

Abstract
Autophagy is a process delivering cytoplasmic components to lysosomes for degradation. Autophagy may, however, play a role in unconventional secretion of leaderless cytosolic proteins. How secretory autophagy diverges from degradative autophagy remains unclear. Here we show that in response to lysosomal damage, the prototypical cytosolic secretory autophagy cargo IL‐1β is recognized by specialized secretory autophagy cargo receptor TRIM16 and that this receptor interacts with the R‐SNARE Sec22b to recruit cargo to the LC3‐II + sequestration membranes. Cargo secretion is unaffected by downregulation of syntaxin 17, a SNARE promoting autophagosome–lysosome fusion and cargo degradation. Instead, Sec22b in combination with plasma membrane syntaxin 3 and syntaxin 4 as well as SNAP‐23 and SNAP‐29 completes cargo secretion. Thus, secretory autophagy utilizes a specialized cytosolic cargo receptor and a dedicated SNARE system. Other unconventionally secreted cargo, such as ferritin, is secreted via the same pathway.
Keywords: autophagy, galectins, inflammasome, lysosome, tuberculosis
Subject Categories: Autophagy & Cell Death, Immunology, Membrane & Intracellular Transport
Introduction
Autophagy is a key intracellular quality control process in eukaryotes with multiple roles in development, normal physiology, and disease (Mizushima et al, 2008; Mizushima & Komatsu, 2011). The autophagy pathway governed by ATG factors is primarily known for its degradative cytoplasmic functions (Mizushima et al, 2011). It maintains cellular energy and nutrient supplies during starvation or absence of growth factors (Galluzzi et al, 2014), controls organellar quality and quantity in the cell, prevents accumulation of large protein aggregates, and possesses antimicrobial and other immune functions (Randow & Youle, 2014; Rubinsztein et al, 2015; Sica et al, 2015; Khaminets et al, 2016). Autophagy's potential for secretion has also been considered (Ponpuak et al, 2015), of particular interest in the context of extracellular immune signaling (Deretic et al, 2013; Ma et al, 2013).
The core autophagy machinery in mammalian cells has several subsystems interconnected via specific molecular interactions into a unifying apparatus (Mizushima et al, 2011). During autophagy, the cytoplasmic cargo is sequestered by specialized organelles termed autophagosomes, which are characterized by the presence of LC3B, one of the mammalian paralogues of yeast Atg8 (mAtg8s). The conversion of LC3 to LC3‐II via its C‐terminal lipidation with phosphatidylethanolamine, catalyzed by the E3 ligase complex (ATG5‐ATG12/ATG16L1), represents a hallmark of nascent autophagic membranes (Kabeya et al, 2000). The initiation of autophagosome formation depends on upstream Ser/Thr protein kinases, including AMPK (Kim et al, 2011, 2013), mTOR (Egan et al, 2011; Kim et al, 2011; Settembre et al, 2012), and ULK1 (Russell et al, 2013), and lipid kinases centered upon phosphatidylinositol 3‐kinase VPS34 complex containing Beclin 1 (Liang et al, 1999) and ATG14L (Sun et al, 2008), to generate phosphatidylinositol 3‐phosphate (PI3P) on autophagic membranes. These membranes likely originate from the endoplasmic reticulum (ER) (Axe et al, 2008), with ER‐to‐Golgi intermediate compartment (ERGIC) playing a key role in LC3 lipidation and formation of autophagosomal membranes (Ge et al, 2014), whereas contributions of the ER–mitochondrial contact sites (Hamasaki et al, 2013), endocytic membranes (Puri et al, 2013; Lamb et al, 2016), and potentially other intracellular compartments (Joachim et al, 2015) play additional roles in redirecting net membrane flow to autophagosomes. The production of PI3P by VPS34 is recognized by WIPI2, which in turn binds to ATG16L (Dooley et al, 2014) of the ATG5‐ATG12/ATG16L1 complex, thus localizing LC3 lipidation (Fujita et al, 2008). ATG16L1 is also a binding partner for FIP200, a component of the ULK1 complex (Fujita et al, 2013; Gammoh et al, 2013; Nishimura et al, 2013; Dooley et al, 2014), ensuring that all core subsystems are coming together. Nascent autophagosomes eventually acquire a Qa‐SNARE, syntaxin 17 (Itakura et al, 2012; Tsuboyama et al, 2016), which binds to the CCD domain of ATG14L to form a stable binary complex with Qbc‐SNARE SNAP‐29 (Diao et al, 2015). The binary SNARE complex stabilization and tethering of PI3P‐containing membranes by ATG14L permit pairing with the lysosomal R‐SNARE VAMP8 to drive a 4‐helix bundle SNARE‐catalyzed fusion with lysosomes and generate autolysosomes where the captured material is degraded (Mizushima et al, 2011; Itakura et al, 2012; Hamasaki et al, 2013; Takats et al, 2014; Diao et al, 2015).
Autophagy can be non‐selective or selective with various degrees of precision (Kimura et al, 2016). During selective autophagy, the cargo to be sequestered by autophagosomes is either labeled with tags, including ubiquitin (Khaminets et al, 2016), phosphorylated ubiquitin (Koyano et al, 2014), or galectins (Thurston et al, 2012; Randow & Youle, 2014). These tags can then be recognized by sequestosome 1‐like receptors (SLRs) (Birgisdottir et al, 2013; Deretic et al, 2013), including sequestosome 1/p62, NDP52, TAXBP1, NBR1, and optineurin (Bjorkoy et al, 2005; Kirkin et al, 2009; Wild et al, 2011; Newman et al, 2012; Thurston et al, 2012; Lazarou et al, 2015). SLRs recognize ubiquitin on autophagic targets via a variety of ubiquitin binding domains (e.g., UBAN, UBA, UBZ) (Khaminets et al, 2016), whereas some of them can bind galectins, which in turn recognize carbohydrates exposed on exofacial leaflets of damaged endomembranes (Thurston et al, 2012; Randow & Youle, 2014). In addition to SLRs (Lazarou et al, 2015), unique autophagy receptors potentially directly recognizing the cargo have been reported (Sandoval et al, 2008; Zhang et al, 2008; Orvedahl et al, 2011; Liu et al, 2012; Khaminets et al, 2015; Murakawa et al, 2015). Among these, NCOA4 plays the role of a receptor for autophagic degradation of ferritin (Dowdle et al, 2014; Mancias et al, 2014). Recently, a new class of autophagic cargo receptors have been described, which not only recognize targets but also assemble the necessary regulators and execution autophagy factors thus acting dually as receptor regulators of autophagy (Kimura et al, 2016). These factors belong to the TRIM protein family (Reymond et al, 2001), whose members in principle contain N‐terminal RING domain, B‐box domains, a coiled‐coil domain, and a C‐terminal domain such as SPRY. TRIMs assemble ULK1 and Beclin 1 complexes and bind to mAtg8s via their LIR (LC3 interaction region) motifs, while recognizing cargo via their SPRY, PYRIN, and potentially other domains (Mandell et al, 2014; Kimura et al, 2015, 2016).
Although autophagy is primarily known as a tributary to the degradative lysosomal pathway (Mizushima et al, 2011), functionally different terminations have been considered for autophagocytosed cytosolic material, leading to secretion of cytokines (Ponpuak et al, 2015) or small molecular weight immune mediators (Ma et al, 2013). In particular, autophagy has been examined for its potential in unconventional secretion of leaderless cytosolic proteins that cannot enter the conventional secretory pathway but play extracellular functions (Duran et al, 2010; Manjithaya et al, 2010; Dupont et al, 2011; Zhang & Schekman, 2013; Ponpuak et al, 2015). In mammalian cells, autophagy has been implicated in secretion of IL‐1β, as a prototypical cargo for unconventional secretion of cytosolic proteins (Zhang & Schekman, 2013), through its dependence on ATG factors including ATG5 (Dupont et al, 2011). Furthermore, carriers positive for the autophagy marker LC3‐II have been identified as IL‐1β‐sequestration membranes en route for secretion (Zhang et al, 2015). However, how IL‐1β is secreted instead of being degraded in autophagic organelles remains unknown. Here we report the key elements of the pathway for unconventional protein secretion through autophagy that separates it from degradative autophagy. This pathway, termed secretory autophagy, requires a specialized receptor recognizing the cytosolic cargo. It also relies on cooperation of this receptor with the R‐SNARE present on LC3‐II+ ERGIC‐derived membranes whereupon the cargo is sequestered. Finally, secretion of the cargo depends on plasma membrane syntaxins instead of delivery to lysosomes via syntaxin 17. The secretory autophagy pathway utilizes a specialized cytosolic cargo receptor regulator from the TRIM family responsive to inducers and agonists of secretion, and a dedicated ERGIC R‐SNARE Sec22b that both interacts with the receptor regulator and engages the plasma membrane Qa‐SNAREs. This system is utilized by a subset of unconventionally secreted leaderless cytosolic proteins.
Results
Screen for TRIMs affecting secretory autophagy of IL‐1β
Diverse lysosome‐damaging and inflammasome‐activating agents can trigger IL‐1β secretion (Schroder & Tschopp, 2010) including silica (Hornung et al, 2008), alum (Hornung et al, 2008), monosodium urate (Martinon et al, 2006), and Leu‐Leu‐O‐Me (LLOMe) (Ito et al, 2015). Lysosomal damage or stress induce autophagy (Maejima et al, 2013; Napolitano & Ballabio, 2016), whereas starvation sensors for induction of autophagy are located on lysosomes (Roczniak‐Ferguson et al, 2012; Settembre et al, 2012; Medina et al, 2015; Napolitano & Ballabio, 2016). We used LLOMe as a standard treatment and confirmed that autophagy factors, represented by LC3B and ATG16L1, are required for efficient IL‐1β secretion in response to lysosomal damage (Figs 1A and EV1A and B) as well as in response to starvation, a common inducer of autophagy both in general and in association with IL‐1β secretion (Dupont et al, 2011; Zhang et al, 2015) (Fig EV1C and D; LDH release was used as a measure of a non‐specific leak from cells). A different mAtg8 tested, GABARAP, known to interact strongly with a number of TRIM proteins (Mandell et al, 2014; Kimura et al, 2015, 2016), was also required for IL‐1β secretion elicited by either LLOMe or starvation (Fig 1B and Appendix Fig S1A–D).
Figure 1. TRIM16 recruits IL‐1β to LC3‐positive carrier membranes for secretion.
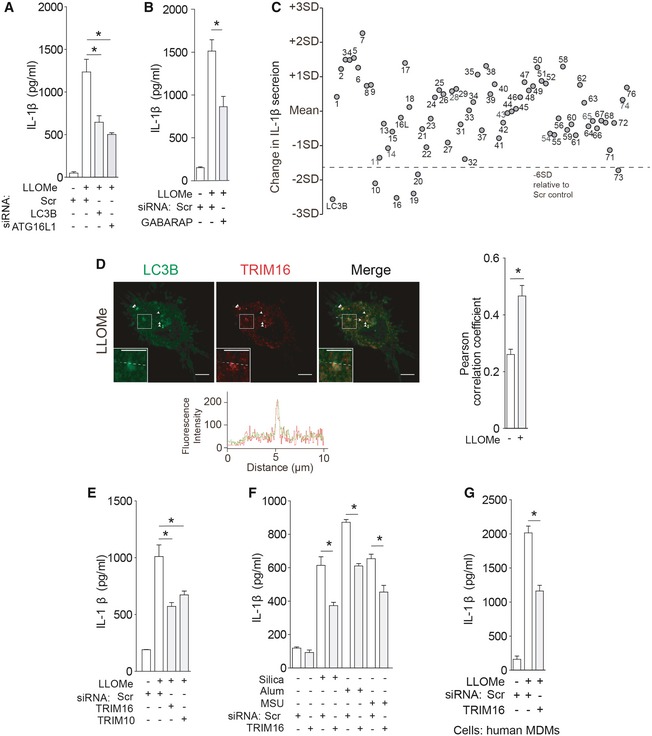
- THP‐1 cells were subjected to knockdowns as indicated and treated with 100 ng/ml LPS overnight, followed by 0.25 mM LLOMe for 3 h, and the levels of IL‐1β were determined in supernatants.
- THP‐1 cells were subjected to GABARAP knockdown and processed as in (A).
- IL‐1β levels in supernatants of THP‐1 cells subjected to TRIM knockdowns and treated with LPS and then with LLOMe.
- Confocal microscopy of THP‐1 cells treated sequentially with LPS and LLOMe and stained for LC3B and TRIM16. Line tracings correspond to arrows. Arrowheads indicate colocalization. Scale bars, 5 μm.
- IL‐1β levels in supernatants of THP‐1 cells subjected to TRIM knockdowns and treated with LPS and then with LLOMe.
- Levels of IL‐1β were determined in supernatants from THP‐1 cells subjected to knockdown as indicated, treated with 100 ng/ml LPS overnight and then with 0.25 mM of Silica, Alum, or monosodium urate (MSU).
- IL‐1β in supernatants of primary human macrophages (MDM) subjected to knockdowns and sequentially treated with LPS and LLOMe.
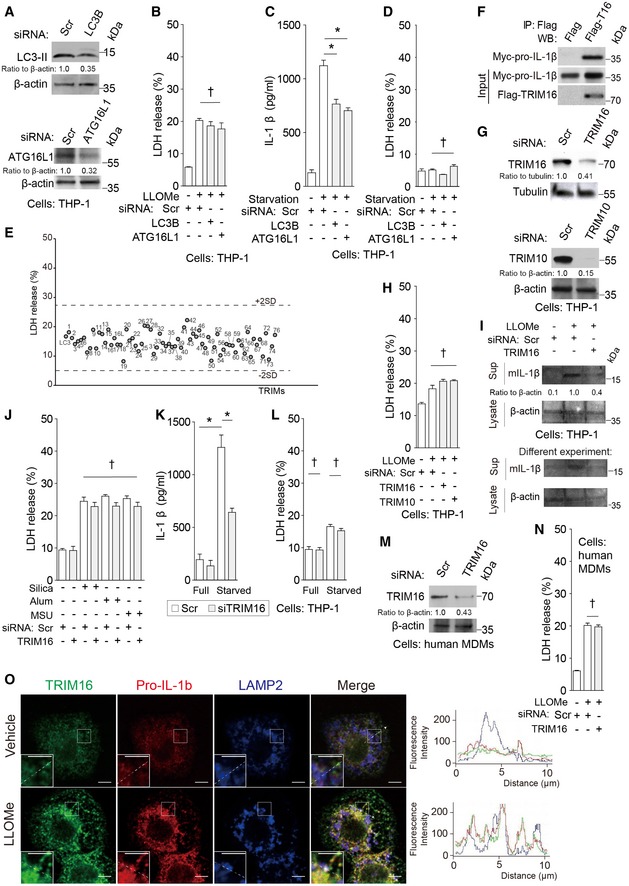
Figure EV1. TRIM16 mediates IL‐1β secretion.
-
AKnockdown efficacies as determined by immunoblots.
-
BLDH release data for samples in Fig 1A.
-
C, DLevels of (C) IL‐1β and (D) LDH release were determined in supernatants from THP‐1 cells subjected to knockdowns as indicated, treated with 100 ng/ml LPS overnight, then starved in EBSS for 3 h.
-
ELDH release results for samples in Fig 1C.
-
FCo‐IP analysis of interactions between flag‐TRIM16 with myc‐pro‐IL‐1β in HEK293T cells.
-
GImmunoblot analyses of knockdown efficacies in THP‐1 cells.
-
HLDH release data for samples in Fig 1E.
-
IImmunoblot analyses of the levels of mIL‐1β in supernatants from THP‐1 cells that were subjected to knockdown as indicated and were sequentially treated with LPS and LLOMe.
-
JLDH release data from samples in Fig 1F.
-
K, LLevels of (K) IL‐1β and (L) LDH release was determined in supernatants from THP‐1 cells subjected to knockdown as indicated, treated with 100 ng/ml LPS overnight, then starved in EBSS for 3 h.
-
MImmunoblot analyses of knockdown efficacies in primary human MDM cells.
-
NLDH release results for samples in Fig 1G.
-
OIntracellular localization analysis of TRIM16, pro‐IL‐1β, and LAMP2 by confocal microscopy. Cells, THP‐1. Line tracings correspond to arrows. Scale bars, 5 μm.
Members of the TRIM family of proteins have been shown to control autophagy acting as selective autophagy receptors and autophagy regulators (Kimura et al, 2016), whereas several of them, including TRIM16 (Munding et al, 2006) and TRIM20 (Kimura et al, 2015), have been implicated in control of inflammasome activation. We thus screened human TRIMs and found that there were a number of TRIMs influencing IL‐1β secretion in response to LLOMe challenge (Figs 1C and EV1E).
TRIM16 acts as a receptor for IL‐1β in secretion
TRIM16 directly interacts with IL‐1β as previously reported (Munding et al, 2006), and this association was verified in our experiments (Fig EV1F). TRIM16 colocalized with LC3B upon LLOMe treatment (Fig 1D and Appendix Fig S1E and F), in keeping with its recently described role in autophagic response to and repair of endomembrane and lysosomal damage (Chauhan et al, 2016). Thus, we hypothesized that TRIM16 might connect lysosomal damage with autophagy‐sponsored unconventional secretion of IL‐1β. We found that TRIM16 was needed for optimal secretion of IL‐1β triggered by LLOMe (Figs 1E and EV1G–I), silica (Hornung et al, 2008), alum (Hornung et al, 2008), monosodium urate (Martinon et al, 2006) (Figs 1F and EV1J), and starvation (Fig EV1K and L). TRIM16 was also required for optimal secretion of IL‐1β from primary macrophages (human peripheral blood monocyte‐derived macrophages, MDM; Figs 1G and EV1M and N). In keeping with these observations, TRIM16 and pro‐IL‐1β colocalized in cells treated with LLOMe (Fig EV1O). A subset of TRIM16+ profiles overlapped with the lysosomal marker LAMP2 (Fig EV1O), in keeping with the recently described role of TRIM16 in autophagic homeostasis of damaged lysosomes (Chauhan et al, 2016). Another TRIM subjected to follow‐up analyses, TRIM10, was confirmed for effects on IL‐1β secretion (Figs 1E and EV1G and H) and showed capacity to interact with TRIM16 (Appendix Fig S1G). Thus, TRIM16, a protein that binds IL‐1β, as well as additional TRIMs affect IL‐1β secretion.
Galectin‐8 cooperates with TRIM16 in IL‐1β secretion in response to lysosomal damage
Galectin‐3 and galectin‐8, the cytosolic lectins which recognize β(1–4)‐linked galactosides normally restricted to organellar lumenal membrane leaflets (Nabi et al, 2015), have been associated with autophagic response to endomembrane damage (Thurston et al, 2012; Fujita et al, 2013; Maejima et al, 2013). Hence, we tested whether these galectins contributed to IL‐1β secretion. Knockdowns of galectin‐8, but not that of galectin‐3, suppressed the secretion of IL‐1β in THP‐1 cells and in primary MDMs upon stimulation (Figs 2A–C and EV2A–E). These relationships were confirmed using bone marrow derived macrophages from galectin‐3 and galectin‐8 knockout mice (Appendix Fig 2A and B). Zhang et al (2015) have recently shown that HSP90 participates in unconventional secretion of IL‐1β. Galectin‐8 showed similar overall intracellular distribution and colocalization with HSP90 in cells treated with LLOMe (Fig EV2F).
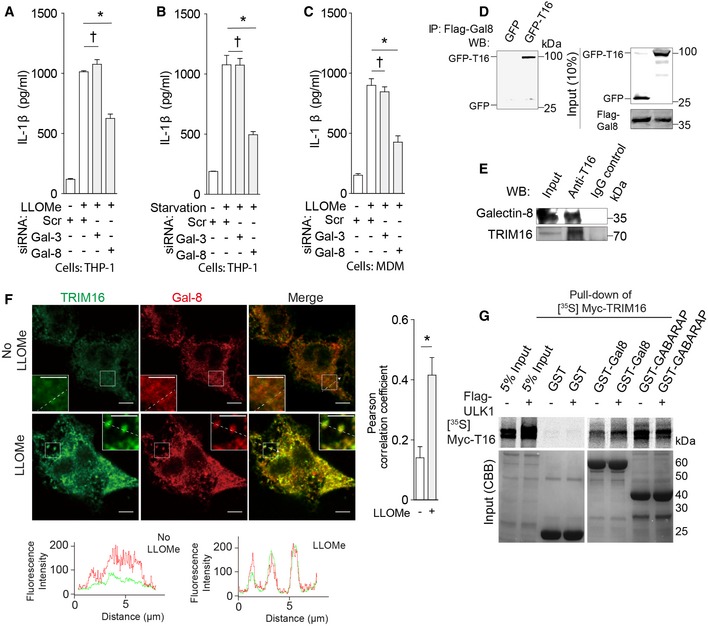
Figure 2. Galectin‐8 participates in IL‐1β secretion and interacts with TRIM16.
-
A, BLevels of IL‐1β in supernatants from THP‐1 cells that were subjected to knockdown as indicated and were treated with LPS, and then (A) treated with LLOMe or (B) starved in EBSS.
-
CIL‐1β in supernatants of primary human macrophages (MDM) subjected to knockdowns and sequentially treated with LPS and LLOMe.
-
DCo‐immunoprecipitation (co‐IP) of flag‐galectin‐8 (Gal‐8) and GFP‐TRIM16 (T16) in HEK293T cells.
-
ECo‐IP analysis of endogenous galectin‐8 and TRIM16 in protein complexes from LLOMe‐treated THP‐1 cells.
-
FConfocal microscopy of THP‐1 cells treated with LPS and LLOMe and stained for TRIM16 and galectin‐8. Line tracings correspond to arrowed dashed lines. Scale bars, 5 μm.
-
GIn vitro translated and radiolabeled [35S] myc‐HA‐TRIM16 was incubated with GST‐galectin‐8 in the presence (+) or absence (−) of flag‐ULK1 and cold ATP, GST pull downs were performed, and [35S] radiolabeled Myc‐HA‐TRIM16 in pulled‐down material detected by SDS‐PAGE and autoradiography. Amounts of GST fusion proteins are shown in Coomassie brilliant blue (CBB)‐stained gels.
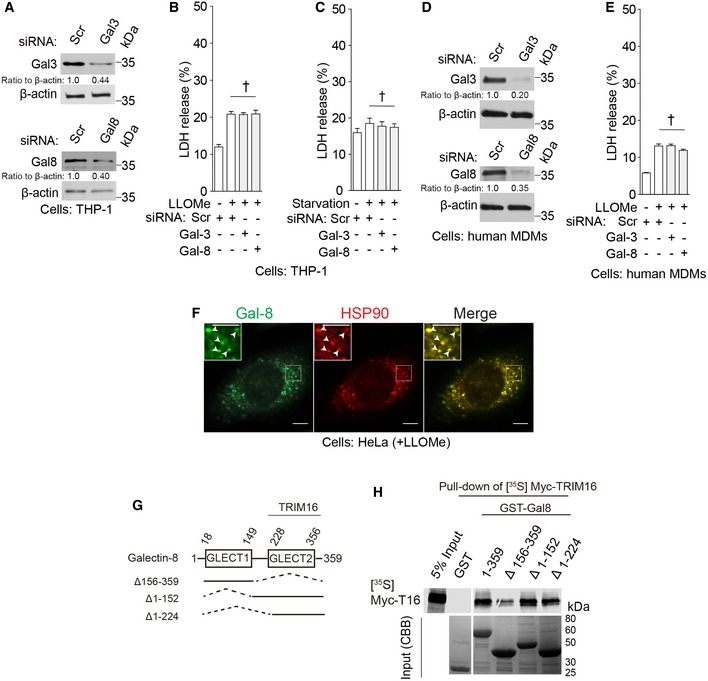
Figure EV2. Galectin‐8 participates in IL‐1β secretion and interacts with TRIM16.
-
AImmunoblot analyses of knockdown efficacies in THP‐1 cells.
- B, C
-
DImmunoblot analyses of knockdown efficacies in primary human MDMs.
-
ELDH release data for samples in Fig 2C.
-
FConfocal microscopy of HeLa cells treated with LLOMe and stained for galectin‐8 and HSP90. Arrowheads in enlarged insets indicate colocalization. Scale bars, 5 μm.
-
GGalectin‐8 domains and deletion constructs used.
-
HIn vitro translated and radiolabeled [35S] myc‐HA‐TRIM16 was incubated with full‐length‐ and deletions of GST‐galectin‐8 in the presence of flag‐ULK1 and cold ATP, and GST pull downs were performed and [35S] radiolabeled Myc‐HA‐TRIM16 detected by SDS‐PAGE and autoradiography. Amounts of GST fusion proteins are shown in Coomassie brilliant blue (CBB)‐stained gels.
A question arose whether galectin‐3 and galectin‐8 could influence the TRIM16‐LC3 colocalization detected in response to LLOMe illustrated in Fig 1D. Neither galectin‐8 knockdown alone (Appendix Fig S2C and D) nor galectin‐3 knockdown alone (Appendix Fig S2E and F) affected colocalization between TRIM16 and LC3B elicited in cells by LLOMe. However, a combined knockdown of galectin‐3 and galectin‐8 reduced the percentage of TRIM16 profiles that were also positive for LC3B (Appendix Fig S2E and F). Thus, galectin‐3 and galectin‐8 showed redundant effects on bulk (i.e., not differentiated for function) TRIM16‐LC3B profiles formed in response to lysosomal damage.
Galectin‐8 and TRIM16 co‐immunoprecipitated in cell extracts (Fig 2D and E) and colocalized in cells treated with LLOMe (Fig 2F). TRIM16 associated with galectin‐8 in GST pull‐down assays (Figs 2G and EV2G and H), and as described for other galectins, that is, galectin‐3 (Chauhan et al, 2016), this association was somewhat enhanced in the presence of ULK1 (Fig 2G). Thus, both the receptor TRIM16 and its interacting partner galectin‐8 recognizing membrane damage are required for efficient secretion of IL‐1β in response to lysosomal damage.
TRIM16 is required for IL‐1β delivery to LC3+ carriers
Autophagy‐dependent IL‐1β secretion can be reconstituted in model cell lines whereby IL‐1β is sequestered by LC3‐II+ carriers en route for secretion (Zhang et al, 2015). These carriers, herein referred to as IL‐1β‐sequestration membranes, have been shown to fractionate as a 25 k pellet during differential centrifugation of membranes isolated from model cells overexpressing pro‐IL‐1β and pro‐caspase‐1 (Zhang et al, 2015). We wondered whether TRIM16, acting as an IL‐1β receptor, might play a role in delivering its cargo to IL‐1β‐sequestration membranes. Endogenous TRIM16 was present along with LC3‐II and IL‐1β in the 25 k membranes prepared from wild‐type HeLa cells reconstituted with flag‐pro‐IL‐1β and myc‐pro‐caspase‐1, as previously described (Fig 3A; Zhang et al, 2015). HeLa cells reconstituted as above were responsive to LLOMe and starvation treatment as evidenced by IL‐1β secretion (Fig EV3A and B). Colocalization between LC3 puncta and IL‐1β profiles was responsive to LLOMe as determined by high content microscopy in cells reconstituted as above (Appendix Fig S3A). This system permitted us to compare wild‐type TRIM16+ HeLa cells and their CRISPR TRIM16 knockout derivative (TRIM16KO cells; Fig EV3C; Chauhan et al, 2016) for their capacity to colocalize LC3 and IL‐1β and transfer IL‐1β to 25 k membranes. Absence of TRIM16 diminished colocalization between IL‐1β and LC3 (Appendix Fig S3A) and abrogated delivery of mature, that is, caspase‐1‐processed, IL‐1β (mIL‐1β) to IL‐1β‐sequestration membranes in cells treated with LLOMe (Fig 3B–D). This was not due to indirect effects inhibiting processing of pro‐IL‐1β (Fig EV3D). Furthermore, when membrane was subjected to further separation by density gradient centrifugation using previously described methods and fractionation schemes (Zhang et al, 2015), despite equal expression of pro‐IL‐1β (Fig 3E), mIL‐1β co‐fractionated with the Sec22b+ LC3‐II+ membranes in TRIM16wt cells, but was no longer associated/focused on these membranes in TRIM16KO cells (Fig 3F and G). Of further note was separation of the membrane peak containing mIL‐1β, TRIM16, Sec22b, and LC3‐II+ from lysosomes (LAMP2) (Fig 3F and G). Thus, TRIM16 is required for IL‐1β delivery to IL‐1β‐sequestration membranes.
Figure 3. TRIM16 recruits IL‐1β to LC3‐positive carrier membranes for secretion and interacts with Sec22b.
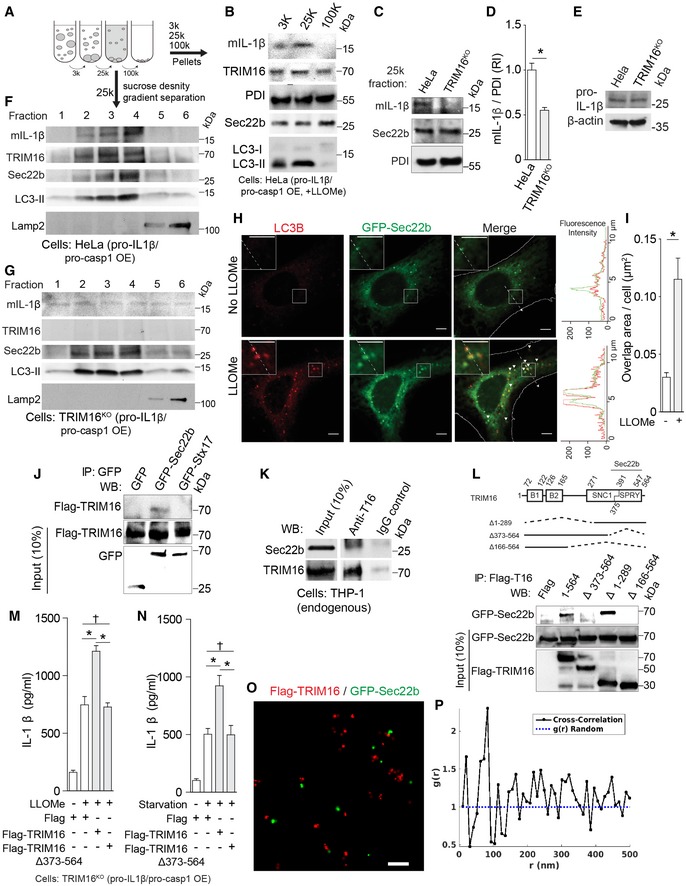
-
A, BImmunoblot analyses of indicated membrane fractions prepared by differential centrifugation. HeLa cells reconstituted with flag‐pro‐IL‐1β and myc‐pro‐caspase‐1 were treated with 1 mM LLOMe for 1 h, and lysates were subjected to differential centrifugations at 3,000 g (3 k), 25,000 g (25 k) and 100,000 g (100 k) as depicted in the cartoon. Arrows, two ways the membranes were analyzed—pellets from velocity sedimentation were analyzed in panels (B–D), or by further sucrose and OptiPrep density separation of 25 k pellets in panels (F, G).
-
CIL‐1β levels in 25 k membrane fractions (pellets) from HeLa cells and their CRISPR knockout derivatives (TRIM16KO) that were reconstituted for IL‐1β secretion and treated with 1 mM LLOMe for 1 h.
-
DQuantification of analyses in (C). RI, relative intensity.
-
EWhole‐cell lysates (immunoblots) analyzed in (F, G).
-
F, GSucrose density gradient analysis of 25 k membranes. Note that mature IL‐1β (mIL‐1β) is recruited to and co‐fractionates with TRIM16+, Sec22b+, and LC3‐II+ membranes in TRIM16wt HeLa cells reconstituted with pro‐IL‐1β and pro‐caspase‐1, but not in TRIM16KO mutant cells.
-
HConfocal images of LC3 in HeLa cells expressing GFP‐Sec22b treated with LLOMe or untreated (control). Arrowheads, colocalization. Scale bars, 5 μm.
-
IGFP‐Sec22b and LC3 overlapping area obtained by high content microscopy.
-
JCo‐IP analysis between flag‐TRIM16 with GFP‐Sec22b or GFP‐syntaxin 17 co‐expressed in HEK293T cells.
-
KCo‐IP analysis of endogenous proteins, TRIM16 and Sec22b, in lysates from THP‐1 cells treated with LLOMe.
-
LCo‐IP analysis of interactions between deletion variants of flag‐TRIM16 with GFP‐Sec22b in HEK293T cells.
-
M, NIL‐1β secretion complementation experiments of TRIM16KO cells with TRIM16 Sec22b‐interacting and its Sec22b‐non‐binding mutant shown.
-
OTwo color super‐resolution image showing co‐clustering of flag‐TRIM16 (red) and GFP‐Sec22b (green). Scale bar, 200 nm.
-
PCross‐correlation analysis of flag‐TRIM16 and GFP‐Sec22b super‐resolution data showing a characteristic separation of ˜70 nm.
Figure EV3. TRIM16 recruits IL‐1β to LC3‐positive carrier membranes for secretion and motifs in TRIM16 and Sec22b.

-
A, BIL‐1β levels in supernatants of HeLa cells reconstituted with flag‐pro‐IL‐1β and myc‐caspase‐1 and treated as indicated.
-
CCRISPR TRIM16 mutant A9 in HeLa cells (Chauhan et al, 2016).
-
DProcessing of pro‐IL1β detected by immunoblotting of lysates from HeLa cells (wild type and TRIM16KO) reconstituted with pro‐IL‐1β and pro‐caspase‐1 by transfection and treated with LLOMe in the presence of autolysosomal/lysosomal degradation inhibitors E64D plus pepstatin. Ratios of mIL‐1β to tubulin levels are shown below the blots.
-
EOverlapping area (high content microscopy and processing) of GFP‐Sec22b and LC3B in HeLa cells fed or starved in EBSS.
-
FAlignments of the SNC1 domain of TRIM16, shown as they are annotated in the NCBI entry NP_006461, where TRIM16 SNC1 domain is compared with R‐SNAREs, including longin‐containing SNAREs, in NCBI CDD:227472 (http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=227472). Note that the SNC1 domain CDD: 227472 includes sequences from both longin and SNARE domains and that only the SNARE domains contain typical “0” ionic layers (the boxed R residues). Underlined residues, features of the SNARE motif in Saccharomyces cerevisiae (Sc) Snc1, with flanking layers maintaining hydrophobic interactions when this SNARE is in the four‐helix bundle SNARE fusion complex; the N‐terminal flanking parts encompass all layers of the SNARE motif of SNC1, whereas the C‐terminal part shown encompasses a half of C‐terminally positioned hydrophobic layers.
-
GDomain organization of TRIM16 and Sec22b. The relationship between the NCBI‐annotated domains (boxes) and predicted coiled‐coil regions of TRIM16 (overlined regions; as annotated in UniProtKB – O95361) are indicated. The relationships between NCBI‐annotated SNC1 motif (underlined region) and the NCBI‐annotated longin and SNARE domains of Sec22b are shown.
-
H, ILDH release controls corresponding to IL‐1β secretion complementation experiments of TRIM16KO cells with TRIM16 Sec22b‐interacting and its Sec22b‐nonbinding mutant shown in Fig 3M and N.
-
JSub‐regions selected for cross‐correlation analysis of flag‐TRIM‐16 and GFP‐Sec22b super‐resolution data.
R‐SNARE Sec22b interacts with TRIM16 and is required for IL‐1β secretion
Sec22b, a longin R‐SNARE (Rossi et al, 2004; Jahn & Scheller, 2006), colocalized with LC3B (Fig 3H), and their colocalization increased with LLOMe treatment (Fig 3I) and starvation (Fig EV3E). Sec22b, previously used as a marker that implicated ERGIC in LC3 lipidation (Ge et al, 2014), was found in cellular membrane fractions including 25 k IL‐1β‐sequestration membranes that also contained TRIM16 (Fig 3A). Sec22b co‐fractionated with TRIM16, mIL‐1β, and LC3‐II membranes (Fig 3F). TRIM16 is unique among the TRIMs by possessing a hybrid longin‐SNARE domain, NCBI CDD:227472 (http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=227472), with mixed features of longin and SNARE domains and predicted helical structure (Fig EV3F and G). The CDD:227472 domain is based on several R‐SNAREs including yeast Snc1, Snc2, Sec22, Nyv1, and Ykt6 and is annotated in the TRIM16 NCBI entry NP_006461 as SNC1; it is co‐terminal but partially overlapping with the SPRY domain of TRIM16 (Fig EV3F and G). TRIM16 was found in macromolecular complexes with the human Sec22b (Fig 3J and K). Mapping of TRIM16 domains required for complex formation with Sec22b indicated that the SPRY domain (partially overlapping with the TRM16 SNC1 domain) was needed for this association (Fig 3L). In contrast to full‐length TRIM16, TRIM16Δ373–564 (the mutant that no longer binds Sec22b) could not complement IL‐1β secretion defect in reconstituted TRIM16KO mutant HeLa cells stimulated by LLOMe or starvation (Figs 3M and N, and EV3H and I). Super‐resolution fluorescence microscopy analysis showed intimate colocalization of Sec22b and TRIM16 in cells with a characteristic separation of 70 nm (Figs 3O and P, and EV3J). Thus, Sec22b is in complexes with TRIM16 and their association is important for IL‐1β secretion.
Knockdowns of Sec22b did not reduce autophagosomal LC3+ puncta formation (Fig EV4A and B), in keeping with prior reports that downregulation of Sec22b does not diminish LC3 puncta albeit it is known to affect lysosomal enzyme delivery to autolysosomes at later, degradative stages (Renna et al, 2011). Nevertheless, Sec22b knockdowns reduced IL‐1β secretion in response to LLOMe and starvation (Figs 4A–C and EV4C–G). A question arose of whether Sec22b could indirectly affect secretion through its actions on Atg9 trafficking as shown in yeast (Nair et al, 2011). When Atg9 knockout MEFs were compared to wild‐type MEFs, no effect on IL‐1β secretion in response to LLOMe or starvation was observed (Fig EV4H–K). Another question arose regarding potential indirect effects of Sec22b on caspase‐1‐dependent processing of pro‐IL‐1β. However, when HeLa cells were transfected with mature IL‐1β (mIL‐1β), thereby bypassing the requirement for pro‐IL‐1β processing by caspase‐1, Sec22b knockdown still reduced levels of secreted IL‐1β (Fig EV4L and M). In a complementary set of experiments, overexpression of Sec22b in HeLa cells (a reconstituted model system that in parallel experiments required TRIM16 for IL‐1β processing and secretion; Appendix Fig S3B and C) transfected with mIL‐1β to bypass the requirement for caspase‐1‐dependent pro‐IL‐1β processing, increased secretion of mature IL‐1β (Fig EV4N–Q). Thus, Sec22b is required for IL‐1β secretion directly and not due to potential indirect effects via Atg9 or effects on caspase‐1‐dependent pro‐IL‐1β processing. The direct function of Sec22b in IL‐1β secretion is in concordance with its association with the IL‐1β receptor TRIM16.
Figure EV4. R‐SNARE Sec22b participates in IL‐1β secretion.
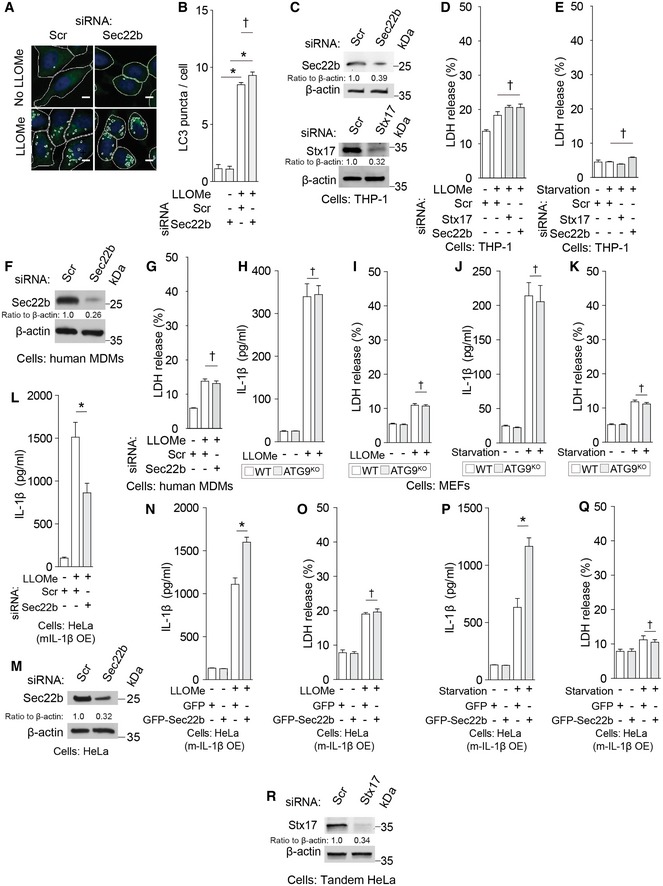
-
AHeLa cells subjected to knockdown as indicated were treated with LLOMe, and high content analysis of LC3 puncta formation in response to LLOMe was performed using a Cellomics HCS scanner (epifluorescence) and iDEV software. Masks, software‐defined objects (primary objects, cell outlines; internal secondary objects, LC3 puncta). Scale bar, 5 μm.
-
BAverage count of LC3 puncta per cell illustrated in (A), carried out in 96‐well plates with 49 fields per well and 500 primary objects per field.
-
CImmunoblot analyses of knockdown efficacies in THP‐1 cells.
- D, E
-
FImmunoblot analyses of knockdown efficacies in human primary macrophages (MDM).
-
GLDH release data for samples in Fig 4C.
-
H–KIL‐1β levels or LDH release in supernatants of either wild‐type or ATG9 knockout MEFs reconstituted with flag‐pro‐IL‐1β and myc‐caspase‐1 and treated as indicated.
-
L, MHeLa cells transfected with mature IL‐1β (mIL‐1β) were subjected to siRNA knockdowns and IL‐1β release measured after LLOMe stimulation; blot shows Sec22b knockdown efficiency.
-
N–QIL‐1β or LDH release from HeLa cells overexpressing GFP‐sec22b or GFP and mature mIL‐1β upon stimulation by LLOMe or starvation.
-
RImmunoblot analyses of knockdown efficacies in HeLa cells stably expressing mRFP‐GFP‐LC3 (tandem HeLa) configured for simultaneous IL‐1β secretion measurements by expressing flag‐pro‐IL‐1β and myc‐caspase‐1.
Figure 4. R‐SNARE Sec22b participates in IL‐1β secretion.
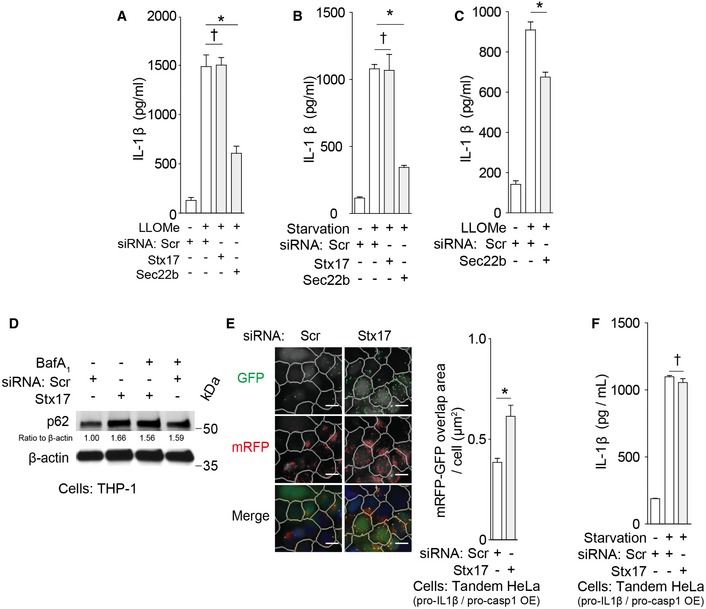
-
A, BIL‐1β levels in supernatants of THP‐1 cells subjected to knockdowns, treated with LPS, and then (A) with LLOMe or (B) starved in EBSS.
-
CIL‐1β levels in supernatants of primary human macrophages (MDM) subjected to knockdowns and sequentially treated with LPS and LLOMe.
-
DImmunoblot analyses of p62 levels in THP‐1 cells subjected to knockdowns as indicated and starved for 3 h.
-
EmRFP‐GFP overlapping areas of tandem HeLa cells reconstituted with flag‐pro‐IL‐1β and myc‐caspase‐1 and starved for 3 h. Mask overlay, iDev software‐defined objects [primary objects, cell outlines; internal secondary objects, GFP (top), mRFP (middle), or their overlapping area (bottom)]. Scale bars, 10 μm.
-
FIL‐1β levels in supernatants of starved tandem HeLa cells reconstituted with flag‐pro‐IL‐1β and myc‐caspase‐1 as in (E).
Syntaxin 17 controlling autophagosome–lysosome fusion is not required for IL‐1β secretion
In contrast to Sec22b dependence, IL‐1β secretion was not affected by knockdowns of syntaxin 17 (Figs 4A and B, and EV4C–E), a SNARE required for formation of degradative autolysosomal compartments (Itakura et al, 2012; Tsuboyama et al, 2016). As expected, knockdowns of syntaxin 17 affected measures of degradative autophagy flux by inhibiting p62 degradation and increasing double‐positive GFP+RFP+ puncta in the tandem mRFP‐GFP‐LC3 HeLa assay (Figs 4D and E, and EV4R), whereas they did not affect IL‐1β secretion in the same cells (Fig 4F). Syntaxin 17 showed no evidence of interactions with TRIM16 (Fig 3F). Thus, Sec22b is required for optimal IL‐1β secretion, whereas syntaxin 17 is not, indicating a separation between secretory and degradative autophagy pathways.
R‐SNARE Sec22b and plasma membrane Qa‐SNAREs syntaxin 3 and syntaxin 4 cooperate in IL‐1β secretion
Sec22b plays primary role in ERGIC trafficking (Jahn & Scheller, 2006) but can interact with plasma membrane syntaxins (Becker et al, 2005; Cebrian et al, 2011; Petkovic et al, 2014) to form SNARE complexes. Hence, we screened relevant Q‐SNAREs involved in fusion processes at the plasma membrane for their potential role in IL‐1β secretion. We tested non‐neuronal plasma membrane syntaxin 2, syntaxin 3, and syntaxin 4, as well as Qbc‐SNAREs SNAP‐23 and SNAP‐29. Knockdowns of Qbc‐SNAREs, SNAP‐23 or SNAP‐29, individually or in combination, significantly inhibited IL‐1β secretion (Figs 5A and B, and EV5A–K, and Appendix Fig S4A–E). SNAP‐23 was observed in Sec22b complexes (Fig EV5L), whereas syntaxin 4 has been previously reported in complexes with Sec22b (Cebrian et al, 2011). Detection of SNAP‐23–Sec22b complexes required NEM, an inhibitor of SNARE complex unwinding by NSF, which was negated upon quenching of NEM with DTT (Fig EV5L). Although knockdowns of individual plasma membrane syntaxins caused only slight reduction in IL‐1β secretion, a combined knockdown of syntaxin 3 and syntaxin 4 blocked IL‐1β secretion (Figs 5C and EV5M–Q). Nair et al (2011) have reported that yeast exocytic SNAREs Sso1 and Sso2 may affect trafficking of autophagy factors via plasma membrane. Thus, we tested whether the exocytic SNAREs used in our study (syntaxin 3 and syntaxin 4) acted indirectly by influencing autophagosome formation. The results shown in Fig EV5R and S indicate that Stx3/Stx4 double knockdown did not affect LC3 puncta formation, whereas TRIM16 knockdown, used as a control (Chauhan et al, 2016), did (Fig 5 and EV5R and S). Thus, plasma membrane syntaxins and Qbc‐SNAREs along with the R‐SNARE Sec22b participate in secretion of IL‐1β. A model for the sequential steps in secretory autophagy terminating with SNARE‐dependent fusion at the plasma membrane is depicted in Fig 5D.
Figure 5. Qbc‐SNAREs and plasma membrane Qa‐SNAREs syntaxin 3 and syntaxin 4 participate in IL‐1β secretion.
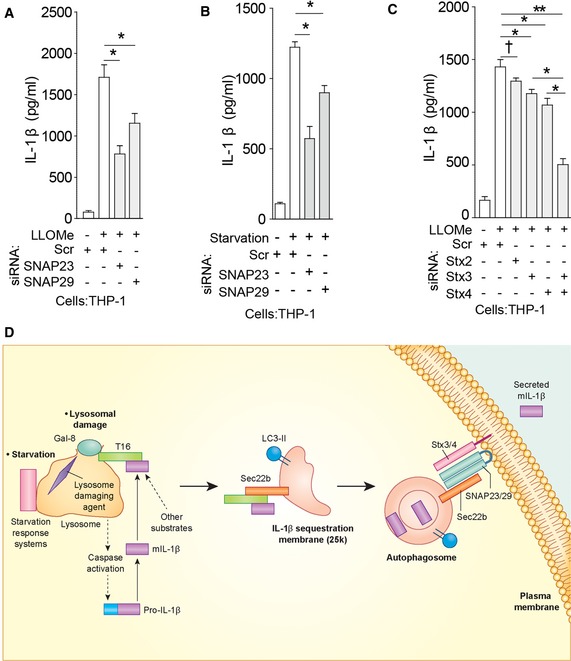
-
A–CIL‐1β levels in supernatants of THP‐1 cells subjected to knockdowns, treated with LPS, and then with LLOMe (A, C) or starved in EBSS (B).
-
DSecretory autophagy pathway. Upon lysosomal damage caused by inflammasome agonists such as silica, alum, monosodium urate, and Leu‐Leu‐O‐Me, inflammasome activation leads to caspase‐1‐dependent processing of cytosolic pro‐IL‐1β into mature IL‐1β (mIL‐1β). The autophagy regulatory systems (starvation/nutrition response systems: mTOR, LYNUS, TFEB) located at the lysosome transduce starvation signals (Napolitano & Ballabio, 2016). Upon lysosomal damage and stress, galectin‐8 recognizes β‐galactosides on the exposed lumenal membrane and/or glycosylated lysosomal lumenal proteins released into the cytosol and activates the galectin‐8–TRIM16 complex. TRIM16 binds secretory autophagy cargo (mIL‐1β and potentially other cargo). TRIM16 forms complexes with Sec22b to transfer the cargo to the autophagy‐induced LC3‐II+ membrane 25 k carriers (a direct interaction between Sec22b and TRIM16 remains to be determined). Depiction of mIL‐1β between the two membranes or within the lumen of the double membrane‐delimited autophagosomal intermediates reflects the sequestration processes that may be involved: IL‐1β is sequestered either through the reported Hsp90‐dependent import across the delimiting membrane into the lumen of carriers topologically corresponding to the intermembrane space in conventional autophagosomes (Zhang et al, 2015), or is sequestered into the inner lumen of conventional double‐membrane autophagosomal organelles, specializing in secretion, simultaneously observed in the same study (Zhang et al, 2015). Not depicted is the compatibility of TRIM16's receptor function with both of the above processes, that is, either a hand‐over of the cargo to Hsp90 or cargo sequestration via TRIM16 acting as a receptor/adaptor directly interacting with mAtg8s as previously reported (Mandell et al, 2014); the latter option is in keeping with the observations that TRIM16 is co‐secreted with IL‐1β (Munding et al, 2006). At this point, Sec22b molecules (acting as R‐SNAREs) on the delimiting membrane facing the cytosol carry out fusion at the plasma membrane in conjunction with the Qbc‐SNAREs SNAP‐23 and SNAP‐29 and plasma membrane Qa‐SNAREs syntaxin 3 and syntaxin 4, thus delivering IL‐1β to the extracellular milieu where it exerts its biological function.
Figure EV5. Qbc‐SNAREs and plasma membrane Qa‐SNAREs syntaxins 3 and 4 participate in IL‐1β secretion.
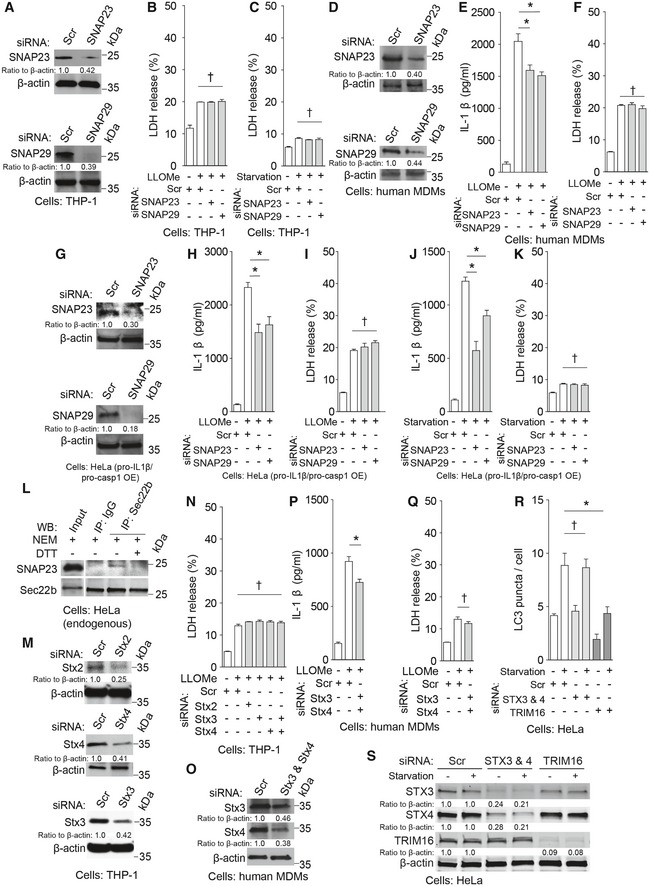
-
AImmunoblot analyses of knockdown efficacies in THP‐1 cells.
- B, C
-
DImmunoblot analyses of knockdown efficacies in primary human MDM cells.
-
E, FLevels of (E) IL‐1β and (F) LDH release determined in supernatants from primary human MDM cells that were subjected to knockdowns as indicated and were sequentially treated with LPS and LLOMe.
-
GImmunoblot analyses of knockdown efficacies in HeLa cells.
-
H, ILevels of (H) IL‐1β and (I) LDH release were determined in supernatants from HeLa cells reconstituted for IL‐1β secretion and treated with LLOMe.
-
J, KLevels of (J) IL‐1β and (K) LDH release were determined in supernatants from HeLa cells reconstituted for IL‐1β secretion and starved in EBSS.
-
LCo‐IP analysis between SNAP‐23 with Sec22b in lysates from HeLa cells treated with NEM and DTT as indicated.
-
MImmunoblot analyses of knockdown efficacies in THP‐1 cells.
-
NLDH release data from Fig 5C.
-
OImmunoblot analyses of knockdown efficacies in primary human MDM cells.
-
P, QLevels of (P) IL‐1β and (Q) LDH release determined in supernatants from primary human MDM cells that were subjected to knockdowns as indicated and were sequentially activated with LPS and treated with LLOMe.
-
RConventional autophagy response to starvation measured by LC3 puncta (HC imaging and analysis) in HeLa cells knocked down as indicated.
-
SKnockdown efficacies in cells corresponding to panel (R).
Secretory autophagy is involved in secretion of other cargo
The above experiments explain how secretory autophagy machinery separates from degradative autophagy specifically in the case of IL‐1β. We wondered whether this might apply to other unconventionally secreted cytosolic proteins. For this, we employed quantitative proteomics based on isobaric tandem mass tags (TMT*) and LC‐MS/MS spectrometry, and compared secreted proteins from Atg5fl/fl (autophagy‐competent) and Atg5fl/fl LysM‐Cre (autophagy‐impaired) macrophages using conditions that yielded the initial connection between IL‐1β secretion and autophagy (Dupont et al, 2011). We identified 74 proteins showing decreased levels in supernatants from Atg5fl/fl LysM‐Cre macrophages compared to those secreted from Atg5fl/fl macrophages (Fig 6A and Table 1), which were below the Atg5−/Atg5+ ratio for lactate dehydrogenase used as a cutoff measure for nonspecific release. Among these, several leaderless cytosolic and thus potentially unconventionally secreted proteins were identified (Fig 6B and Table 1), including ferritin (represented by ferritin heavy chain‐1 species, FTH1; Table 1).
Figure 6. Secretory autophagy plays a role in unconventional secretion of ferritin.
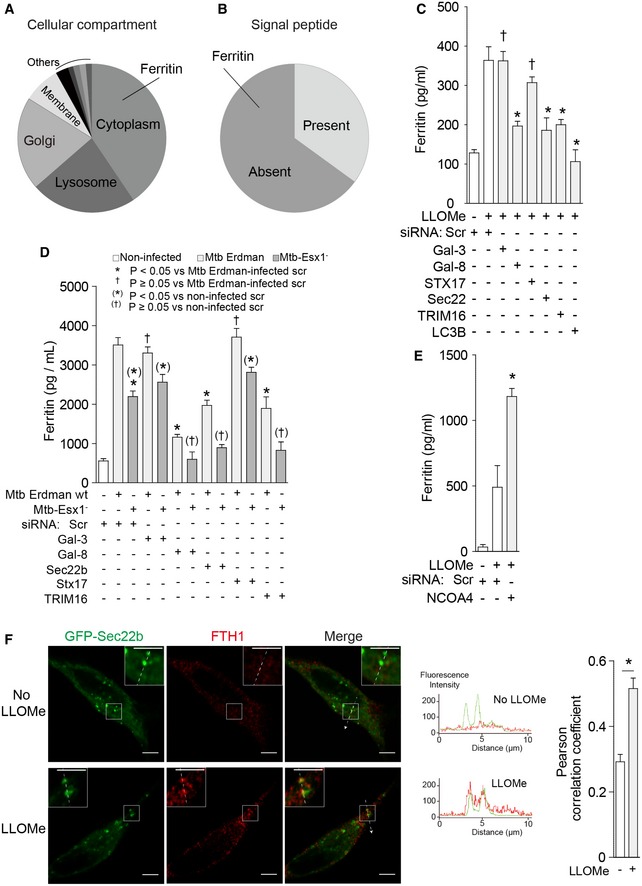
-
A, BSummary of candidate secretory autophagy cargos identified by quantitative proteomics based on isobaric tandem mass tags (listed in Table 1) and sorted by cellular compartments in (A) or the presence/absence of leader signal peptide in (B).
-
CFerritin levels in supernatants of THP‐1 cells subjected to knockdowns as indicated and sequentially treated with LPS and LLOMe.
-
DFerritin levels in supernatants of THP‐1 cells subjected to knockdowns as indicated and infected with M. tuberculosis as indicated.
-
ELevels of ferritin were determined in supernatants from THP‐1 cells that were subjected to knockdown as indicated and were sequentially treated with LPS and LLOMe.
-
FConfocal microscopy of HeLa cells expressing GFP‐Sec22b were treated with LLOMe, and stained for FTH1. Line tracings correspond to arrows. Scale bars, 5 μm.
Table 1.
Quantitative proteomic analysis of Atg5‐dependent secretome
| Gene symbol | Entrez gene ID | Description | Atg5−/+ ratio |
|---|---|---|---|
| Lyz2 | 17105 | Lysozyme C‐2 | −0.7 |
| Ctsc | 13032 | Cathepsin C (dipeptidyl‐peptidase 1) | −0.6 |
| Lipa | 16889 | Lysosomal acid lipase/cholesteryl ester hydrolase | −0.6 |
| Ctsd | 13033 | Cathepsin D | −0.5 |
| Gm2a | 14667 | Ganglioside GM2 activator | −0.5 |
| Hexa | 15211 | Beta‐hexosaminidase subunit alpha | −0.5 |
| Psap | 19156 | Prosaposin isoform A preproprotein | −0.5 |
| Hnrnph2 | 56258 | Heterogeneous nuclear ribonucleoprotein H2 | −0.5 |
| Asah1 | 11886 | Acid ceramidase | −0.4 |
| Cdc42 | 12540 | Isoform 2 of cell division control protein 42 homolog | −0.4 |
| Cfl1 | 12631 | Cofilin‐1 | −0.4 |
| Ctsb | 13030 | Cathepsin B | −0.4 |
| Fth1 | 14319 | Ferritin heavy chain | −0.4 |
| Grn | 14824 | Granulin | −0.4 |
| Hexb | 15212 | Beta‐hexosaminidase subunit beta | −0.4 |
| Il1rn | 16181 | Isoform 1 of interleukin‐1 receptor antagonist protein | −0.4 |
| Tgm2 | 21817 | Protein‐glutamine gamma‐glutamyltransferase 2 | −0.4 |
| Tpd52 | 21985 | Isoform 2 of Tumor protein D52 | −0.4 |
| Galns | 50917 | N‐acetylgalactosamine‐6‐sulfatase | −0.4 |
| Plek | 56193 | Pleckstrin | −0.4 |
| Ctsz | 64138 | Cathepsin Z | −0.4 |
| Plbd2 | 71772 | Isoform 1 of putative phospholipase B‐like 2 | −0.4 |
| Scpep1 | 74617 | Serine carboxypeptidase 1 | −0.4 |
| Dpp7 | 83768 | Dipeptidyl‐peptidase 2 | −0.4 |
| Gpnmb | 93695 | Transmembrane glycoprotein NMB | −0.4 |
| Gnptab | 432486 | Gnptab protein | −0.4 |
| Creg1 | 433375 | Protein CREG1 | −0.4 |
| Arf2 | 11841 | ADP‐ribosylation factor 2 | −0.3 |
| Coro1a | 12721 | Coronin‐1A | −0.3 |
| Eef1a1 | 13627 | Elongation factor 1‐alpha 1 | −0.3 |
| Msn | 17698 | Moesin | −0.3 |
| Naga | 17939 | Alpha‐N‐acetylgalactosaminidase | −0.3 |
| Efhd2 | 27984 | EF‐hand domain‐containing protein D2 | −0.3 |
| Gns | 75612 | N‐acetylglucosamine‐6‐sulfatase | −0.3 |
| Glrx | 93692 | Glutaredoxin‐1 | −0.3 |
| Ehd4 | 98878 | EH‐domain‐containing 4‐KJR (Fragment) | −0.3 |
| Pld4 | 104759 | Isoform 1 of phospholipase D4 | −0.3 |
| Rpl12 | 269261 | 60S ribosomal protein L12 | −0.3 |
| Aldoa | 11674 | Fructose‐bisphosphate aldolase A | −0.2 |
| Akr1b3 | 11677 | Aldose reductase | −0.2 |
| Cct7 | 12468 | T‐complex protein 1 subunit eta | −0.2 |
| Cd14 | 12475 | Monocyte differentiation antigen CD14 | −0.2 |
| Ctss | 13040 | Cathepsin S | −0.2 |
| Eef2 | 13629 | Elongation factor 2 | −0.2 |
| Eif4a1 | 13681 | Eukaryotic initiation factor 4A‐I | −0.2 |
| Eno1 | 13806 | Alpha‐enolase | −0.2 |
| Rack1 | 14694 | Guanine nucleotide‐binding protein subunit beta‐2‐like 1 | −0.2 |
| Gpi1 | 14751 | Glucose‐6‐phosphate isomerase | −0.2 |
| Grb2 | 14784 | Isoform 1 of growth factor receptor‐bound protein 2 | −0.2 |
| Hnrnpk | 15387 | Isoform 1 of heterogeneous nuclear ribonucleoprotein K | −0.2 |
| Lamp1 | 16783 | Lysosomal membrane glycoprotein 1, isoform CRA_a | −0.2 |
| Pfn1 | 18643 | Profilin‐1 | −0.2 |
| Pgk1 | 18655 | Phosphoglycerate kinase 1 | −0.2 |
| Ctsa | 19025 | Cathepsin A | −0.2 |
| Ppp1 cc | 19047 | Isoform gamma‐1 of serine/threonine protein phosphatase PP1‐gamma catalytic subunit | −0.2 |
| Rpl18 | 19899 | 60S ribosomal protein L18 | −0.2 |
| Ostf1 | 20409 | Osteoclast‐stimulating factor 1 | −0.2 |
| Tkt | 21881 | Transketolase | −0.2 |
| Tln1 | 21894 | Talin‐1 | −0.2 |
| Tpi1 | 21991 | Triosephosphate isomerase 1 | −0.2 |
| Tubb4a | 22153 | Tubulin beta‐4 chain | −0.2 |
| Tubb5 | 22154 | Tubulin beta‐5 chain | −0.2 |
| Uba52 | 22186 | Ubiquitin A‐52 residue ribosomal protein fusion product 1 | −0.2 |
| Uba1 | 22201 | Ubiquitin‐like modifier‐activating enzyme 1 | −0.2 |
| Vim | 22352 | Vimentin | −0.2 |
| Twf2 | 23999 | Isoform 1 of Twinfilin‐2 | −0.2 |
| Prdx5 | 54683 | Isoform mitochondrial of peroxiredoxin‐5, mitochondrial | −0.2 |
| Rplp1 | 56040 | 60S acidic ribosomal protein P1 | −0.2 |
| Sh3bgrl | 56726 | SH3 domain‐binding glutamic acid‐rich‐like protein | −0.2 |
| Rap1a | 109905 | Ras‐related protein Rap‐1A | −0.2 |
| Gusb | 110006 | Beta‐glucuronidase | −0.2 |
| Ppp2r4 | 110854 | Serine/threonine protein phosphatase 2A regulatory subunit B' | −0.2 |
| Ppia | 268373 | Peptidyl‐prolyl cis‐trans isomerase | −0.2 |
| Vcp | 269523 | Transitional endoplasmic reticulum ATPase | −0.2 |
Proteins in culture supernatants of cells induced for autophagy by starvation in EBSS for 1 h (with additional pretreatments and treatments as described in Materials and Methods) were quantified and identified by isobaric tandem mass tags (TMT) and LC‐MS/MS spectrometry. Secreted proteins from Atg5fl/fl (autophagy‐competent) and Atg5fl/fl LysM‐Cre (autophagy‐impaired) macrophages were compared (log fold changes) by the relative abundance of TMT tags. Lactate dehydrogenase was used as a cutoff for reporting and as a measure of cell death (< 10%), which did not exceed that of previously reported (Dupont et al, 2011) and was not statistically significant between samples.
Secretory autophagy pathway is involved in secretion of ferritin
Ferritin is a cytoplasmic protein secreted from macrophages through an unconventional secretory pathway that has remained undefined (Cohen et al, 2010). Ferritin has also been identified as a cargo for delivery to autophagosomes for degradation (Dowdle et al, 2014; Mancias et al, 2014). We thus wondered if secretion of ferritin may utilize the system analogous to that identified here for IL‐1β. Knockdowns of TRIM16, galectin‐8 (but not galectin‐3), and Sec22b (but not syntaxin 17) suppressed secretion of ferritin in response to LLOMe (Figs 6C and EV6A and B). Secretion of ferritin was, like that of IL‐1β, dependent on LC3B (Fig 6C). This pattern of dependence for ferritin secretion on TRIM16, galectin‐8, and Sec22b was furthermore observed in response to a physiological endomembrane injury during infection of macrophages with Mycobacterium tuberculosis Erdman (Figs 6D and EV6C–G). As expected, ferritin secretion was reduced when M. tuberculosis Esx‐1 mutant, which permeabilizes phagosomal membrane less efficiently (Manzanillo et al, 2012), was used instead of the parental wild‐type Erdman strain (Figs 6D and EV6C, F and G). In contrast to TRIM16, a knockdown of NCOA4, which is a ferritin receptor for autophagic degradation of this cargo (Dowdle et al, 2014; Mancias et al, 2014), did not decrease ferritin secretion (Figs 6E and EV6H and I). This indicates that NCOA4 does not take part in secretory autophagy of ferritin. Mirroring this observation, knockdown of p62 previously reported to act as a receptor for degradative autophagy of IL‐1β (Shi et al, 2012) did not decrease IL‐1β secretion in response to inflammasome agonist (Fig EV6J–L). Furthermore, FTH1 colocalized with Sec22b (Fig 6F) and was found in coimmunoprecipitates with TRIM16, galectin‐8 and Sec22b (Fig EV6M). Thus, secretory autophagy machinery involved in unconventional secretion of IL‐1β also plays a role in unconventional secretion of ferritin.
Figure EV6. Secretory autophagy machinery plays a role in unconventional secretion of ferritin.

-
AImmunoblot analysis of the levels of FTH1 in supernatants from THP‐1 cells that were subjected to knockdown as indicated and were sequentially treated with LPS and LLOMe.
-
BFerritin levels in supernatants of primary human MDM macrophages subjected to knockdowns as indicated and treated sequentially with LPS and LLOMe.
-
CFerritin levels in supernatants of THP‐1 cells subjected to knockdowns as indicated and infected with M. tuberculosis Erdman as indicated.
-
D, E(D) Ferritin levels and (E) LDH release in supernatants of primary human MDM cells subjected to knockdowns as indicated and infected with M. tuberculosis as indicated.
-
F, G(F) Ferritin levels and (G) LDH release in supernatants of primary human MDM cells infected with M. tuberculosis as indicated.
-
HLDH release data from Fig 6E.
-
I, JImmunoblot analyses of knockdown efficacies in THP‐1 cells.
-
K, LLevels of (K) IL‐1β and (L) LDH release determined in supernatants from THP‐1 cells subjected to knockdowns as indicated and were sequentially treated with LPS and LLOMe.
-
MCo‐IP analysis of interaction between FTH1 with GFP‐TRIM16, GFP‐galectin‐8, and GFP‐Sec22b.
Discussion
This work defines an ordered set of molecular events, protein sorting, and intracellular trafficking processes for secretory autophagy, as depicted in the model in Fig 5D. The galectin‐8‐TRIM16, TRIM16‐Sec22b, and Sec22b‐QabcSNAREs complexes represent interconnected intermediates that act in a sequential manner to respond to triggers, and to initiate and execute the unconventional secretion of the cargo. This pathway utilizes specialized receptors that are different from those engaged in degradative autophagy. TRIM16 acts as a secretory autophagy receptor in keeping with its reported co‐secretion with IL‐1β (Munding et al, 2006) and pivots cargo to secretory autophagy through differential utilization of SNAREs. The autophagic secretory pathway avoids autolysosomal degradation, because it is independent of syntaxin 17, a SNARE that directs autophagosomes via a syntaxin 17‐VAMP8‐SNAP‐29‐driven complex to fuse with lysosomes leading to cargo degradation (Itakura et al, 2012). Instead, TRIM16, the first identified member of what we postulate to be a class of secretory autophagy receptors, interacts with Sec22b leading to Sec22b‐QabcSNARE‐sponsored fusion at the plasma membrane.
The role of Sec22b in unconventional secretion of IL‐1β provides a novel function to a hitherto incompletely defined but long‐anticipated non‐ERGIC roles for the Sec22b SNARE (Becker et al, 2005), and adds to other processes for this SNARE at the plasma membrane previously investigated under conditions different than those in our study (Petkovic et al, 2014) or on membranes derived from the plasma membrane (Cebrian et al, 2011). Of note, by utilizing Sec22b and ERGIC, or ERGIC‐precursor or ERGIC‐derived membranes, as a sorting station for both the biosynthetic ER‐to‐Golgi pathway (Xu et al, 2000) and for secretory autophagy, the cell can control conventional and unconventional secretion from the same trafficking locale.
Both the conventional autophagosomal membranes earmarked for fusion with the lysosomes and those that are destined for secretion may be derived from a subset of ERGIC‐derived autophagosomal precursors (Ge et al, 2014). Other contributing sources of autophagic membrane may modulate this (Hamasaki et al, 2013; Puri et al, 2013; Joachim et al, 2015; Lamb et al, 2016). The sorting step between LC3+ Sec22b+ IL‐1β secretory carriers and ERGIC‐derived (Ge et al, 2014) autophagosomal membranes destined for degradative autophagosomes could involve differential syntaxin 17 insertion (Itakura et al, 2012; Tsuboyama et al, 2016) or alternatively its regulation, if syntaxin 17 is present at all on IL‐1β secretory carriers, which has not been excluded in our study (Itakura et al, 2012; Hamasaki et al, 2013; Takats et al, 2014; Diao et al, 2015).
A key separation step in SNARE utilization between the secretory and degradative autophagy involves a handover of the cargo from the receptor (TRIM16) to precursor membranes whereby IL‐1β is sequestered within the LC3+ Sec22b+ carrier membranes. This can occur either through conventional autophagosomes, or through a recently described HSP90‐dependent process of IL‐1β translocation into the lumen of the delimiting ERGIC‐derived membrane (Zhang et al, 2015). Both of these options are reflected in the depiction of secretory carrier intermediates bound for fusion and release at the plasma membrane (Fig 5D). In our study, we have characterized the above pathway functionally and biochemically, as well as at the fluorescence microscopy level. A full morphological identification of secretory autophagy cargo carriers and their distinction from degradative autophagic organelles at the ultrastructural level is a task that will require separate in‐depth analyses and defines a limitation of our present work.
Although a role for TRIM16 in IL‐1β secretion was suggested in a previous study, the underlying mechanism had not been uncovered (Munding et al, 2006). TRIM16 interactions with autophagy factors and a specific SNARE apparatus, as a mode of delivery of IL‐1β to the secretory autophagy pathway can explain these previously observed phenomena. A unique feature of TRIM16 is the presence of the SNC1/Longin‐like domain and association of a region including it with the longin R‐SNARE Sec22b. Since TRIM16 binds IL‐1β (Munding et al, 2006), this makes it a perfect medium to transfer IL‐1β to the correct sorting compartment for secretory autophagy. Note, however, that although super‐resolution microscopy and co‐IP analyses indicate that TRIM16 and Sec22b are in close proximity and form macromolecular complexes, biochemical evidence for their direct interaction is not available at present. Another important feature of TRIM16 is its ability to bind galectins, which is enhanced by the presence of ULK1, as previously shown for galectin‐3 (Chauhan et al, 2016) and observed here for galectin‐8. This in turn allows synchronization of caspase‐1‐dependent processing of pro‐IL‐1β in response to inflammasome activation, with the induction of secretory autophagy via lysosomal stress circuitry known to control autophagy (Roczniak‐Ferguson et al, 2012; Settembre et al, 2012; Medina et al, 2015; Napolitano & Ballabio, 2016).
When compared to galectin‐3, which showed no effects on secretion in the present study and is important for autophagic homeostasis of damaged lysosomes (Chauhan et al, 2016) involving the process of lysophagy (Thurston et al, 2012; Fujita et al, 2013; Maejima et al, 2013), galectin‐8 may appear as a galectin specializing in secretory autophagy and as a point of divergence between secretory and degradative autophagy. Galectin‐8 contributes (as opposed to galectin‐3) to these sorting steps most likely by helping separate the TRIM16 pools participating in secretory autophagy studied in this work from the TRIM16 pools participating in lysosomal homeostasis/lysophagy (Chauhan et al, 2016). However, galectin‐8 also has the acknowledged functions in degradative autophagy. For example, galectin‐8 is known to play a role in control of intracellular Salmonella (Thurston et al, 2012), which occurs through a process termed xenophagy. Thus, the main point of divergence between degradative and secretory autophagy may not be galectins, despite their contributions, but rather it is based on the observed differential utilization of SNAREs: syntaxin 17 for degradative autophagy vs. Sec22b/syntaxin 3 or syntaxin 4/SNAP‐23 or SNAP‐29 for secretory autophagy.
The utilization of autophagy for regulated secretion of IL‐1β into the extracellular space where it performs its function provides a cell with the opportunity to throttle inflammatory outputs. Although secretory autophagy promotes IL‐1β secretion (Dupont et al, 2011; Zhang et al, 2015), degradative autophagy can reduce levels of IL‐1β (Harris et al, 2011), trim down inflammasome components (Shi et al, 2012; Kimura et al, 2015), or eliminate endogenous inflammasome agonists (Nakahira et al, 2011; Zhou et al, 2011). Mechanistically, this allows fine‐tuning of IL‐1β outputs, and possibly outputs of other biologically active inflammasome effectors (Schroder & Tschopp, 2010; van de Veerdonk et al, 2011; Lamkanfi & Dixit, 2014), to balance protection vs. excessive pathology. Other inflammation‐associated unconventionally secreted proteins such as serum ferritin (Cohen et al, 2010) seem to share autophagy as a regulatory output for either secretion, as shown in this work, or for ferritin degradation (Asano et al, 2011; Dowdle et al, 2014; Mancias et al, 2014).
The placement of TRIM16‐regulated autophagic processes at the intersection between lysosomal repair (Chauhan et al, 2016) and IL‐1β secretion (this study) is not coincidental. The central role of lysosomes in control of autophagy by mTOR and TFEB is well known (Napolitano & Ballabio, 2016). The new regulatory factors including galectins and TRIM16 as revealed in the present study and elsewhere (Chauhan et al, 2016) are also positioned on or relocate to lysosomes where they can sense damage or signaling changes. From this common station, these factors can direct either autophagic degradation/repair (Maejima et al, 2013; Chauhan et al, 2016) or secretion of inflammatory mediators such as IL‐1β (Dupont et al, 2011; Zhang et al, 2015). This is aligned with the known links between lysosomal damage and inflammasome activation (Martinon et al, 2006; Hornung et al, 2008; Schroder & Tschopp, 2010; Ito et al, 2015) and represents a previously unanticipated decision node: On the one hand, autophagy acts to eliminate or repair damaged lysosomal and other endomembranes (Maejima et al, 2013; Chauhan et al, 2016) and thus reduces the endogenous sources of pro‐inflammatory stimuli, adding to the repertoire of inflammasome agonists that are downregulated by autophagy (Nakahira et al, 2011; Zhou et al, 2011). On the other hand, when lysosomal repair is not possible or damage exceeds the autophagic capacity to maintain lysosomal homeostasis in the cell, IL‐1β secretion facilitated by the autophagic systems, now in its secretory mode, serves to elicit tissue‐level homeostatic responses.
The secretory autophagy pathway can also be triggered by starvation signals, normally transduced via lysosomally located mTOR and TFEB (Napolitano & Ballabio, 2016). It is not clear at present how starvation and lysosomal damage/stress overlap, but, since TRIM16 interacts with and affects mTOR, TFEB, and calcineurin (Chauhan et al, 2016), candidate molecular connections already exist. As to the lysosomal damage triggers, the pathway may proceed sequentially as follows (Fig 5D): Galectin‐8 recognizes exofacially located β‐galactosides once they are exposed to the cytosol following membrane damage. This activates the galectin‐8–TRIM16 complex. TRIM16 acts as a receptor for secretory autophagy cargo, for example, mIL‐1β. Next, TRIM16 interacts (directly or indirectly) with Sec22b and transfers the secretory cargo to the LC3‐II+ membrane carriers. The cargo may be sequestered between the two membranes through a unique HSP90‐dependent transport process recently described (Zhang et al, 2015), compatible with TRIM16 colocalization with HSP90 following lysosomal damage. However, cargo may also be captured within the lumen of the double‐membrane‐delimited autophagosomal intermediates, as reported in the same study (Zhang et al, 2015). This may occur via TRIM16 acting as a receptor/adaptor, since TRIM16 directly interacts with mAtg8s (Mandell et al, 2014). The latter option is in keeping with the observations that TRIM16 is co‐secreted with IL‐1β (Munding et al, 2006).
At the end stages of the pathway, Sec22b (acting as an R‐SNARE) on the cytofacial membrane of the carriers engages in membrane fusion at the plasma membrane in combination with the Qbc‐SNAREs SNAP‐23 and SNAP‐29 and plasma membrane Qa‐SNAREs syntaxin 3 and syntaxin 4, thus delivering IL‐1β to the extracellular milieu where it exerts its biological function. Incidentally, SNAP‐29 plays a role in autophagosome–lysosome fusion (Itakura et al, 2012; Hamasaki et al, 2013; Takats et al, 2013; Diao et al, 2015). However, it is also known to interact with plasma membrane syntaxins (Steegmaier et al, 1998) and thus far it has been noted for its regulatory, and even inhibitory roles (Su et al, 2001; Morelli et al, 2014). However, in most cases, it plays a role in fusion events (Xu et al, 2014). To make it even more complicated, SNAP‐29 activity is controlled by recently described O‐Glc‐NAc modifications, which are modulated by starvation (Guo et al, 2014). Nonetheless, SNAP‐29 is required in C. elegans for transport of the apically and basolaterally directed cargos and when it is absent cargo‐containing vesicles accumulate in the cytoplasm (Sato et al, 2011), whereas in humans, SNAP‐29 deficiency has secretory defects (Sprecher et al, 2005).
TRIM16 is known to interact with at least one of the mAtg8s, GABARAP (Mandell et al, 2014). Of note, TRIM16 does not interact with sequestosome‐1/p62 (Mandell et al, 2014), a classical degradative autophagy receptor (Bjorkoy et al, 2005), whereas many other TRIMs do (Mandell et al, 2014). The absence of p62 interaction may indicate a special position of TRIM16 among TRIMs to guide autophagic intermediates to secretion. The shared requirement for TRIM16, galectin‐8, and Sec22b in unconventional secretion of ferritin underscores this notion and indicates a broader role of this secretory autophagy system. However, additional TRIMs have been identified in our screen for IL‐1β secretion, and diverse cytosolic substrates have been found within the ATG5‐dependent secretome, and thus, we predict that other TRIMs as well as other types of specialized receptors contribute to secretory autophagy.
In conclusion, secretory autophagy represents an alternative termination of the autophagosomal pathway enabling a range of leaderless cytosolic proteins to express their extracellular activities of importance in health homeostasis and disease states. The present definition of its molecular and cellular machinery in mammalian cells opens prospects for further study and applications.
Materials and Methods
Antibodies and reagents
Flag (Sigma 1804; WB 1:1,000; IF 1:100–1:250; IP 1–2 μg/ml); LC3B (Sigma L7543; WB 1:1,000); syntaxin 17 (Sigma HPA001204; 1:1,000); TRIM16 (Bethyl A301‐160A; WB 1:1,000); IL‐1β (R&D AF‐201‐NA; 1:1,000–1:3,000); syntaxin 2 (Enzo ADI‐VAP‐SV065‐E; 1:4,000); galectin‐8 (Santa Cruz sc‐28254; WB 1:1,000; IF 1:100); TRIM16 (Santa Cruz sc‐79770; IF 1:100); LC3B (MBL PM036; IF 1:250–1:500); ATG16L1 (MBL PM040; 1:1,000); GFP (Abcam ab290; IP 0.5 μg/ml; WB 1:2,000–1:5,000); IL‐1β (Abcam ab9722; 1:1,000); Sec22b (Abcam ab181076; 1:1,000); PDI (Abcam ab2792; 1:1,000); galectin‐3 (Santa Cruz sc‐32790; WB 1:1,000); SNAP‐23 (Abcam ab131242; 1:1,000); SNAP‐29 (Abcam ab138500; 1,1,000); syntaxin 3 (Abcam ab188583; WB 1:1,000); syntaxin 4 (Abcam ab185556; WB 1:1,000); actin (Abcam ab8226; 1:4,000); tubulin (Abcam6046; 1:1,000); FTH1 (Rockland 200‐401‐090‐0100; WB 1:2,000; IF 1:250); FTH1(CST 3998S; WB 1:1,000); NCOA4 (Sigma SAB1404569; WB 1:1,000); TRIM10 (Abcam ab151306; 1:1,000); HSP90 (Cell Signaling CST4874; 1:200); GABARAP (MBL M1353; 1:1,000); Myc (Cell Signaling CST2278; 1:1,000). The reagents used were Ultrapure LPS (InvivoGen), Cytotoxic LDH assay (Promega), LLOMe (a lysosome disruption agent (Thiele & Lipsky, 1990), Sigma), Bafilomycin A1 (Baf A1, InvivoGen), N‐ethylmaleimide (NEM, Sigma), DL‐dithiothreitol (DTT, Sigma), nigericin (Sigma), monosodium urate (MSU, ENZO), and Alum (Thermo Scientific).
IL‐1β and ferritin measurement
For IL‐1β secretion from THP‐1 cells, cells that had been subjected to the differentiation with 50 nM PMA overnight were treated with 100 ng/ml LPS overnight and then treated with 250 μM LLOMe (referred to as sequential LPS and LLOMe treatment) or other stimuli indicated in the figure legends, for 3 h or as indicated (representing sequential LPS‐other stimuli treatment). For human primary MDM cells and murine bone marrow macrophage cells, cells were treated with 100 ng/ml LPS overnight and then treated with stimuli indicated in the figure legends for 3 h. For HeLa cells and 293T cells, IL‐1β secretion system was reconstructed by co‐transfection with flag‐pro‐IL‐1β and myc‐pro‐caspase‐1 and then IL‐1β secretion was stimulated either by 1 mM LLOMe for 1 h or by starvation in EBSS for 3 h. IL‐1β of supernatants were measured using HEK‐Blue IL‐1β Cells (InvivoGen). Alternatively, cell‐free supernatants were assayed by immunoblotting after methanol precipitation as described in Appendix Supplementary Materials and Methods. For ferritin secretion, cells were treated sequentially with LPS and LLOMe. Ferritin in supernatants was measured using Human Ferritin ELISA Kit (Sigma RAB0197).
TRIM family screen
THP‐1 cells were cultured in 96‐well plates containing SMARTpool siRNA (GE Healthcare) and RNAiMax (Life Technologies). Upon differentiation by overnight incubation with PMA, culture media were changed, and after 48 h after plating, cells were treated with LPS overnight and then treated with 250 μM LLOMe for 3 h. Supernatants of samples were collected, and IL‐1β measurements were performed as described above. Cutoff for hits, < −6 SDs change relative to the mean of stimulated control.
High content image analysis
High content imaging analysis was performed using a Cellomics VTI HCS scanner and iDEV software (Thermo Fisher Scientific). Automated epifluorescence image collection was performed from 49 ≤ fields in 96‐well plates until a minimum of 500 cells per well plate was acquired. Epifluorescence images were machine‐collected and machine‐analyzed using predetermined scanning parameters and object mask definitions. Hoechst 33342 staining was used for automatic focusing and cellular outlines based on background staining of the cytoplasm, and the mean count of LC3 puncta per cell was determined.
Differential centrifugation and membrane fractionation
The procedure was performed as described previously (Ge et al, 2013; Zhang et al, 2015). Cells were harvested in B1 buffer (20 mM HEPES‐KOH, pH 7.2, 400 mM sucrose, 1 mM EDTA) containing protease and phosphatase inhibitors (Roche) and 0.3 mM DTT, and homogenized by passing through a 22G needle until ~85% lysis analyzed by Trypan blue staining. Homogenates were then subjected to sequential differential centrifugation at 3,000 g (10 min), 25,000 g (20 min), and 100,000 g (30 min, TLA100.3 rotor, Beckman) to collect the pelleted membranes. The pellets were suspended in B88 buffer (20 mM Hepes, pH 7.2, 150 mM potassium acetate, 5 mM magnesium acetate, 250 mM sorbitol). Samples were normalized based on the levels of phosphatidylcholine determined as previously described (Ge et al, 2013). 2× SDS loading buffer was added and samples heated in boiling water for 10 min and analyzed by immunoblotting.
For membrane fractionation, protocols by Zhang et al (2015) were followed. The 25,000 g membrane pellet was suspended in 0.75 ml 1.25 M sucrose buffer and overlaid with 0.5 ml 1.1 M and 0.5 ml 0.25 M sucrose buffer. Centrifugation was performed at 120,000 g for 2 h (TLS 55 rotor, Beckman), after which layer fraction (L) at the interface between 0.25 M and 1.1 M sucrose was separated using 22G syringe needle. The L fraction was suspended in 1 ml 19% OptiPrep (Sigma‐Aldrich) for a step gradient containing 0.5 ml 22.5%, 1 ml 19%, 0.9 ml 16%, 0.9 ml 12%, 1 ml 8%, 0.5 ml 5%, and 0.2 ml 0% OptiPrep each. Each density of OptiPrep was prepared by diluting 50% OptiPrep (20 mM tricine‐KOH, pH 7.4, 42 mM sucrose and 1 mM EDTA) with a buffer containing 20 mM tricine‐KOH, pH 7.4, 250 mM sucrose and 1 mM EDTA. The OptiPrep gradient was centrifuged at 150,000 g for 3 h (SW 55 Ti rotor, Beckman), and subsequently, fractions of 0.5 ml each were collected from the top. Fractions were diluted eightfold by 3.5 ml B88 buffer (20 mM HEPES‐KOH, pH 7.2, 250 mM sorbitol, 150 mM potassium acetate, and 5 mM magnesium acetate). Membranes were collected by centrifugation at 100,000 g for 1 h and resuspended in 80 μl B88 buffer.
Super‐resolution fluorescence microscopy analysis
Hela cells transfected with flag‐TRIM16 and GFP‐Sec22b constructs were plated on 25 mm round #1.5 coverslips (Warner instruments), allowed to adhere overnight, and were treated with LLOMe. Super‐resolution microscopy was performed as previously reported (Valley et al, 2015) with full details described in Appendix Supplementary Materials and Methods.
Mass spectrometry of proteins in cell culture supernatants
For the screen of Atg5‐dependent secretory substrates, bone marrow‐derived macrophages (BMM) from Atg5fl/fl LysM‐Cre mice and their Cre‐negative littermates were treated with 100 ng/ml LPS overnight and then treated with 20 μM nigericin in EBSS for 1 h as described previously (Dupont et al, 2011). Supernatants were collected, centrifuged at 500 g for 5 min at 4°C, filtrated (0.45 μm pore size), and concentrated with Amicon ultrafiltration units (3 kDa MWCO, Millipore). 100 μg of proteins from each sample was precipitated with acetone. Samples were then reduced, alkylated, and digested by trypsin overnight. The peptide pools were further acidified and cleaned by C18 spin columns prior to labeling with Tandem Mass Tags (TMT 6plex, ThermoScientific). Purified and dried peptides were solubilized in 100 μl 200 mM HEPES buffer pH 8. The TMT tags were dissolved in absolute ethanol and added to the peptide pools, after 1.5 h, the reaction was quenched by the addition of 10% hydroxylamine, followed by pooling, drying, and another round of C18 spin column cleanup. The TMT multiplexed samples were reconstituted in LC mobile phase A (0.1% formic acid, 99.9% water) and injected onto an in‐house pulled and packed C18 reverse phase analytical column where the inner diameter was 100 μM and the column length was 20 cm. The mass spectrometer was a LTQ Orbitrap Velos operating in a High/High schema, where precursor and HCD fragment ion spectra were analyzed in by the orbitrap mass analyzer. Peptides were identified using Sequest on a Sorcerer platform and the tandem mass tags quantified by Scaffold Q+ (Proteome Software). The Atg5−/Atg5+ ratio for lactate dehydrogenase (−0.1) served as a cutoff measure for non‐specific release. Information regarding the subcellular distribution of proteins was collected from Uniprot and COMPARTMENTS (http://compartments.jensenlab.org).
Statistical analyses
Data are expressed as means ± SEM (n ≥ 5; except for immunoblot quantifications where n ≥ 3). All data were parametric and based on prior published sample size. The variance between the statistically compared groups was assumed as equal. All morphologic data were collected and statistically processed by operator‐independent computer algorithms (iDev program), with the exceptions as indicated. Either a two‐tailed Student's t‐test or ANOVA was used. Statistical significance was defined as P < 0.05.
Author contributions
TK, JJ, and CMA designed, carried out, and interpreted experiments and contributed to the overall design of the study (TK contributed to writing of the original version of the manuscript and JJ to revisions). SK, SWC, YG, AJ, and FF designed, carried out, and interpreted experiments. MM, RP, and SJ carried out experiments. ND helped design experiments. KAL designed and interpreted experiments. TJ designed and interpreted experiments and contributed to the overall design of the study. VD conceived and supervised the project, designed experiments, interpreted data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Christian Ungermann and Jesse Hay for critical comments, Marisa Ponpuak for experimental contributions, and Michael Wester for contributions to the super‐resolution data analysis. We thank Jeffrey Cox for M. tuberculosis Erdman and its Esx1 − mutant, Skip Virgin for Atg5fl/fl LysM‐Cre mice, Shizuo Akira and Sharon Tooze for Atg9 and matching wild‐type MEFs, and Diane Lidke for consultation. This work was supported by grants AI042999, AI111935 and UH2AI122313 from the NIH. T.K. was supported by Manpei Suzuki Diabetes Foundation and Uehara Memorial Foundation. F.F. and K.A.L. were supported by the UNM Center for Spatiotemporal Modeling of Cell Signaling (NIH 5P50GM085273) and the UNM Cancer Center's Bioinformatics & High‐Dimensional Data Analysis Shared Resource (NIH/NCI P30CA118100). T.J. was supported by grant 196898 from the Norwegian Research Council and grant 71043‐PR‐2006‐0320 from the Norwegian Cancer Society.
The EMBO Journal (2017) 36: 42–60
References
- Asano T, Komatsu M, Yamaguchi‐Iwai Y, Ishikawa F, Mizushima N, Iwai K (2011) Distinct mechanisms of ferritin delivery to lysosomes in iron‐depleted and iron‐replete cells. Mol Cell Biol 31: 2040–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3‐phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182: 685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Volchuk A, Rothman JE (2005) Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. Proc Natl Acad Sci USA 102: 4022–4026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif – crucial for selective autophagy. J Cell Sci 126: 3237–3247 [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, Enninga J, Moita LF, Amigorena S, Savina A (2011) Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 147: 1355–1368 [DOI] [PubMed] [Google Scholar]
- Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun JA, Johansen T, Deretic V (2016) TRIMs and galectins globally cooperate and TRIM16 and galectin‐3 co‐direct autophagy in endomembrane damage homeostasis. Dev Cell 39: 13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LA, Gutierrez L, Weiss A, Leichtmann‐Bardoogo Y, Zhang DL, Crooks DR, Sougrat R, Morgenstern A, Galy B, Hentze MW, Lazaro FJ, Rouault TA, Meyron‐Holtz EG (2010) Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 116: 1574–1584 [DOI] [PubMed] [Google Scholar]
- Deretic V, Saitoh T, Akira S (2013) Autophagy in infection, inflammation, and immunity. Nat Rev Immunol 13: 722–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, Pfuetzner RA, Brunger AT, Zhong Q (2015) ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520: 563–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12‐5‐16L1. Mol Cell 55: 238–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, Cantwell J, Luu C, Cornella‐Taracido I, Harrington E, Fekkes P, Lei H, Fang Q, Digan ME, Burdick D, Powers AF et al (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo . Nat Cell Biol 16: 1069–1079 [DOI] [PubMed] [Google Scholar]
- Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V (2011) Autophagy‐based unconventional secretory pathway for extracellular delivery of IL‐1beta. EMBO J 30: 4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V (2010) Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol 188: 527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ (2011) Phosphorylation of ULK1 (hATG1) by AMP‐activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19: 2092–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S, Haraguchi T, Guan JL, Iwai K, Tokunaga F, Saito K, Ishibashi K, Akira S, Fukuda M, Noda T, Yoshimori T (2013) Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 203: 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Levine B, Kroemer G (2014) Metabolic control of autophagy. Cell 159: 1263–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammoh N, Florey O, Overholtzer M, Jiang X (2013) Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex‐dependent and ‐independent autophagy. Nat Struct Mol Biol 20: 144–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Melville D, Zhang M, Schekman R (2013) The ER‐Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2: e00947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Zhang M, Schekman R (2014) Phosphatidylinositol 3‐kinase and COPII generate LC3 lipidation vesicles from the ER‐Golgi intermediate compartment. eLife 3: e04135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovacs AL, Zhang Z, Feng D, Chen S, Zhang H (2014) O‐GlcNAc‐modification of SNAP‐29 regulates autophagosome maturation. Nat Cell Biol 16: 1215–1226 [DOI] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T (2013) Autophagosomes form at ER‐mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]
- Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC (2011) Autophagy controls IL‐1beta secretion by targeting pro‐IL‐1beta for degradation. J Biol Chem 286: 9587–9597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Mizushima N (2012) The hairpin‐type tail‐anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151: 1256–1269 [DOI] [PubMed] [Google Scholar]
- Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, Morita R (2015) Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun 6: 7360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH (2006) SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol 7: 631–643 [DOI] [PubMed] [Google Scholar]
- Joachim J, Jefferies HB, Razi M, Frith D, Snijders AP, Chakravarty P, Judith D, Tooze SA (2015) Activation of ULK Kinase and Autophagy by GABARAP Trafficking from the Centrosome Is Regulated by WAC and GM130. Mol Cell 60: 899–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M, Katona I, Qualmann B, Weis J, Reggiori F, Kurth I, Hubner CA, Dikic I (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522: 354–358 [DOI] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, Johansen T, Deretic V (2015) TRIM‐mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol 210: 973–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Mandell M, Deretic V (2016) Precision Autophagy directed by Receptor‐Regulators. J Cell Sci 125: 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33: 505–516 [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- Lamb CA, Nuhlen S, Judith D, Frith D, Snijders AP, Behrends C, Tooze SA (2016) TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J 35: 281–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157: 1013–1022 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672–676 [DOI] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- Ma Y, Galluzzi L, Zitvogel L, Kroemer G (2013) Autophagy and cellular immune responses. Immunity 39: 211–227 [DOI] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, Yoshimori T (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell MA, Jain A, Arko‐Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Munch J, Kirchhoff F, Simonsen A, Wei Y, Levine B, Johansen T, Deretic V (2014) TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell 30: 394–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjithaya R, Anjard C, Loomis WF, Subramani S (2010) Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol 188: 537–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS (2012) Mycobacterium tuberculosis activates the DNA‐dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11: 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241 [DOI] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto‐Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17: 288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self‐digestion. Nature 451: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- Morelli E, Ginefra P, Mastrodonato V, Beznoussenko GV, Rusten TE, Bilder D, Stenmark H, Mironov AA, Vaccari T (2014) Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila . Autophagy 10: 2251–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munding C, Keller M, Niklaus G, Papin S, Tschopp J, Werner S, Beer HD (2006) The estrogen‐responsive B box protein: a novel enhancer of interleukin‐1beta secretion. Cell Death Differ 13: 1938–1949 [DOI] [PubMed] [Google Scholar]
- Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H, Taneike M, Misaka T, Omiya S, Shah AM, Yamamoto A, Nishida K, Ohsumi Y, Okamoto K, Sakata Y, Otsu K (2015) Bcl‐2‐like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun 6: 7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi IR, Shankar J, Dennis JW (2015) The galectin lattice at a glance. J Cell Sci 128: 2213–2219 [DOI] [PubMed] [Google Scholar]
- Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ (2011) SNARE proteins are required for macroautophagy. Cell 146: 290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G, Ballabio A (2016) TFEB at a glance. J Cell Sci 129: 2475–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AC, Scholefield CL, Kemp AJ, Newman M, McIver EG, Kamal A, Wilkinson S (2012) TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1 bp1/Ndp52 and non‐canonical NF‐kappaB signalling. PLoS One 7: e50672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, Virgin HW, Mizushima N (2013) FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep 14: 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, Luby‐Phelps K, Xavier RJ, Xie Y, Levine B (2011) Image‐based genome‐wide siRNA screen identifies selective autophagy factors. Nature 480: 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkovic M, Jemaiel A, Daste F, Specht CG, Izeddin I, Vorkel D, Verbavatz JM, Darzacq X, Triller A, Pfenninger KH, Tareste D, Jackson CL, Galli T (2014) The SNARE Sec22b has a non‐fusogenic function in plasma membrane expansion. Nat Cell Biol 16: 434–444 [DOI] [PubMed] [Google Scholar]
- Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V (2015) Secretory autophagy. Curr Opin Cell Biol 35: 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC (2013) Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 154: 1285–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, Youle RJ (2014) Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15: 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renna M, Schaffner C, Winslow AR, Menzies FM, Peden AA, Floto RA, Rubinsztein DC (2011) Autophagic substrate clearance requires activity of the syntaxin‐5 SNARE complex. J Cell Sci 124: 469–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A (2001) The tripartite motif family identifies cell compartments. EMBO J 20: 2140–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak‐Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5: ra42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi V, Banfield DK, Vacca M, Dietrich LE, Ungermann C, D'Esposito M, Galli T, Filippini F (2004) Longins and their longin domains: regulated SNAREs and multifunctional SNARE regulators. Trends Biochem Sci 29: 682–688 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Bento CF, Deretic V (2015) Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J Exp Med 212: 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Saegusa K, Sato K, Hara T, Harada A, Sato K (2011) Caenorhabditis elegans SNAP‐29 is required for organellar integrity of the endomembrane system and general exocytosis in intestinal epithelial cells. Mol Biol Cell 22: 2579–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010) The inflammasomes. Cell 140: 821–832 [DOI] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu‐Asab M, Fitzgerald KA, Sher A, Kehrl JH (2012) Activation of autophagy by inflammatory signals limits IL‐1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13: 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica V, Galluzzi L, Bravo‐San Pedro JM, Izzo V, Maiuri MC, Kroemer G (2015) Organelle‐specific initiation of autophagy. Mol Cell 59: 522–539 [DOI] [PubMed] [Google Scholar]
- Sprecher E, Ishida‐Yamamoto A, Mizrahi‐Koren M, Rapaport D, Goldsher D, Indelman M, Topaz O, Chefetz I, Keren H, O'Brien TJ, Bercovich D, Shalev S, Geiger D, Bergman R, Horowitz M, Mandel H (2005) A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet 77: 242–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmaier M, Yang B, Yoo JS, Huang B, Shen M, Yu S, Luo Y, Scheller RH (1998) Three novel proteins of the syntaxin/SNAP‐25 family. J Biol Chem 273: 34171–34179 [DOI] [PubMed] [Google Scholar]
- Su Q, Mochida S, Tian JH, Mehta R, Sheng ZH (2001) SNAP‐29: a general SNARE protein that inhibits SNARE disassembly and is implicated in synaptic transmission. Proc Natl Acad Sci USA 98: 14038–14043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q (2008) Identification of Barkor as a mammalian autophagy‐specific factor for Beclin 1 and class III phosphatidylinositol 3‐kinase. Proc Natl Acad Sci USA 105: 19211–19216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takats S, Nagy P, Varga A, Pircs K, Karpati M, Varga K, Kovacs AL, Hegedus K, Juhasz G (2013) Autophagosomal Syntaxin17‐dependent lysosomal degradation maintains neuronal function in Drosophila . J Cell Biol 201: 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takats S, Pircs K, Nagy P, Varga A, Karpati M, Hegedus K, Kramer H, Kovacs AL, Sass M, Juhasz G (2014) Interaction of the HOPS complex with syntaxin 17 mediates autophagosome clearance in Drosophila . Mol Biol Cell 25: 1338–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele DL, Lipsky PE (1990) Mechanism of L‐leucyl‐L‐leucine methyl ester‐mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci USA 87: 83–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F (2012) Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482: 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboyama K, Koyama‐Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354: 1036–1041 [DOI] [PubMed] [Google Scholar]
- Valley CC, Liu S, Lidke DS, Lidke KA (2015) Sequential superresolution imaging of multiple targets using a single fluorophore. PLoS One 10: e0123941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA (2011) Inflammasome activation and IL‐1beta and IL‐18 processing during infection. Trends Immunol 32: 110–116 [DOI] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Joglekar AP, Williams AL, Hay JC (2000) Subunit structure of a mammalian ER/Golgi SNARE complex. J Biol Chem 275: 39631–39639 [DOI] [PubMed] [Google Scholar]
- Xu H, Mohtashami M, Stewart B, Boulianne G, Trimble WS (2014) Drosophila SNAP‐29 is an essential SNARE that binds multiple proteins involved in membrane traffic. PLoS One 9: e91471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch‐Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL (2008) Mitochondrial autophagy is an HIF‐1‐dependent adaptive metabolic response to hypoxia. J Biol Chem 283: 10892–10903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang M, Schekman R (2013) Cell biology. Unconventional secretion, unconventional solutions. Science 340: 559–561 [DOI] [PubMed] [Google Scholar]
- Zhang M, Kenny S, Ge L, Xu K, Schekman R (2015) Translocation of interleukin‐1beta into a vesicle intermediate in autophagy‐mediated secretion. eLife 4: e11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File