

Abstract
Mutations in the APP and PSEN genes have provided direct evidence for the central role of aberrant amyloid β (Aβ) peptide production in familial Alzheimer's disease (AD). Newly identified risk factors will further help us to unravel how derailed physiological and cell biological processes lead to identical pathogenesis in late‐onset AD (LOAD). Ubelmann et al now unveil in this issue how two of such risk factors, Bin1 and CD2AP, regulate the encounter of APP and BACE1 in axonal and dendritic endosomes, emphasizing endosomal transport balance as a critical factor in AD pathogenesis 1.
Subject Categories: Membrane & Intracellular Transport, Molecular Biology of Disease, Neuroscience
Abnormal accumulation of toxic Aβ peptides in extracellular senile plaques is a major hallmark of AD. These peptides result from dual successive proteolytic cleavage of the amyloid precursor protein (APP), first by BACE1, releasing the bulk of the APP ectodomain, and secondly by the γ‐secretase complex, releasing C‐terminal cytosolic domains and generating N‐terminal hydrophobic Aβ peptides. In rare familial cases of AD (FAD), the disease is caused by autosomal dominant mutations in the genes encoding APP and presenilins (PSENs), the catalytic components of γ‐secretase. That both substrate and a pivotal enzyme involved in Aβ generation are hit in FAD provided a major linchpin for the central role of Aβ in disease initiation and progression, as formulated in the amyloid cascade hypothesis. However, over 95% of AD cases are sporadic, challenging scientists for several decades how and which physiological processes in the brain are compromised during aging resulting in identical neuropathological outcomes and disease.
Genome‐wide association studies (GWAS) and whole‐genome and whole‐exome sequencing have identified novel loci with an increased risk for late‐onset AD, further supporting a strong genetic component 2. Although these loci confer very low AD risk or are very rare, they are turning out to be of value in identifying pathways and delineating biological processes that are more vulnerable in this age‐related neurodegeneration. Herein, cholesterol homeostasis, Aβ clearance pathways (through microglia), and endosomal transport regulation are recurrently surfacing. These unbiased analyses surprisingly converge with pathological observations of endolysosomal abnormalities in early, preclinical stages of the disease 3. Variants of endocytic regulators with increased risk for late‐onset AD include sortilin‐related receptor 1 (SORL1), phosphatidylinositol‐binding clathrin assembly protein (PICALM), bridging integrator 1 (BIN1), and CD2‐associated protein (CD2AP), and their molecular involvement in Aβ generation is gradually revealed.
In this issue, Ubelmann et al report on the function of Bin1 and CD2AP asking how these two molecules independently regulate APP and BACE1 transport and how they affect Aβ production. Importantly, the authors address these questions mainly in primary hippocampal neurons allowing them to differentiate between selective axonal and dendritic transport regulation in these highly and stereotyped polarized neurons.
Notably, they focus on intracellular Aβ42 generation and developed for this purpose an imaging approach that allows them to specifically evaluate axonal and dendritic compartments. Using this strategy, they unravel a polarization effect of these risk factors on Aβ42 levels: Bin1 more pronouncedly controlled axonal Aβ42, while CD2AP expression more selectively steered Aβ42 production in dendrites and cell bodies 1. Looking at APP processing, they conclude that downregulation of Bin1 and CD2AP enhances APP processing at distinct steps (Fig 1), thus promoting Aβ production. Bin1 downregulation stimulates BACE1 processing in axons, while CD2AP enhances γ‐secretase cleavage in dendrites. Therefrom, the authors focus on looking at the trafficking of BACE1 and APP using live imaging and fluorescent pulse chase approaches and ask how Bin1 and CD2AP knockdown affect their endocytic transport. The two proteins again displayed a polarized effect: Bin1 supports axonal recycling of BACE1, while CD2AP regulates APP sorting toward degradation in dendrites (Fig 1).
Figure 1. Polarized effects of Bin1 and CD2AP expression on intracellular Aβ42 pools.
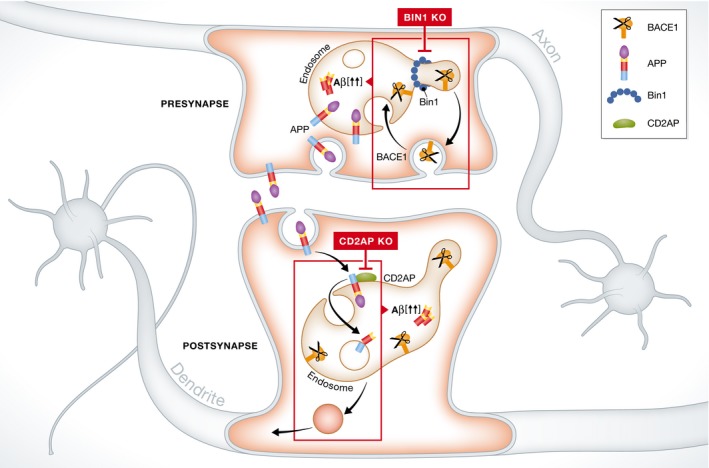
Bin1 regulates endocytic recycling of BACE1 particularly in axons/presynapse, while CD2AP controls delivery of APP to intraluminal vesicles and lysosomal degradation in dendrites/postsynapse. Knocking down Bin1 increases BACE1 in axonal endosomes, while silencing CD2AP expression blocks sorting of APP to ILVs in dendritic endosomes (rectangles in top and bottom parts, respectively). In both events, production of endosomal Aβ42 is increased.
Mechanistically, Bin1, also known as amphiphysin 2, contains an N‐terminal Bin–amphiphysin–Rvs (BAR) domain and a C‐terminal Src homology 3 (SH3) domain implicated in membrane bending, tubulation, and recruitment of the dynamin fission machinery. Knocking down Bin1 prevents BACE1 from being recruited to recycling tubules resulting in its retention in Rab5‐positive axonal endosomes. This appears to be selective for BACE1, leaving APP unaffected. CD2AP on the other hand seems to regulate sorting to intraluminal vesicles (ILVs) as its knockdown traps APP on the limiting membrane of maturing endosomes. For both LOAD risk factors, the net event is that APP and BACE1 co‐reside for an extended period. As this increases amyloidogenic processing, these results suggest that major Aβ production occurs at the limiting endosomal membrane, likely reflecting the presence of active pools of BACE1 as well as γ‐secretase. In agreement, a recent study showed predominant localization of PSEN2/γ‐secretase at the limiting membrane of late endosomes/lysosomes 4. Other factors that increase the residence time of APP at the limiting membrane of neuronal endosomes have identical outcomes. These include, but are not limited to, interference with APP ubiquitination and deficiencies in (early) ESCRT‐mediated sorting to ILVs such as decreased levels of phosphatidylinositol 3‐phosphate (PI3P), its effectors Hrs and Tsg101 5, and retromer components 6. Together these studies suggest that this endosomal sorting mechanism may be more vulnerable in the pathogenesis of LOAD and may, in later stages, affect overall degradation including autophagy and clearance of Aβ and tau aggregates 3.
This study adds to the converging evidence that local transport mechanisms keep APP segregated from its processing enzymes, starting from the TGN, and that the proteins remain separated through distinct internalization routes 7 until they reach endosomes where Bin1 and CD2AP regulate their encounter. In this way, these endocytic regulators control the levels of intracellular Aβ. While FAD‐linked mutations in PSENs dramatically increase intracellular Aβ42/40 ratios 4, whether these pools can act as toxic seeds and whether they contribute as well to LOAD pathogenesis are exciting topics for future research. Further, this work also highlights the importance of studying LOAD risk factors in the context of polarized neuronal trafficking as a prerequisite to evaluate their impact in in vivo models and in sporadic cases. Conversely, the in‐depth study of such risk factors will also improve our knowledge on the regulation of endosomal trafficking in neurons in general.
The findings also raise important new questions. Are these endocytic LOAD risk factors mainly acting in neurons or are they also involved in microglial Aβ clearance or disease propagation? Noteworthy, both Bin1 and CD2AP have been implicated in tau‐induced toxicity in a Drosophila model 8, 9, and recently, loss of Bin1 was shown to promote tau pathology propagation through increasing endocytosis of tau seeds 10. Is their expression profile affected during aging or more selectively aberrantly expressed in LOAD‐affected brain regions such as prefrontal and entorhinal cortex? Resolving the impact of single nucleotide polymorphisms (SNP) on the expression/function of these risk factors is therefore a key question that needs to be addressed. This vital knowledge may offer exciting new clues to disease mechanisms, but also provides the basis to develop appropriate in vivo models that faithfully mimic late‐onset AD pathogenesis, which are urgently needed for the development of drug therapies.
See also: Ubelmann et al (January 2017)
Contributor Information
Ragna Sannerud, Email: ragna.sannerud@cme.vib-kuleuven.be.
Wim Annaert, Email: wim.annaert@cme.vib-kuleuven.be.
References
- 1. Ubelmann F, Burrinha T, Salavessa L et al (2017) EMBO Rep 18: 102–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Karch CM, Goate AM (2015) Biol Psychiatry 77: 43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peric A, Annaert W (2015) Acta Neuropathol 129: 363–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sannerud R, Esselens C, Ejsmont P et al (2016) Cell 166: 193–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morel E, Chamoun Z, Lasiecka ZM et al (2013) Nat Commun 4: 2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhalla A, Vetanovetz CP, Morel E et al (2012) Neurobiol Dis 47: 126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sannerud R, Declerck I, Peric A et al (2011) Proc Natl Acad Sci USA 108: E559–E568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chapuis J, Hansmannel F, Gistelinck M et al (2013) Mol Psychiatry 18: 1225–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shulman JM, Chipendo P, Chibnik LB et al (2014) Hum Mol Genet 23: 870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calafate S, Flavin W, Verstreken P et al (2016) Cell Rep 17: 931–940 [DOI] [PubMed] [Google Scholar]