouse) knock‐in mutation in the endogenous Yap locus. Surprisingly, although nuclear localization of Yap protein was observed in tissues and cultured cells harboring mutant Yap‐S112A, knock‐in mice were phenotypically normal probably due to compensatory decrease in YAP protein levels 33. These findings suggest a homeostatic regulation that maintains physiological YAP activity by yet unidentified mechanisms.
In addition to serine phosphorylation, phosphorylation of YAP at tyrosine residues by Src or c‐Abl kinase modulates YAP transcriptional activity 34, 35. Tyrosine phosphatase PTPN14 is able to inhibit YAP transcriptional activity by inducing cell density‐dependent translocation of YAP from the nucleus to cytoplasm 36. A more recent report showed that monomethylation of YAP at K494 by Set7, a SET domain‐containing lysine (K) methyltransferase, promotes cytoplasmic retention and inhibition of YAP‐dependent growth 37. Taken together, these observations suggest that control of YAP subcellular localization is the sum of multiple regulatory layers and is critical for transactivation. Thus, identifying the molecular regulatory mechanisms of YAP localization will be important to fully understand how the organ size and occurrence of tumors are controlled.
NLK regulates activities of several transcription factors, including TCF/LEF1, NICD, and FOXO, which are key players in diverse signaling pathways 18, 19, 20. Consistently, NLK knockout mice show varying degrees of morphological abnormalities, such as growth retardation, neurological abnormalities, and hematopoietic defects 38. Here, we identified NLK as a novel kinase for YAP. Our results show that NLK phosphorylates Ser128 of YAP and plays a crucial role in regulating YAP localization and activation. The phospho‐mimetic form of YAP‐S128 showed strong nuclear localization and led to enhanced wound healing ability, whereas substitution of Ser128 with alanine showed the opposite effects. NLK depletion led to significant reduction in the YAP‐dependent reporter activity. In the accompanying paper, Dr. Guan and colleagues report similar findings that NLK mediates YAP phosphorylation at Ser128 and nuclear localization by inhibiting association with 14‐3‐3 upon osmotic stress 39. Together with their results, our findings suggest that NLK is required for YAP nuclear localization and transcriptional activity in various cellular processes.
We expanded the role of NLK in the regulation of YAP activity in vivo. Knockdown of Nemo (Drosophila NLK) in the fruit fly wing imaginal discs reduced expression of the Yorkie (Drosophila YAP) target genes expanded and DIAP1, while Nemo overproduction produced the opposite effect. The localization of NLK was changed from nuclei to cytoplasm, and the level was reduced along with increasing cell density. It has been shown that dimerization and nuclear localization of NLK are necessary for its activity 25. Further work is required to explore how the cell density affects NLK nuclear localization and level. It is possible that regulation of subcellular localization and level of NLK at different cell densities could be another mechanism to secure YAP localization and regulation of its transcriptional activity; nuclear NLK phosphorylates Ser128 of YAP and prevents phosphorylation of Ser127/Ser397 by LATS1/2, which allows YAP to act as a transcriptional activator in nuclei of low‐density cells.
Using phospho‐specific antibody and in vitro kinase assay, we observed that NLK phosphorylated YAP at Ser128, which is adjacent to the LATS1/2‐mediated phosphorylation site Ser127. More importantly, LATS1/2‐mediated phosphorylation at Ser127/Ser397 was inhibited by YAP phosphorylation at Ser128. As shown in Fig 2A, Ser128 is evolutionarily conserved from fruit flies to humans. A previous report examined potential phosphorylation sites within Drosophila Yorkie and observed reduced phosphorylation at Ser168, corresponding to Ser127 of human YAP, in a double‐mutant harboring S169D and S172D 28. This study suggested that phosphorylation at Ser169/Ser172 could reduce Ser168 phosphorylation, thereby promoting Yorkie activity. However, they did not identify endogenous kinase(s) responsible for phosphorylation at Ser169 and Ser172. Although it is unclear whether or not NLK affects phosphorylation at Ser131 (Ser172 of Drosophila Yorkie), our findings provide strong evidence that NLK is an endogenous kinase for phosphorylation at Ser128 that promotes YAP nuclear localization and transcriptional activity. Therefore, we speculate that NLK and LATS1/2 might compete for adjacent phosphorylation residues. Currently, we do not know how the phosphorylation of Ser397 is regulated by NLK, but we think that decrease in phosphorylation on S397 might be due to enhanced nuclear localization of YAP by NLK (Figs 3D and E, and EV4A), which sequesters YAP from phosphorylation by LATS1/2. Overall, our study uncovers a previously unknown kinase involved in regulation of YAP activity and sheds new light on the evolutionarily conserved regulatory mechanism of the Hippo signaling pathway.
Materials and Methods
Plasmids, siRNAs, and CRISPR
Full‐length human YAP was PCR‐amplified using cDNA of HEK293T cells and inserted into pCMV4‐FLAG (Sigma), pEGFP (Clontech), pCS2‐HA3 (constructed in our laboratory), and pGEX‐4T (GE Healthcare) expression vectors. Point mutation constructs were generated by a PCR‐based site‐directed mutagenesis method using Pfu polymerase (Promega) and KOD hot start DNA polymerase (Novagen). MST1, WW45, and TEAD1 coding sequences obtained by PCR were subcloned into pEGFP‐C1. FLAG‐tagged NLK and its mutant constructs were kindly provided by Dr. Ishitani (Kyushu University, Japan). HA‐tagged 14‐3‐3 was a gift from Dr. Hong (Korea University, Korea). CTGF‐luciferase was generously provided from Dr. Guan (University of California San Diego, USA). 8xGTIIC‐luciferase was a gift from Dr. Lim (Korea Advanced Institute of Science and Technology, Korea). ON‐TARGET plus siRNAs targeting human and mouse NLK were purchased by Dharmacon. siGSK3β for human duplexes was purchased by Samchully and its sequence is as follows: sense: 5′‐CCCAAATGTCAAACTACCA‐3′ antisense: 5′‐TGGTAGTTTGACATTTGGG‐3′. To generate a guide RNA Vector for NLK knockout using CRSPR‐Cas9 system, we inserted two different 20‐mer guide RNA sequences (#1 5′‐AAAATGATGGCGGCTTACAA‐3′ and 5′‐TTGTAAGCCGCCATCATTTT‐3′, #2 5′‐ACACCATCTTCATCCGGGGT‐3′ and 5′‐ ACCCCGGATGAAGATGGTGT‐3′) into pSpCas9(BB)‐2A‐Puro (a gift from Feng Zhang, Addgene plasmid # 62988).
Cell culture and transfection
HEK293, HEK293T, HeLa, and NIH3T3 cells were maintained in Dulbecco's modified Eagles' medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1× antibiotic–antimycotic (Gibco) and maintained at 37°C in a humidified 5% CO2 incubator. MCF10A cells were maintained in DMEM/F‐12 medium supplemented with 5% horse serum, 20 ng/ml of epidermal growth factor, 0.5 μg/ml of hydrocortisone, 10 μg/ml insulin, 100 ng/ml of cholera toxin (Sigma), 1× penicillin, and streptomycin (Gibco). Cell transfection was performed using Lipofectamine 2000 and LTX (Invitrogen), Turbofect (Fermentas) or calcium phosphate method.
Immunoprecipitation and immunoblotting
Cell lysates were prepared in lysis buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 1 mM EDTA, 2 mM EGTA, 50 mM β‐glycerophosphate, 50 mM NaF, 1 mM sodium vanadate, 2 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml of leupeptin, and 1% Triton X‐100). Lysates were centrifuged at 14,200 g for 10 min at 4°C, and the supernatant was collected and used for immunoprecipitation and immunoblotting. For immunoprecipitation, 400–800 μg of cell lysate was incubated with antibodies indicated in the figures and protein A/G plus agarose beads (Santa Cruz Biotechnology). Beads were washed five times with lysis buffer and centrifuged at 3,000 g for 3 min between each wash. Equal amounts of protein were boiled with Laemmli sample buffer and resolved on 6, 8, or 10% SDS–PAGE and transferred to PVDF membrane (Pall). Phos‐tag™ acrylamide was purchased from Wako Pure Chemical Industries, and Phos‐tag™ SDS–PAGE gel was prepared according to the manufacturer's protocol.
Mouse monoclonal anti‐GST (B‐14), anti‐YAP (63.7), anti‐GFP (B‐2), anti‐NLK (B‐5), pan 14‐3‐3 (H‐8), anti‐HA (F‐7), and goat polyclonal lamin B (M‐20) antibodies were obtained from Santa Cruz. Rabbit polyclonal anti‐YAP phospho‐128 antibody was generated using recombinant YAP peptide PQHVRAHS(pS)PASLQLG and affinity purified (AbClon). Rabbit polyclonal anti‐HA (#A190‐108A) antibody was purchased from Bethyl Laboratories. Mouse monoclonal anti‐FLAG M2 antibody was obtained from Stratagene. Mouse monoclonal anti‐β‐catenin (#610154), anti‐GSK3β (#610201), and anti‐Tead1 (#610923) antibodies were purchased from BD Transduction Laboratories. Rabbit polyclonal anti‐β‐tubulin (#GTX107175) antibody was obtained from GeneTex. Rabbit polyclonal anti‐YAP, anti‐phospho‐YAP (Ser127), and anti‐phospho‐YAP (Ser397) antibody were purchased from Cell Signaling Technology. Mouse monoclonal anti‐β‐actin and rabbit polyclonal anti‐NLK (#N8288) antibodies were purchased from Sigma. Mouse monoclonal anti‐active‐β‐catenin (anti‐ABC) (#05‐665) antibody was obtained from Millipore.
Immunofluorescence analysis
HEK293T, NIH3T3, HeLa, and MCF10A cells were seeded onto glass coverslips in 6‐well plates, fixed for 20 min in 4% paraformaldehyde in PBS, and then permeabilized for 15 min with 0.1% Triton X‐100 in PBS at room temperature. Samples were washed three times with PBS, blocking with 5% BSA (Bio basic) in PBS for 1 h, and then incubated with the indicated primary antibodies for overnight at 4°C. Samples were then rinsed three times with 1% BSA in PBS and incubated with the secondary antibodies Alexa‐Fluor antibodies 488 or 633 (Invitrogen) for 2 h. For nuclei staining, Hoechst 33342 solution or DAPI were added to each sample and incubated at 5 min at room temperature. Localization of proteins was analyzed using fluorescence microscopy [Cell Observer (Zeiss)] and confocal microscopy [TCS SP8 (Leica)]. The values for the histogram were calculated using ImageJ software.
Cellular fractionation
To separate cytoplasmic and nuclear proteins, cells were washed in phosphate‐buffered saline (PBS) and suspended in hypertonic buffer (50 mM HEPES (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM Na3VO4, 50 mM NaF, 1 mM PMSF, 1 μg/ml of leupeptin). After being incubated on ice for 30 min, cells were sheared by being passed 25 times through a 26‐gauge needle. The lysates were centrifuged at 800 g for 5 min at 4°C to obtain the cytosolic and membrane fraction (supernatant) and nuclear fraction (pellet). The nuclear pellet was washed three times with hypotonic buffer and resuspended in hypotonic buffer containing 1% Triton X‐100 and 150 mM NaCl for 30 min on ice. The supernatant of lysed pellet, which has nuclear proteins, was isolated after centrifugation at 12,000 g for 20 min at 4°C.
Preparation of recombinant proteins and in vitro kinase assay
pGEX‐YAP was transformed into E. coli BL21 (DE3). For induction of GST fusion protein, 0.2 mM IPTG (Calbiochem) was added to the YTA medium. Purification of GST fusion proteins was performed according to standard procedures (GE Healthcare). For in vitro kinase assays, WT or KM FLAG‐NLK was transfected into HEK293T cells, followed by immunoprecipitation using anti‐FLAG monoclonal antibody coupled to protein A/G plus agarose beads. Immunoprecipitates were incubated in kinase buffer containing GST‐fused proteins, 10 μCi of [γ‐32P]ATP, 10 mM HEPES (pH 7.4), 1 mM DTT, and 5 mM MgCl2 at 25°C for 5 min. Samples were resolved by SDS–PAGE, and phosphorylated proteins were detected by autoradiography. In vitro kinase assays without using radioisotope were performed as described previously 25.
Analysis by nano‐LC‐ESI‐MS/MS
The protein separated by SDS–PAGE was excised from the gel and digested with endoproteinase chymotrypsin (Roche) in incubation buffer (100 mM Tris–HCl, 10 mM CaCl2, pH 7.6) at 25°C for overnight. Following digestion, chymotryptic peptides were extracted with 5% formic acid in 50% acetonitrile solution at room temperature for 20 min. The supernatants were dried and desalted using C18 ZipTips (Millipore, MA) before LC‐ESI‐MS/MS analysis. Nano‐LC (EksigentnanoLC Ultra 2D, Eksigent Technologies, Dublin, CA) separation was conducted under a linear gradient from 3 to 40% solvent B (0.1% formic acid in 100% acetonitrile) with a flow rate of 250 nl/min for 60 min. The column was directly connected to LTQ linear ion‐trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with a nano‐electrospray ion source. All spectra were acquired in data‐dependent scan mode. Each full MS scan was followed by five MS/MS scan corresponding from the most intense to the fifth intense peaks of full MS scan. The acquired LC‐ESI‐MS/MS fragment spectra were searched in the BioWorksBrowserTM (version Rev. 3.3.1 SP1, Thermo Fisher Scientific, San Jose, CA) with the SEQUEST search engines against the data in FASTA format generated from YAP (NCBI accession number NM_001282101.1) in National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/).
Luciferase assay
HEK293T cells were seeded in 12‐well plates 1 day before transfection. A mixture of 8xGTIIC promoter‐luciferase reporter, pRL‐TK‐Renilla, and the indicated plasmids was co‐transfected into HEK293T cells. Twenty‐four hours after transfection, cells were lysed and luciferase activity was measured using a dual‐luciferase reporter assay system (#E1960; Promega) according to the manufacturer's instructions. Luciferase activity was measured by a GLOMAX 20/20 luminometer (Promega). Transfection efficiency was normalized to thymidine kinase promoter‐driven Renilla luciferase (pRL‐TK) activity as the internal control.
Phosphatase treatment
Cell lysates were prepared for PPase lysis buffer (25 mM Tris–HCl, pH 8.0, 150 mM NaCl, 10% NP‐40, 10% glycerol, 0.25% deoxycholic acid, 1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml of leupeptin). Lysates were incubated with or without λ‐PPase (New England Biolabs) in PPase buffer at 30°C for 30 min.
Real‐time PCR analysis
For quantitative real‐time PCR, total RNA was isolated using TRIZOL reagent (Invitrogen) according to the manufacturer's protocol. cDNA was synthesized from total RNA using ImProm‐II™ Reverse Transcriptase (Promega) with random primer. Real‐time PCR was carried out by an ABI prism 7000 Sequence Detector (Applied Biosystem) with SYBR Green PCR Master Mix (Applied Biosystem). PCR products were designed to amplify specific mRNA fragments and as a displayed unique dissociation (melting) curve. PCR conditions were 95°C (10 min) and 40 cycles at 95°C (30 s) and 60°C (1 min). The threshold cycle (Ct) value for each gene was normalized to the Ct value for β‐actin or GAPDH. Relative mRNA expression was calculated using the ΔΔCt method. Primers used for human cell lines: ANKRD1, 5′‐CACTTCTAGCCCACCCTGTGA‐3′ and 5′‐CCACAGGTTCCGTAATGATTT‐3′; CTGF, 5′‐CCAATGACAACGCCTCCTG‐3′ and 5′‐GAGCTTTCTGGCTGCACCA‐3′; NLK, 5′‐CCAGTGACTTTGAGCCTGTC‐3′ and 5′‐GATGGCTGAGCAACAGTGG‐3′; and β‐ACTIN, 5′‐GCGGGAAATCGTGCGTGACATT‐3′ and 5′‐GATGGAGTTGAAGGTAGTTTCGTG‐3′.
Primers used for mouse cell lines: Amotl2, 5′‐AGAGATTGGAATCGGCAAAC‐3′ and 5′‐TTCTCCTGTTCCTGTTGCTG‐3′; Ctgf, 5′‐CAAGGACCGCACAGCAGTT‐3′ and 5′‐AGAACAGGCGCTCCACT CTG‐3′; Cyr61 5′‐ATGATGATCCAGTCCTGCAA‐3′ and 5′‐TAGGCTGTACAGTCGGAACG‐3′; Gapdh 5′‐TGTTCCTACCCCCAATGTGT‐3′ and 5′‐TGTGAGGGAGATGCTCAGTG‐3′.
Migration assay
HeLa cells (3 × 104 cells) were plated in the upper chamber of cell culture inserts (SPL, 8‐μm membrane pore size) with serum‐free media. Cell growth medium containing 5% serum was added to the lower well. After 24‐h incubation, cells were fixed for 2 min in 3.7% formaldehyde in PBS and then permeabilized for 20 min with 100% methanol at room temperature. After removal of non‐migratory cells, migrated cells were stained with 0.5% crystal violet. Migrated cells were counted using an inverted microscope, and the values for histogram were calculated using ImageJ software.
Wound healing assay
HEK293 and NLK KO pool cells were transfected with control or YAP mutants in 24‐well plates and cultured until full confluency. Cells were scratched by 200‐μl pipette tip and then washed with media to remove cell debris, and the media was replaced. Photographs of the wound region were taken at 0 and 24 h. Cell free area was measured using TScratch software 40.
Experiments with Drosophila
Analysis of the effects of manipulations of Nemo levels on Yorkie target gene expression was performed following 41, whereas the posterior‐specific hedgehog‐Gal4 (hh‐Gal4) or engailed‐Gal4 (en‐Gal4) drivers were used to induce overexpression or downregulation of nemo using the UAS‐nmo 17 or UAS‐nmo‐RNAi lines (104885KK or 3002GD 42), respectively. UAS‐yki[S168A] was used as described 28. In third instar larval wing imaginal discs, these posterior‐specific manipulations were compared to the unaffected anterior halves of the discs, which served as internal controls. expanded‐LacZ 43 and DIAP1 were used to monitor Yorkie target gene expression. Mouse anti‐DIAP1 27, and anti‐Ci, anti‐β‐gal, and anti‐Hh antibodies were used as described 41. Quantification of fluorescent intensity after confocal microscopy was performed with ImageJ.
Statistical analysis
All data analysis in this study was carried out using GraphPad Prism7 (San Diego, CA, http://www.graphpad.com) for Mac OS. Quantifications were done from at least three independent experiment groups. Statistical analysis between groups was assessed by two‐tailed Student's t‐test to determine significance. P‐values < 0.05 were considered significant. Error bars on all graphs are presented as standard error of the mean (SEM) unless otherwise indicated.
Author contributions
SM and WK carried out most of the experiments and wrote the manuscript; TL and BC performed imaging analysis; SK, YK, YS, OB, JK performed experiments during the preparation of revision; MK and HK contributed to intellectual inputs; and VLK (who also performed the Drosophila experiments) and EJ supervised the entire project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Bruce Hay (California Institute of Technology) for anti‐DIAP1 antibody and Bloomington and VDRC stock centers for Drosophila lines. This work was supported by the grants from the National Research Foundation (2013R1A1A2064012) and the National R&D Program for Cancer Control, Ministry of Health & Welfare, and Republic of Korea (1420060) to E. Jho, as well as the grant from Swiss National Science Foundation (31003A_138350) to V.L.K.
EMBO Reports (2017) 18: 61–71
See also: AW Hong et al (January 2017) and A Hergovich (January 2017)
Contributor Information
Vladimir L Katanaev, Email: vladimir.katanaev@unil.ch.
Eek‐hoon Jho, Email: ej70@uos.ac.kr.
References
- 1. Halder G, Johnson RL (2011) Hippo signaling: growth control and beyond. Development 138: 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan D (2010) The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harvey KF, Pfleger CM, Hariharan IK (2003) The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114: 457–467 [DOI] [PubMed] [Google Scholar]
- 4. Jia J, Zhang W, Wang B, Trinko R, Jiang J (2003) The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev 17: 2514–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pantalacci S, Tapon N, Leopold P (2003) The Salvador partner Hippo promotes apoptosis and cell‐cycle exit in Drosophila . Nat Cell Biol 5: 921–927 [DOI] [PubMed] [Google Scholar]
- 6. Udan RS, Kango‐Singh M, Nolo R, Tao C, Halder G (2003) Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol 5: 914–920 [DOI] [PubMed] [Google Scholar]
- 7. Wu S, Huang J, Dong J, Pan D (2003) Hippo encodes a Ste‐20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114: 445–456 [DOI] [PubMed] [Google Scholar]
- 8. Edgar BA (2006) From cell structure to transcription: Hippo forges a new path. Cell 124: 267–273 [DOI] [PubMed] [Google Scholar]
- 9. Huang J, Wu S, Barrera J, Matthews K, Pan D (2005) The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122: 421–434 [DOI] [PubMed] [Google Scholar]
- 10. Yu FX, Zhao B, Guan KL (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L et al (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta‐TRCP). Genes Dev 24: 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183 [DOI] [PubMed] [Google Scholar]
- 14. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D (2007) Elucidation of a universal size‐control mechanism in Drosophila and mammals. Cell 130: 1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM et al (2008) TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 22: 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brott BK, Pinsky BA, Erikson RL (1998) Nlk is a murine protein kinase related to Erk/MAP kinases and localized in the nucleus. Proc Natl Acad Sci USA 95: 963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verheyen EM, Mirkovic I, MacLean SJ, Langmann C, Andrews BC, MacKinnon C (2001) The tissue polarity gene nemo carries out multiple roles in patterning during Drosophila development. Mech Dev 101: 119–132 [DOI] [PubMed] [Google Scholar]
- 18. Ishitani T, Ninomiya‐Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N, Waterman M, Bowerman B, Clevers H, Shibuya H et al (1999) The TAK1‐NLK‐MAPK‐related pathway antagonizes signalling between beta‐catenin and transcription factor TCF. Nature 399: 798–802 [DOI] [PubMed] [Google Scholar]
- 19. Ishitani T, Hirao T, Suzuki M, Isoda M, Ishitani S, Harigaya K, Kitagawa M, Matsumoto K, Itoh M (2010) Nemo‐like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nat Cell Biol 12: 278–285 [DOI] [PubMed] [Google Scholar]
- 20. Kim S, Kim Y, Lee J, Chung J (2010) Regulation of FOXO1 by TAK1‐Nemo‐like kinase pathway. J Biol Chem 285: 8122–8129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuan HX, Wang Z, Yu FX, Li F, Russell RC, Jewell JL, Guan KL (2015) NLK phosphorylates Raptor to mediate stress‐induced mTORC1 inhibition. Genes Dev 29: 2362–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sudol M, Bork P, Einbond A, Kastury K, Druck T, Negrini M, Huebner K, Lehman D (1995) Characterization of the mammalian YAP (Yes‐associated protein) gene and its role in defining a novel protein module, the WW domain. J Biol Chem 270: 14733–14741 [DOI] [PubMed] [Google Scholar]
- 23. Ishitani T, Ishitani S, Matsumoto K, Itoh M (2009) Nemo‐like kinase is involved in NGF‐induced neurite outgrowth via phosphorylating MAP1B and paxillin. J Neurochem 111: 1104–1118 [DOI] [PubMed] [Google Scholar]
- 24. Klein PS, Melton DA (1996) A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA 93: 8455–8459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishitani S, Inaba K, Matsumoto K, Ishitani T (2011) Homodimerization of Nemo‐like kinase is essential for activation and nuclear localization. Mol Biol Cell 22: 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Djiane A, Zaessinger S, Babaoglan AB, Bray SJ (2014) Notch inhibits Yorkie activity in Drosophila wing discs. PLoS ONE 9: e106211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoo SJ, Huh JR, Muro I, Yu H, Wang L, Wang SL, Feldman RM, Clem RJ, Muller HA, Hay BA (2002) Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat Cell Biol 4: 416–424 [DOI] [PubMed] [Google Scholar]
- 28. Oh H, Irvine KD (2009) In vivo analysis of Yorkie phosphorylation sites. Oncogene 28: 1916–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA (2006) Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA 103: 12405–12410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moroishi T, Hansen CG, Guan KL (2015) The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Basu S, Totty NF, Irwin MS, Sudol M, Downward J (2003) Akt phosphorylates the Yes‐associated protein, YAP, to induce interaction with 14‐3‐3 and attenuation of p73‐mediated apoptosis. Mol Cell 11: 11–23 [DOI] [PubMed] [Google Scholar]
- 32. Zhang L, Tang F, Terracciano L, Hynx D, Kohler R, Bichet S, Hess D, Cron P, Hemmings BA, Hergovich A et al (2015) NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr Biol 25: 296–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Q, Zhang N, Xie R, Wang W, Cai J, Choi KS, David KK, Huang B, Yabuta N, Nojima H et al (2015) Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev 29: 1285–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML (2001) TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes‐associated protein localized in the cytoplasm. Genes Dev 15: 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Levy D, Adamovich Y, Reuven N, Shaul Y (2008) Yap1 phosphorylation by c‐Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell 29: 350–361 [DOI] [PubMed] [Google Scholar]
- 36. Wang W, Huang J, Wang X, Yuan J, Li X, Feng L, Park JI, Chen J (2012) PTPN14 is required for the density‐dependent control of YAP1. Genes Dev 26: 1959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oudhoff MJ, Freeman SA, Couzens AL, Antignano F, Kuznetsova E, Min PH, Northrop JP, Lehnertz B, Barsyte‐Lovejoy D, Vedadi M et al (2013) Control of the hippo pathway by Set7‐dependent methylation of Yap. Dev Cell 26: 188–194 [DOI] [PubMed] [Google Scholar]
- 38. Kortenjann M, Nehls M, Smith AJ, Carsetti R, Schuler J, Kohler G, Boehm T (2001) Abnormal bone marrow stroma in mice deficient for nemo‐like kinase, Nlk. Eur J Immunol 31: 3580–3587 [DOI] [PubMed] [Google Scholar]
- 39. Hong AW, Meng Z, Yuan HX, Plouffe SW, Moon S, Kim W, Jho E, Guan KL (2017) Osmotic stress‐induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep 18: 72–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Geback T, Schulz MM, Koumoutsakos P, Detmar M (2009) TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 46: 265–274 [DOI] [PubMed] [Google Scholar]
- 41. Katanaev VL, Solis GP, Hausmann G, Buestorf S, Katanayeva N, Schrock Y, Stuermer CA, Basler K (2008) Reggie‐1/flotillin‐2 promotes secretion of the long‐range signalling forms of Wingless and Hedgehog in Drosophila . EMBO J 27: 509–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S et al (2007) A genome‐wide transgenic RNAi library for conditional gene inactivation in Drosophila . Nature 448: 151–156 [DOI] [PubMed] [Google Scholar]
- 43. Boedigheimer M, Laughon A (1993) Expanded: a gene involved in the control of cell proliferation in imaginal discs. Development 118: 1291–1301 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File