

Abstract
Cells are constantly threatened by multiple sources of genotoxic stress that cause DNA damage. To maintain genome integrity, cells have developed a coordinated signalling network called DNA damage response (DDR). While multiple kinases have been thoroughly studied during DDR activation, the role of protein dephosphorylation in the damage response remains elusive. Here, we show that the phosphatase Cdc14 is essential to fulfil recombinational DNA repair in budding yeast. After DNA double‐strand break (DSB) generation, Cdc14 is transiently released from the nucleolus and activated. In this state, Cdc14 targets the spindle pole body (SPB) component Spc110 to counterbalance its phosphorylation by cyclin‐dependent kinase (Cdk). Alterations in the Cdk/Cdc14‐dependent phosphorylation status of Spc110, or its inactivation during the induction of a DNA lesion, generate abnormal oscillatory SPB movements that disrupt DSB‐SPB interactions. Remarkably, these defects impair DNA repair by homologous recombination indicating that SPB integrity is essential during the repair process. Together, these results show that Cdc14 promotes spindle stability and DSB‐SPB tethering during DNA repair, and imply that metaphase spindle maintenance is a critical feature of the repair process.
Keywords: Cdc14, DNA repair, double‐strand break, homologous recombination, spindle pole body
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Cell Cycle; DNA Replication, Repair & Recombination
Introduction
Maintenance of genomic integrity is an essential part of cellular physiology. Genotoxic insults that induce DNA lesions must be repaired in order to avoid propagation of mutations that can lead to the appearance of a malignant transformation. DNA damage can arise following a wide range of stimuli, including ionizing radiation, ultraviolet radiation, replication stress, chemicals and reactive oxygen species generated during the cell metabolism. Of the various forms of damage that are caused by these mutagens, the most hazardous is arguably the DNA double‐strand break (DSB), generated when the two complementary DNA strands of the double helix are severed simultaneously. The capacity to deal with these DNA lesions is critical for cellular survival and for the maintenance of genome stability, since errors during the repair process might result in mutations, cancer or even cell death. To combat this threat, cells have evolved a series of mechanisms collectively known as DDR for DNA damage response to detect the lesion, signal its presence and promote its repair.
DSBs may be repaired by either non‐homologous end joining (NHEJ) (Burma et al, 2006) or homologous recombination (HR) (Paques & Haber, 1999). Repair by NHEJ is generally considered to be an error‐prone pathway because it requires direct rejoining of the broken DSB ends. By contrast, HR involves a genomic search for sequences to be used as template for repair and is considered an error‐free repair pathway (Branzei & Foiani, 2008; Mathiasen & Lisby, 2014; Ceccaldi et al, 2016). At the DNA level, HR requires 5′‐to‐3′ resection of the DSB ends to generate a single‐stranded DNA tail (ssDNA). This ssDNA is used to stimulate homologous pairing and strand invasion with a DNA template that serves as donor, forming a structure called the displacement loop (D‐loop). The D‐loop can be extended by DNA polymerases, copying the information that might be missing at the break site. In order to resolve this DNA structure, cells can use different strategies: (i) synthesis‐dependent strand annealing (SDSA), by which the invading strand of the DNA can be displaced and re‐annealed to the other broken chromosome end; or (ii) formation of a double Holliday junction (dHJ), whereby the displaced strand of the D‐loop anneals with ssDNA on the other end of the break (second end capture) and the 3′ end primes DNA synthesis.
Many steps of the HR pathway require cyclin‐dependent kinase (Cdk) activity. Early steps are particularly sensitive to Cdk1 inhibition because of its role in DSB‐end resection (Chen et al, 2011). In later steps, Cdk1 is important as it recruits Rad52 to sites of DNA damage (Barlow & Rothstein, 2009) and promotes the correct function of the Srs2 helicase during SDSA (Saponaro et al, 2010). It has also been recently shown that the Cdk controls the decision to use NHEJ or HR depending on the cell cycle stage at which the lesion occurs (Zhang et al, 2009). Consequently, repair by HR is favoured during late S and G2 phases, while NHEJ is preferentially used in G1 (Aylon et al, 2004). Cdk1 function therefore influences the cellular response to DNA damage at multiple steps of the repair process. However, it is currently unknown whether specific phosphatases of Cdk phosphorylation are also involved in the regulation of the DNA damage response.
The dual‐specificity Cdc14 phosphatase was originally identified in Saccharomyces cerevisiae for its role in reversing Cdk1 phosphorylation during mitotic exit (Visintin et al, 1998). Further studies on Cdc14 orthologues from yeast to humans have characterized additional roles for this family of phosphatases in cytokinesis (Meitinger et al, 2010, 2012), chromosome segregation (Clemente‐Blanco et al, 2009; Mocciaro & Schiebel, 2010), transcription (Clemente‐Blanco et al, 2009, 2011; Guillamot et al, 2011), centrosome duplication (Mocciaro & Schiebel, 2010), ciliogenesis (Clement et al, 2011) and in resolving linked DNA intermediates (Blanco et al, 2014; Eissler et al, 2014; Garcia‐Luis et al, 2014). In budding yeast, Cdc14 activity is regulated by dynamic changes in the subcellular localization throughout the cell cycle. Cdc14 is sequestered in the nucleolus until early anaphase, when it migrates to the nucleus and cytoplasm, a process regulated by the FEAR and MEN networks, respectively (Stegmeier et al, 2002). Surprisingly, Clp1/Flp1, the Schizosaccharomyces pombe Cdc14 orthologue, and Cdc14B, its mammalian counterpart, exit the nucleolus during interphase upon DNA replication stress or damage, implicating Cdc14 phosphatases in response to genotoxic insults (Diaz‐Cuervo & Bueno, 2008; Mocciaro & Schiebel, 2010). Despite the evidences of an evolutionary conserved function of Cdc14 in response to DNA damage, there is not a consensus agreement about the molecular function of the phosphatase during DDR activation. Flp1 exclusion from the nucleolus after a DNA replication arrest induced by the addition of hydroxyurea (HU) is crucial to promote a fully checkpoint activation (Diaz‐Cuervo & Bueno, 2008). Similarly, Cdc14B translocation from the nucleolus to the nucleoplasm in response to genotoxic stress is responsible for Plk1 degradation by the ubiquitin ligase APC/CCdh1. This results in the stabilization of the DNA damage checkpoint activator Claspin and the cell cycle inhibitor Wee1, with the subsequent initiation of the G2 checkpoint (Bassermann et al, 2008). Intriguingly, by using both chicken and human cells, Mocciaro et al have shown that Cdc14A/B‐KO mutants arrest efficiently in G2 with normal levels of Chk1 and Chk2 activation in response to irradiation. However, γ‐H2A.X foci and DSBs persist longer in Cdc14A‐KO or Cdc14B‐KO cells than controls, suggesting that both Cdc14 phosphatases are required for efficient DNA repair (Mocciaro et al, 2010). Supporting this work, another study has shown that Cdc14b‐deficient mouse embryonic fibroblasts cells accumulate more endogenous DNA damage, and more cells enter senescence in response to exogenous DNA damage, suggesting that the function of this phosphatase is restricted to enhance efficient DNA damage repair (Wei et al, 2011).
Here, we show that in budding yeast, Cdc14 is transiently released from the nucleolus in response to genotoxic stress to enhance recombinational repair. This function is attained by stimulating the recruitment of the broken DNA to the vicinity of SPBs (spindle pole bodies) in a process that requires the SPB component Mps3 and a competent DNA damage checkpoint activation. Inactivation of Cdc14 during the induction of the DNA lesion causes continuous misalignment of the metaphase spindle, increases oscillatory SPB movements and impairs DSB‐SPB tethering, suggesting a role of the phosphatase in DNA repair by promoting spindle stability. In a screen looking for Cdc14 substrates at the SPBs during the induction of a DNA lesion, we identified Spc110, the intra‐nuclear receptor for the γ‐tubulin complex. Remarkably, the steady‐state phosphorylation of Spc110 during the induction of a DNA lesion is essential to promote DSB‐SPB interaction and DNA repair by HR. Taken together, our results point to the function of Cdc14 in DNA repair by promoting SPB stabilization and DSB‐SPB interaction and suggest that the relocation of damage sites to the SPBs proximities stimulates homologous recombination in a naturally occurring repair process that minimizes genome instability.
Results
Cells lacking Cdc14 activity exposed to genotoxic stress are compromised in cell viability but retain DNA damage checkpoint proficiency
As a first approach to analyse the role of Cdc14 in the DNA damage response in S. cerevisiae, we tested the effect of the DNA‐damaging agent methyl methanesulphonate (MMS) on the growth of cells carrying the temperature‐sensitive allele cdc14‐1 (Fig 1A). Because Cdc14 is an essential gene in S. cerevisiae, we carried out tests on a range of temperatures. At 30°C, both the cdc14‐1 mutant and its isogenic wild‐type strain grew in the absence of DNA damage. On the contrary, a severe defect in cell growth was observed when the mutant was plated on MMS, indicating that Cdc14 function is important when cells are exposed to DNA damage (Fig 1A). To further characterize the essential role of Cdc14 when grown on different genotoxic compounds, we plated both wild‐type and cdc14‐1 backgrounds in the presence of the UV‐mimic 4‐nitroquinoline‐1‐oxide (4NQO), the ribonucleotide reductase inhibitor hydroxyurea (HU), the radiomimetic drug phleomycin and the microtubule‐destabilizing drug benomyl at the semipermissive temperature of 30°C (Fig 1B). Remarkably, cdc14‐1 cells presented a substantial sensitivity in all media tested, extending the essential role of this phosphatase to a great variety of DNA damage stresses.
Figure 1. Cdc14 is required for intra‐chromosomal DNA repair by HR.
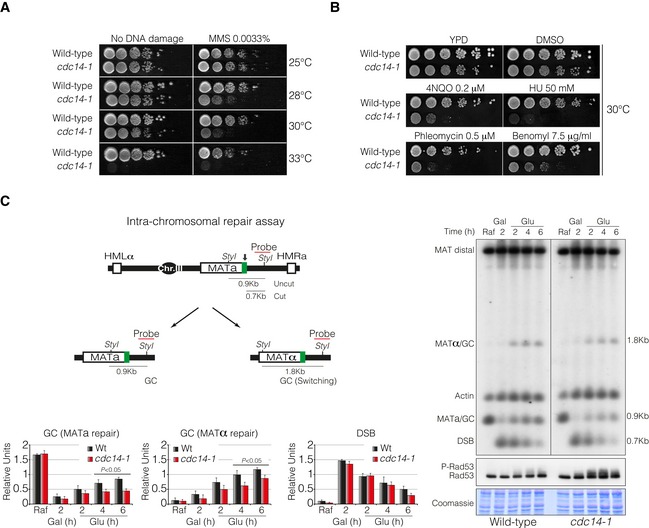
- Tenfold serial dilutions from overnight cultures of wild‐type and cdc14‐1 cells dropped and grown on solid rich media or media containing MMS at 25, 28, 30 or 33°C. Note that cdc14‐1 cells exhibit growth sensitivity to MMS at 28 and 30°C compared to wild‐type cells.
- Tenfold serial dilution from mid‐log phase cultures of wild‐type and cdc14‐1 cells grown on solid rich media or media containing mock DMSO (as non‐treated control), 4NQO, HU, phleomycin and benomyl at 30°C. Note that cdc14‐1 cells present a marked sensitivity to all DNA damage agents tested.
- Left panel: Schematic representation showing relevant genomic structure of the strain used to assess intra‐chromosomal repair. The location of a MAT‐specific probe and the restriction endonuclease cleavage sites used for Southern blot analysis to detect repair product formation are indicated. Arrow depicts the localization of the double‐strand break. Right panel: Physical analysis of intra‐chromosomal repair in wild‐type and cdc14‐1 cultures at the semipermissive temperature. After DSB formation by the expression of the HO, glucose was added to repress it, thus allowing repair with HM donor sequences. Genomic DNA was digested with StyI, separated on agarose gel and blotted. Blots were hybridized with a probe corresponding to the MATa‐distal sequence. A second probe for the actin gene was used to control the amount of genomic DNA loaded at each time point. Immunoblot analysis of Rad53 during mating‐type switching experiments is shown. Coomassie blue staining is depicted as a loading control. Graphs show quantification of gene conversion (GC) leading to the re‐establishment of MATa or switching to MATα and DSB formation. The data were normalized with the actin as a loading control. Graphs show the mean ± SD from three independent experiments. Replicates were averaged and statistical significance of differences assessed by a two‐tailed unpaired Student's t‐test.
Cells containing a defect either in DNA damage checkpoint or in DSB repair fail to proliferate after induction of DNA damage. To test for a putative function of Cdc14 in DNA damage checkpoint activation, we took advantage of strains containing the homothallic endonuclease HO gene under the control of the inducible galactose promoter GAL1. Addition of galactose to the media induces expression of the nuclease and the subsequent production of a single DSB in the genome at the MAT locus on chromosome III. We generated a DNA break by continuous expression of the HO endonuclease in both wild‐type and cdc14‐1 strains and tested their efficiency to block in G2/M due to the DNA damage checkpoint activation. Southern blots of both wild‐type and cdc14‐1 showed the same kinetics of DSB formation and stabilization during the entire experiment (Fig EV1A). After 8 h in the presence of galactose, both cultures presented more than 90% of mono‐nucleated G2/M arrested cells, indicating that Cdc14 is not required for activation of the DNA damage checkpoint pathway induced by a single DSB. This finding suggests that the sensitivity to genotoxic compounds observed in cdc14‐1 mutants could be a consequence of defective repair rather than a checkpoint deficiency.
Figure EV1. Cdc14 is required for recombinational DNA repair.
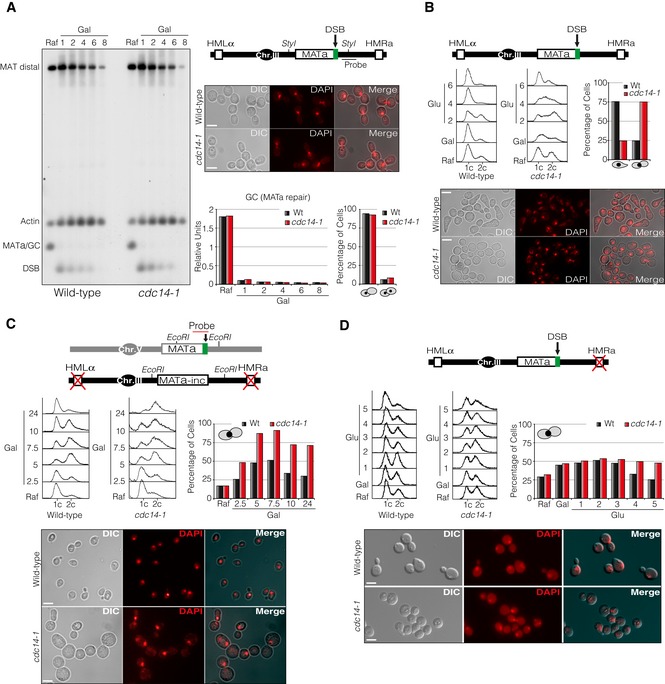
- Constitutive expression of HO produces a constant DSB at the MAT locus in wild‐type and cdc14‐1 cultures at semipermissive temperature. Southern blot of wild‐type and cdc14‐1 cells showing the efficiency and stability of the DSB. A diagram with the genomic information, the restriction sites used and the location of the MAT‐distal probe is shown. Micrographs depict cultures blocked in G2/M 8 h after DSB induction. Scale bar: 5 μm. Graphs show quantification of gene conversion (GC) leading to the re‐establishment of MATa and the percentage of cells blocked in G2/M after 8 h from the galactose addition.
- Analysis of mating‐type switching through formation of G1 shmoo cells in wild‐type and cdc14‐1 cultures at semipermissive temperature. Glucose was added to the cultures after DSB formation to repress HO, thus allowing repair with HM donor sequences (top diagram). FACS analysis was performed on cells collected at each time point indicated. Graph shows percentage of cells blocked at G1 (shmoos) or mono‐nucleated arrested G2/M (by DAPI staining) 4 h after glucose addition. Micrographs show cultures 4 h after incubation with glucose. Scale bar: 5 μm.
- Wild‐type and cdc14‐1 mutant strains carrying the ectopic gene conversion assay depicted were grown overnight and exposed to expression of HO through addition of galactose, thus producing a DSB on chromosome V (top diagram). Samples were taken at 0, 2.5, 5, 7.5, 10 and 24 h. FACS analysis of each time point is shown. Graphs show percentage of mono‐nucleated G2/M cells of each sample. Micrographs show cultures 24 h after DSB induction. Scale bar: 5 μm.
- Wild‐type and cdc14‐1 mutant strains harbouring the repair pathway choice assay described (top diagram) were grown overnight and exposed to expression of HO through addition of galactose for 1.5 h. Samples were taken before and at 1, 2, 3, 4 and 5 h after supplementing with glucose. FACS analysis of each time point is shown. Quantification of mono‐nucleated G2/M arrested cells of wild‐type and cdc14‐1 cells during the time course is shown. Micrographs show cultures 5 h after glucose addition. Scale bar: 5 μm.
Cdc14 is required for intra‐ and inter‐chromosomal DNA repair by homologous recombination but is not involved in DSB repair pathway choice
To determine whether Cdc14 was directly implicated in DNA repair and to investigate which specific process in the DSB response pathway was controlling, we examined DNA repair by using strains harbouring the HO‐induced DSB system in three different conditions: intra‐chromosomal DNA repair, inter‐chromosomal gene conversion and repair pathway choice. It has been proposed that intra‐chromosomal DNA repair at the MAT locus occurs mainly by SDSA (reviewed in Haber, 2012). To check whether Cdc14 plays a role in this repair pathway, we induced the DSB formation by supplementing the media with galactose for 2 h. HO was subsequently repressed by glucose addition, and the kinetics of the repair process by HR with the donor sequence HMLα or HMRa was followed (Fig 1C, diagram). Physical analysis by Southern blot showed that cdc14‐1 cells repaired the DSB slower and less efficiently than wild‐type cells when occurred by restoration of the original MATa or switched to MATα allele (Fig 1C). We also observed that the checkpoint effector kinase Rad53 was significantly phosphorylated during repair in cdc14‐1 mutants (Fig 1C). This was not the case for wild‐type cells where the DNA damage checkpoint kinases are not active during intra‐chromosomal repair (Pellicioli et al, 2001; Kim et al, 2011). Supporting this data, wild‐type controls presented 75% of the cells blocked in G1 with shmoo morphology by 4 h of glucose addition (Fig EV1B). Shmoo formation is a good indicator of successful repair as it requires the appearance of cells with the opposite mating type in the culture. By contrast, cdc14‐1 mutant cells presented only 25% of shmoo cells, while an increased number of mono‐nucleated G2/M arrested cells were observed (Fig EV1B). These results demonstrate that SDSA‐mediated DSB repair is defective when Cdc14 function is impaired.
In order to assess whether Cdc14 was also required for gene conversion events when the donor sequence is located on a different chromosome, that is inter‐chromosomal repair, we used a genetic background containing a MATa sequence in chromosome V that can be cleaved by HO and repaired with an uncleavable MATa‐inc sequence located on chromosome III (Ira et al, 2003). This strain lacks the donor HM loci and the MATa‐inc mutation renders the repair product insensitive to subsequent HO cleavage (Fig 2A, diagram). By using this system, a single reparable DSB by gene conversion is generated which can occur either with or without an accompanying crossover (Mazon & Symington, 2013). As the restriction fragments have an altered size when a crossover is formed, one can detect these events during the repair process. Gene conversion with no associated crossovers was the predominant form of repair in both wild‐type and cdc14‐1 cells. However, wild‐type cells repaired the break with faster kinetics and more efficiently than cdc14‐1 mutants (Fig 2A). We detected no significant differences in the proportion of gene conversion associated with crossing‐over between both strains among cells that repaired the DNA lesion (Fig 2A). Accordingly, while wild‐type cells repaired the DNA break and re‐entered the cell cycle 10 h after the induction of the break, cdc14‐1 mutants presented a significant delay in cell cycle progression with up to 75% of mono‐nucleated dumbbell cells (Fig EV1C). Confirming our previous observations, we found high levels of Rad53 phosphorylation in both wild‐type and cdc14‐1 strains, ruling out a function of the phosphatase in the DNA damage checkpoint activation (Fig 2A). These data confirm that Cdc14 activity is also required to promote ectopic recombination with homologous sequences.
Figure 2. Cdc14 is required for inter‐chromosomal DNA repair by HR but is not involved in DSB repair pathway choice.
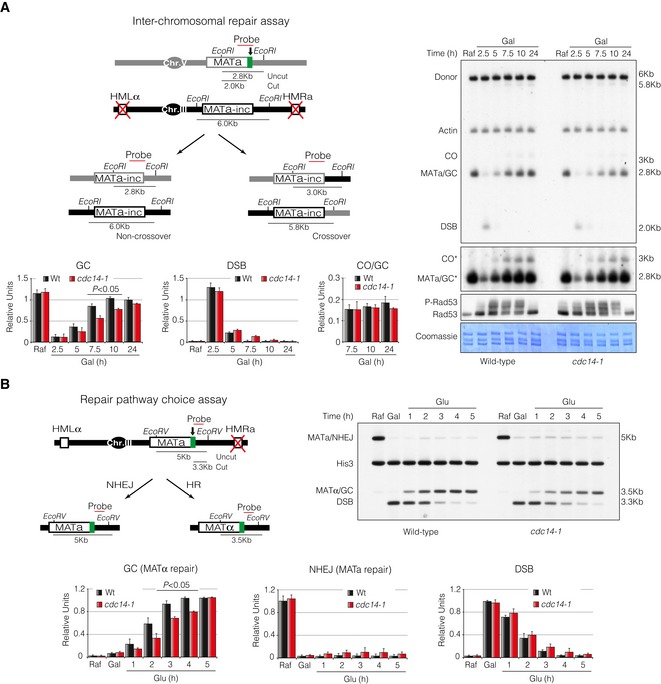
- Left panel: Schematic diagram displaying relevant genomic structure of the strains used to measure inter‐chromosomal DNA repair efficiency. The location of a MAT‐specific probe and the restriction endonuclease cleavage sites used for Southern blot analysis to detect repair product formation are indicated. Note that crossover and non‐crossover products have different restriction fragment sizes that can be differentiated in a Southern blot assay. Arrow depicts the localization of the double‐strand break. Right panel: Physical analysis of wild‐type and cdc14‐1 mutant strains carrying the inter‐chromosomal gene conversion assay. Cells were grown overnight before adding galactose at semipermissive temperature to induce HO expression, thus producing a DSB on chromosome V. Samples were taken at different time points, and genomic DNA was extracted, digested with EcoRI and analysed by Southern blot. Blots were hybridized with a MATa‐only DNA sequence, and an actin probe was used as a loading control. Asterisk denotes an overexposed film to visualize crossover formation. Immunoblot of Rad53 was performed as previously described. Coomassie staining is shown as a control for loading. Graphs show the mean ± SD of gene conversion, DSB induction and crossover versus non‐crossover ratio from three independent experiments. All data were normalized using actin as a loading control. Replicates were averaged and statistical significance of differences assessed by a two‐tailed unpaired Student's t‐test.
- Left panel: Schematic representation depicting the genomic structure of the strains used to evaluate repair pathway choice. The location of a MAT‐specific probe and the restriction endonuclease cleavage sites used for Southern blot analysis to detect repair product formation are indicated. Note that the use of NHEJ or HR generates different product that can be recognized in Southern blots by the different disposition of the restriction sites for the endonucleases used. Arrow depicts the localization of the double‐strand break. Right panel: Physical analysis of wild‐type and cdc14‐1 cells harbouring the repair pathway choice assay. Cells were grown overnight and HO induction was attained through addition of galactose for 1.5 h at semipermissive temperature. After induction of the HO, glucose was added to repress the HO expression and samples were taken to analyse repair efficiency. DNA was extracted, digested with EcoRV, separated on agarose gels and blotted. Blots were hybridized with a probe corresponding to the MATa‐only DNA sequence. A second probe to the HIS3 gene was used to control the amount of genomic DNA loaded at each time point. Graphs show quantification of mating‐type switching by gene conversion (HR), restoration of MATa (NHEJ) and the kinetics of the DSB induction. All data were normalized with the HIS3 gene. Graphs show the mean ± SD from three independent experiments. Replicates were averaged and statistical significance of differences assessed by a two‐tailed unpaired Student's t‐test.
If Cdc14 function is required at the initial steps during repair, this might translate into alteration of DSB repair pathway choice in cdc14‐1 mutants. To address this subject, we used a strain where repair of the HO‐induced break by HR is obligatorily associated with mating‐type switching from MATa to MATα, while repair by NHEJ retains the mating type as MATa (Fig 2B, diagram). We expressed the HO endonuclease for 90 min in logarithmically growing cells and observed the dynamics of the repair process 5 h after repressing the HO expression by glucose addition (Fig 2B). As before, cdc14‐1 cells presented a delay in the appearance of the switched product when compared to wild‐type cells, indicating a failure in DNA repair by gene conversion. Supporting this observation, an extended G2/M arrest with an increased number in mono‐nucleated cells was detected in cdc14‐1 cultures (Fig EV1D). We could not detect a significant accumulation of NHEJ repair product when comparing both strains suggesting that Cdc14 does not contribute to DSB repair pathway choice but rather affects downstream events during recombinational repair.
Cdc14 is released from the nucleolus in response to DNA damage
During interphase, Cdc14 is kept inactive by its binding to the nucleolar component of the RENT complex Net1. Upon entry into anaphase, two regulatory pathways known as FEAR and MEN promote Net1 phosphorylation and in turn Cdc14 release and activation (Azzam et al, 2004). Our previous results demonstrated that Cdc14 is required for DSB repair by HR, implying that the phosphatase must be activated in response to DNA damage. We therefore sought to investigate whether the generation of DNA damage promotes nucleolar Cdc14 exclusion and activation. To confirm this, asynchronous cultures were treated with the DNA damage agent phleomycin and samples were taken at intervals to check for the localization pattern of Cdc14‐YFP. After treating the cells with the drug, in addition to the strong nucleolar localization, a fraction of the protein was transiently detected throughout the nucleoplasm (Fig 3A). This partial Cdc14 release was not sufficient to promote mitotic entry, as most cells remained blocked in metaphase with short spindles as denoted by the presence of two Cnm67‐RFP foci (used as a SPBs marker) located at the mother cell and aligned to the axial plane of the cell (Fig 3A). Moreover, Rad53 was phosphorylated throughout the entire experiment denoting a fully active DNA damage checkpoint (Fig 3A). Supporting the microscope observation of a nucleolar Cdc14 release, we observed that Net1 became phosphorylated in response to DNA damage, a prerequisite that facilitates the phosphatase shuttling and activation (Fig 3A). Surprisingly, the pattern of Net1 phosphorylation bands in response to DNA damage resembled those observed in non‐DNA damage synchronized cultures at the anaphase stage when Cdc14 is fully released from the nucleolus by the MEN pathway (Fig 3B). This suggests that Net1 phosphorylation is not sufficient to sustain a fully Cdc14 nucleolar release during DNA damage response and that additional mechanisms might be controlling Cdc14 localization throughout the DDR execution.
Figure 3. Cdc14 is released from the nucleolus in response to DNA damage.
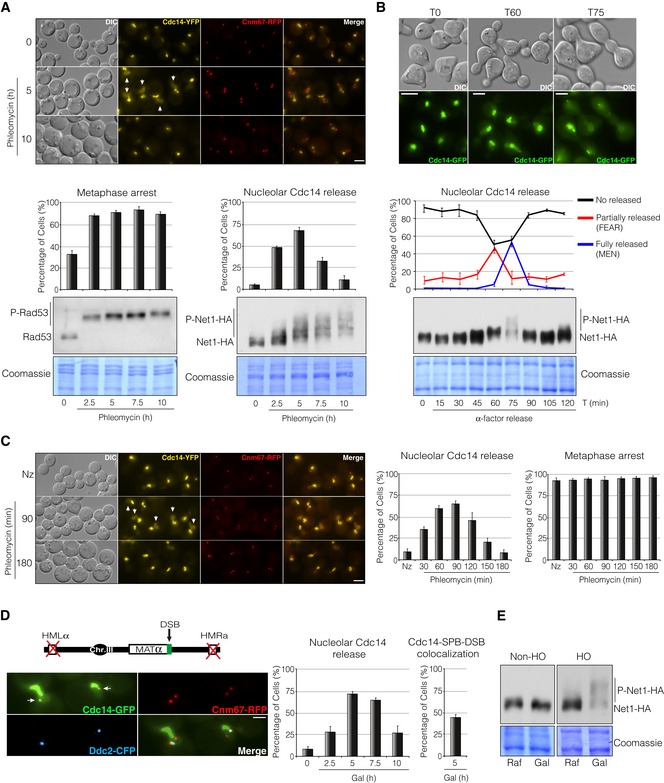
- Live‐cell imaging of Cdc14‐YFP and Cnm67‐RFP asynchronous cells treated with the DNA damage agent phleomycin. Cells were grown overnight and phleomycin was added to a final concentration of 1 μM. Samples were taken at different intervals to determine Cdc14 localization. Cnm67 was used as a SPB reporter to determine spindle length. Scale bar: 3 μm. Graphs represent the average percentage ± SD from three independent experiments of cells arrested in metaphase (left graph) and with Cdc14 signal at the nucleoplasm (right graph). Western blot analysis to analyse Rad53 and Net1 phosphorylation levels under these conditions is shown. Note that Rad53 is rapidly phosphorylated in response to phleomycin and maintained throughout the entire experiment. Detection of Net1 phosphorylation levels was achieved by using Phos‐tag polyacrylamide gels. Coomassie staining is shown as a loading control.
- Cell cycle profile of Cdc14 localization and Net1 phosphorylation in the absence of DNA damage. A strain containing Cdc14‐GFP and Net1‐HA was blocked in G1 by using the pheromone alpha‐factor and released into the cell cycle. Samples were taken at different time points to analyse Cdc14 spatial localization and Net1 phosphorylation levels. A representative picture of non‐released (T0), partially released (T60) and fully released (T75) Cdc14 from the nucleolus is shown. Scale bar: 3 μm. Graph represents the percentage ± SD from three independent experiments of cells with no released, partially released (FEAR) and totally released (MEN) Cdc14 from the nucleolus along the cell cycle. Western blot analysis showing Net1 phosphorylation through the cell cycle is included. The separation of the phospho‐bands was achieved by using Phos‐tag polyacrylamide gels as in (A). Coomassie staining is depicted as a loading control.
- Cells expressing Cdc14‐YFP and Cnm67‐RFP were grown overnight and blocked in G2/M by using nocodazole prior to phleomycin treatment (1 μM). Samples were taken every 30 min to follow Cdc14 re‐localization. The efficiency of the arrest was determined as in (A). Scale bar: 3 μm. Graphs represent the percentage ± SD of cells with Cdc14 nucleolar exclusion (left graph) and cells arrested in metaphase (right graph) from three independent experiments.
- Live‐cell imaging of Cdc14 localization in the presence of a non‐reparable DSB at the MAT locus. Cells were grown overnight and the HO endonuclease was expressed by adding galactose to the media, thus producing a DSB on chromosome III. Cdc14, the SPB component Cnm67 and the DNA damage checkpoint protein Ddc2 (used as a DSB reporter) were labelled with the green, red and cyan fluorescent proteins, respectively. Micrographs display the maximum projection of nine planes showing Cdc14 co‐localizing with the DSB and the SPBs in response to DNA damage (arrows). Scale bar: 3 μm. Graphs represent the percentage ± SD from three independents experiment of cells with nucleoplasmic Cdc14 along the HO endonuclease induction (left graph) and the percentage of Cdc14‐SPB‐DSB co‐localization after 5 h in galactose (right graph).
- Western blot showing Net1‐6HA phosphorylation state before and after the generation of a DSB induced by the HO expression. A strain lacking the HO endonuclease under the galactose promoter was used as undamaged control. The phospho‐bands were resolved by using Phos‐tag polyacrylamide gels as in (A and B). Coomassie staining is depicted as a loading control.
To rule out an indirect cell cycle effect in the Cdc14 nucleolus/nucleoplasm shuttling, we induced DNA damage by adding phleomycin to previously nocodazole‐arrested metaphase cells. As before, Cdc14‐YFP was transiently observed into the nucleoplasm upon induction of DNA damage (Fig 3C). Again, nucleolar Cdc14 release was not enough to override the G2/M arrest as denoted by the presence of short metaphase spindles along the entire experiment (Fig 3C).
Next, we checked whether a single DSB generated by expressing the HO endonuclease was enough to promote Cdc14 relocation from the nucleolus. To prevent repair of the break, we used a strain in which both HM loci were deleted, thus maximizing DSB persistence. As expected, upon HO expression a fraction of Cdc14‐GFP was transiently released from the nucleolus into the nucleoplasm as previously observed (Fig 3D). A comparable localization was observed when using a Cdc14 tagged with the YFP epitope (Fig EV2A). Surprisingly, a distinct Cdc14 focus was also noticed in 40% of the cells overlapping with the Ddc2‐CFP signal (used as a DSB reporter) and with the SPB component Cnm67‐RFP at 5 h after HO induction (Figs 3D and EV2A). This confirms that a single DSB induced by the HO endonuclease is sufficient to promote Cdc14 nucleolar/nucleoplasm re‐localization. Accordingly, Net1 phosphorylation levels were increased during the induction of the endonuclease HO by galactose addition (Fig 3E). Galactose treatment in a non‐HO background did not increase Net1 phosphorylation levels, demonstrating that Net1 phospho‐shift was dependent on the formation of a DNA lesion (Fig 3E). These results demonstrate that Cdc14 is partially excluded from the nucleolus and thus activated during the execution of the DDR.
Figure EV2. Single or multiple DSBs generated outside the MAT locus are recruited to the SPBs.
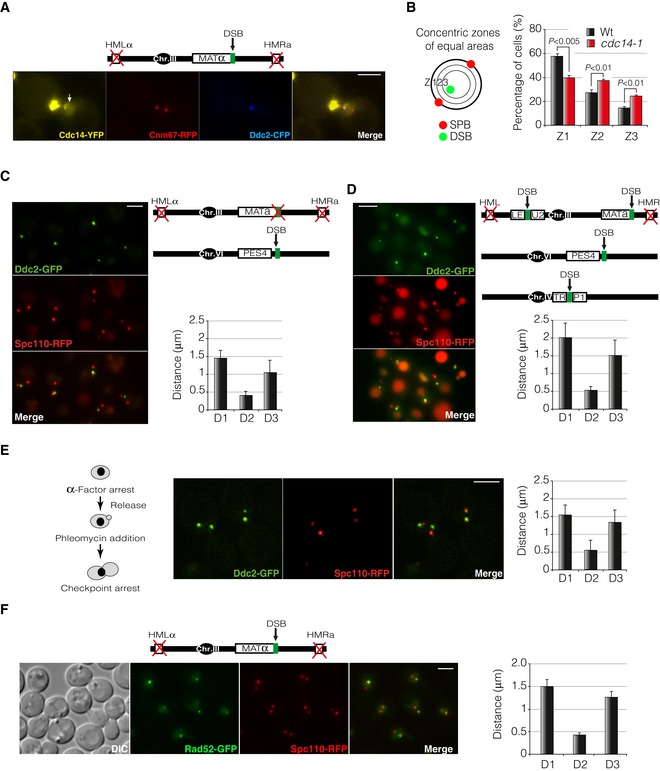
- Cdc14 is released from the nucleolus into the nucleoplasm in response to a single DSB induced by the HO endonuclease. A strain carrying Cdc14‐YFP was grown in raffinose before inducing a non‐reparable DSB by expressing the HO endonuclease. Cnm67‐RFP and Ddc2‐CFP were used as SPB and DSB reporters, respectively. Arrow depicts Cdc14 signal at the nucleoplasm. Scale bar: 3 μm.
- Schematic representation of the approach used to analyse DSB recruitment to the nuclear rim. The Ddc2‐GFP foci proximity to a circumference connecting both SPBs was calculated by determining the position of the DSB into three concentric zones of equal area. Graphs represent the percentage of Ddc2 foci falling into each zone. Values represent the mean ± SD of three independent experiments. At least 100 cells per experiment were scored. P‐values were calculated using a two‐tailed unpaired Student's t‐test.
- Diagram depicting the genomic background used to induce a single DSB outside the MAT locus. Spc110‐RFP and Ddc2‐GFP were used as SPB and DSB markers, respectively. D1: inter‐SPB distance; D2: DSB distance to proximal SPB; D3: DSB distance to distal SPB. Graph represents the mean ± SD of D1, D2 and D3 distances of at least 100 cells scored using the maximum projection of three z‐planes. A representative picture is shown. Scale bar: 3 μm.
- Diagram showing the genomic background used to induce multiple DSBs. Spc110‐RFP and Ddc2‐GFP were used as SPB and DSB markers, respectively. D1: inter‐SPB distance; D2: DSB distance to proximal SPB; D3: DSB distance to distal SPB. Graph represents the mean ± SD of D1, D2 and D3 distances of at least 100 cells scored using the maximum projection of three z‐planes. A representative picture is shown. Scale bar: 3 μm.
- Diagram illustrating the workflow used to analyse DSB‐SPB recruitment of DNA lesions induced by phleomycin treatment. Phleomycin was added to a final concentration of 1 μM. Spc110‐RFP and Ddc2‐GFP were used as SPB and DSB reporters, respectively. D1: inter‐SPB distance; D2: DSB distance to proximal SPB; D3: DSB distance to distal SPB. Graph represents the mean ± SD of D1, D2 and D3 distances of at least 100 cells scored using the maximum projection of seven z‐planes. A representative picture is shown. Scale bar: 3 μm.
- Rad52 foci are recruited to the SPBs in response to a non‐reparable DSB generated by expressing the HO endonuclease. Rad52‐GFP and Spc110‐RFP were used as DSB and SPB reporters, respectively. Graph represents the mean ± SD of D1, D2 and D3 distances of at least 100 cells scored using the maximum projection of nine z‐planes. Scale bar: 3 μm.
Cdc14 activity is necessary to promote DSB‐SPB recruitment in response to DNA damage
It has been proposed that recruitment of DSBs to the nuclear periphery is a physiological mechanism that enhances legitimate DNA repair by HR. Unrepairable or slowly repaired DSBs re‐localize to the nuclear periphery through their interaction with the nuclear pore component Nup84 (Nagai et al, 2008) or the nuclear envelope protein Mps3 (Oza & Peterson, 2010; Horigome et al, 2014). To check whether cells lacking Cdc14 activity were impaired in DSB recruitment to the nuclear periphery, we determined the Ddc2 foci proximity to a circumference connecting both SPBs after the induction of a non‐reparable HO endonuclease break in wild‐type and cdc14‐1 mutant strains (Fig EV2B). We scored the presence of DSBs in three concentric zones of equal area as described in Horigome et al (2014). While a wild‐type strain expressing the HO presented 60% of cells with Ddc2‐GFP overlapping with the outermost rim (Zone 1), only 40% of cdc14‐1 cells showed Ddc2‐GFP foci at this position, indicating that Cdc14 activity is required to promote recruitment of DNA lesions to the nuclear periphery. As mentioned before, under this experimental condition most of the cells presented a constant recruitment of Ddc2‐GFP to the nuclear rim specifically at the proximities of the SPBs (using Cnm67 as SPB marker) (Fig 4A). To confirm DSB‐SPB association and its dependency of Cdc14 activity, we measured three independent parameters: (i) the inter‐SPB distance; (ii) the distance between the Ddc2 focus and the proximal SPB; and (iii) the distance between the Ddc2 focus and the distal SPB (herein referred to as D1, D2 and D3 distances, respectively) (Fig 4A). As predicted, in wild‐type cells the D2 distance was lower than in cdc14‐1 cells (0.46 μm versus 0.74 μm), suggesting an active Cdc14‐dependent DSB‐SPB recruitment upon DNA damage (Fig 4A). To confirm this observation, we evaluated the dynamics of DSB‐SPB interaction by time‐lapse experiments in living cells. As before, wild‐type cells presented a low and constant D2 distance over the time, indicating a stable DSB‐SPB tethering. Inversely, cdc14‐1 mutants showed an increase in D2 coupled with a decrease in D3 distance, denoting a weaker DSB‐SPB affinity (Fig 4B and Movie EV1). DSB‐SPB association is not exclusive of DSBs produced at the MAT locus, since a DNA break generated near the PES4 locus (chromosome VI) or the induction of multiple HO cleavage sites located at MAT, LEU2, PES4 and TRP1 loci (chromosomes III, VI and IV respectively) was also recruited to the SPBs with the same efficiency (Fig EV2C and D). Moreover, DSB‐SPB recruitment is not exclusive of DNA breaks produced by the HO endonuclease as DNA lesions generated by the genotoxic agent phleomycin were as well tethered to the SPBs (Fig EV2E). To confirm that Ddc2‐GFP is a suitable reporter to analyse DSB‐SPB recruitment, we validated D1, D2 and D3 distances in a wild‐type strain using Rad52‐GFP as DSB marker. The same D1, D2 and D3 distributions were obtained as before, confirming the reproducibility of the results (Fig EV2F). Additionally, to further verify the accuracy of DSB‐SPB recruitment, we used a strain containing a unique I‐SceI cut‐site marked by a tandem array of TetO on chromosome V. By expressing a TetI‐RFP fusion protein, the localization of the DSB appeared as a single red focus (Lisby et al, 2003). Additionally, Rad52 was labelled with the cyan fluoresce protein (CFP) in order to confirm previous measurements and Cnm67‐GFP was used as a DSB reporter. We compared D1, D2 and D3 profiles in wild‐type and cdc14‐1 mutant cells. No significant differences were obtained for the three parameters between both backgrounds in undamaged conditions. However, 2.5 h after expressing the endonuclease, a reduction in D2 distance was observed in wild‐type cells for both TetI‐RFP and Rad52‐CFP signals (Fig 4C). As expected and confirming previous observation, no reduction in D2 distance was observed in cells lacking Cdc14 activity for both TetI‐RFP and Rad52‐CFP markers (Fig 4C). This result confirms that DSBs are preferentially bound to the SPB proximities and that Cdc14 activity is essential to encourage DSB‐SPB tethering.
Figure 4. Cdc14 stimulates DSBs recruitment to the SPBs in response to DNA damage.
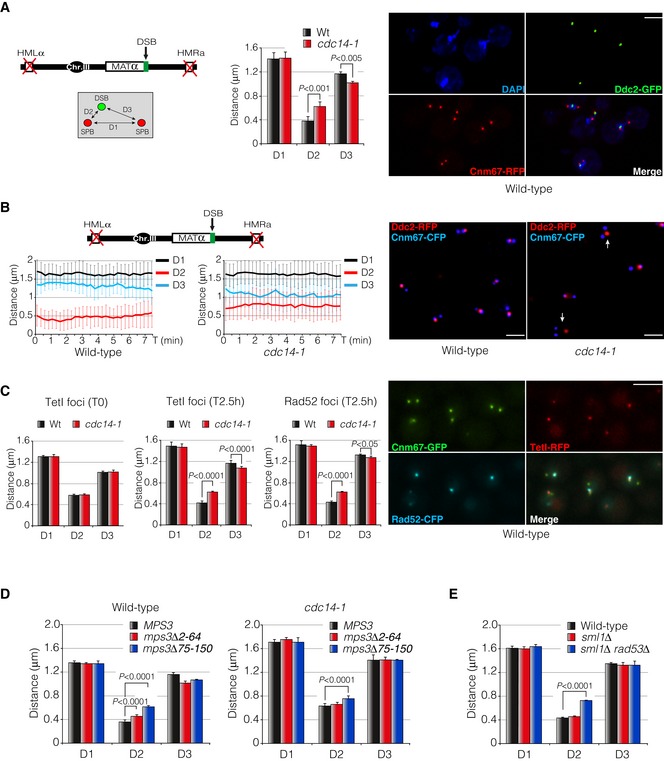
- Diagram showing the genomic background and the approximation used to measure DSB‐SPB interaction. Cnm67‐RFP and Ddc2‐GFP were used as SPB and DSB markers, respectively. D1: inter‐SPB distance; D2: DSB distance to proximal SPB; D3: DSB distance to distal SPB. Graph represents the mean ± SD of D1, D2 and D3 distances from three independent experiments. At least 100 cells per experiment were scored using the maximum projection of three z‐planes. P‐values were calculated using a two‐tailed unpaired Student's t‐test. A representative picture of a wild‐type strain depicting DSB‐SPB interaction is shown. Scale bar: 3 μm.
- Time‐lapse experiments in living cells showing the efficiency of DSB‐SPB interaction upon induction of a single DSB. Cnm67‐CFP and Ddc2‐RFP were used as SPB and DSB markers, respectively. D1, D2 and D3 distances in wild‐type and cdc14‐1 cells were tracked using the maximum projection of three z‐planes images at 15‐s intervals over period of 7.5 min. A total of 32 time‐lapses for each strain were measured. Graphs represent the average distance ± SD. A representative picture is shown. Examples of cells defective in DSB‐SPB interaction are marked with arrows. Scale bar: 3 μm.
- An RFP‐marked I‐SceI recognition site is re‐localized to the SPBs after expressing the endonuclease. Cells harbouring a TetO array adjacent to the I‐SceI site and a plasmid containing the endonuclease under the control of the galactose promoter were grown overnight in SC‐Ade prior to galactose induction. To check reproducibility with previous results, a Rad52‐CFP was used. Cnm67 was labelled with the GFP to be used as a SPB reporter. D1, D2 and D3 distances were measured in wild‐type and cdc14‐1 mutants before and after endonuclease expression by using both TetI‐RFP and Rad52‐CFP foci using the maximum projection of nine z‐planes images. Graphs represent the average ± SD of three independent experiments. P‐values were calculated using a two‐tailed unpaired Student's t‐test. A representative picture of a wild‐type strain depicting TetI‐Rad52‐SPB interaction is shown. Scale bar: 3 μm.
- DSB‐SPB tethering requires the N‐terminus domain of Mps3. Cells expressing a wild‐type Mps3, a mps3Δ2‐64 and a mps3Δ75–150 fused to the RFP as the sole source of the protein were scored for their ability to enhance DSB‐SPB tethering in both wild‐type and cdc14‐1 backgrounds. Ddc2‐YFP was used as a DSB reporter. Measurements were carried out using a maximum projection of nine z‐planes images. Graphs represent the average distribution ± SD of D1, D2 and D3 of three independent experiments. At least 100 cells for each experiment were scored. P‐values were calculated using a two‐tailed unpaired Student's t‐test.
- DSB‐SPB interaction involves a proficient DNA damage checkpoint activation. D1, D2 and D3 distances were scored in wild‐type, sml1Δ and sml1Δ rad53Δ backgrounds using Ddc2‐GFP and Spc110‐RFP as DSB and SPB markers, respectively. Measurements were performed by using a maximum projection of nine z‐planes images. Graphs denote the average distribution ± SD of D1, D2 and D3 from three independent experiments where at least 100 cells for each sample were scored. P‐values were calculated using a two‐tailed unpaired Student's t‐test.
The greater DSB‐SPB distance observed in cdc14‐1 cells may also be explained by a change in the chromatin compaction. To rule out this possibility, we checked for defects in DNA condensation in the absence of Cdc14 during the DDR activation. This was performed by measuring the kinetochore–SPB distance (using Nuf2‐CFP and Ddc2‐RFP as kinetochore and DSB reporters, respectively) after induction of a single DSB by the HO endonuclease (Fig EV3A). No differences in kinetochore–DSB length were observed when comparing a wild‐type strain with a cdc14‐1 mutant indicating that chromatin compaction is not affected when Cdc14 function is impaired during the DNA damage response. Lack of DSB‐SPB interaction in the absence of Cdc14 might well be explained by a deficiency in generating pulling forces throughout kinetochore microtubules emanating from the SPBs. To address this, we checked for the relative localization between kinetochores and SPBs in response to DNA damage. After generating a DSB by inducing the HO expression, the kinetochore reporter Nuf2‐GFP was placed along the two SPBs (Fig EV3B and Movie EV2). However, no significant differences were observed in the distribution of Nuf2 between wild‐type and cdc14‐1 cells. This demonstrates that Cdc14 is not involved in pulling kinetochores regions close to the SPBs during the DNA damage response.
Figure EV3. Chromatin compaction or kinetochore closeness to SPBs is not affected in cells lacking Cdc14 activity in response to a DSB.
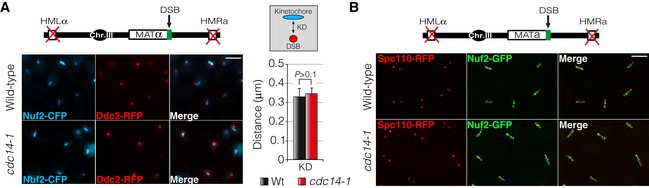
- Chromatin compaction during the DDR is not affected in the absence of Cdc14. A single DSB was generated by expressing the HO endonuclease in both wild‐type and cdc14‐1 mutant cells. Nuf2‐CFP and Ddc2‐RFP were used as kinetochore and DSB markers, respectively. Micrographs were generated by using the maximum projection of nine z‐planes images. Scale bar: 3 μm. The kinetochore–DSB distance in both wild‐type and cdc14‐1 backgrounds was measured and plotted. Graph depicts the mean ± SD of three replicates of at least 100 cells each. P‐value was calculated using a two‐tailed unpaired Student's t‐test.
- Cdc14 is not required to promote DSB‐SPB interaction by generating pulling forces that move kinetochores regions close to the SPBs during the DNA damage response. A single DSB was produced by inducing the HO endonuclease expression in both wild‐type and cdc14‐1 mutant cells. Spc110‐RFP and Nuf2‐GFP were used to visualize the SPBs and the kinetochore, respectively. Micrographs were created by using the maximum projection of nine z‐planes images. Scale bar: 3 μm.
DSB‐SPB tethering stimulated by Cdc14 requires the N‐terminus domain of Mps3 and a proficient DNA damage checkpoint activity
Mps3 has been described to be essential to support the binding of DNA lesions to the nuclear envelope. It was recently demonstrated that deletion of the amino‐terminus of Mps3 is not essential for SPB duplication nor for its own localization to the nuclear envelope, but is required for the tethering of DSBs to the nuclear rim (Oza et al, 2009). To address whether the DSB‐SPB interaction previously observed was dependent on an intact Mps3 allele, we generated strains expressing wild‐type and two truncated versions of the N‐terminus domain of the protein (mps3Δ2‐64 and mps3Δ75–150) fused to RFP as the sole source of the protein. These mutations do not affect viability or SPB duplication (Bupp et al, 2007; Conrad et al, 2007; Fig EV4C). As anticipated, a full‐length Mps3‐GFP was observed at the nuclear envelope and accumulated at the SPBs (Spc110‐RFP) upon induction of a single DSB (Fig EV4A). Under this situation, and confirming our previous observation, DSBs (using Ddc2‐YFP as reporter) were preferentially located close to the SPB signal of Mps3 (Fig EV4B). Moreover, a similar distribution of D1, D2 and D3 was observed as previously reported, ratifying DSB‐SPB interaction (Fig 4D). Mutants lacking residues 2‐64 (mps3Δ2‐64) slightly impaired DSB‐SPB tethering when compared with a full‐length Mps3 (D2 from 0.37 to 0.45 μm). However, removal of the amino acids 75–150 (mps3Δ75–150) drastically affected DSB‐SPB interaction (D2 from 0.37 to 0.62 μm; Fig 4D). Disruption of Cdc14 activity in mps3Δ75–150 cells slightly exacerbates the lack of DSB‐SPB interaction, indicating that Cdc14 cooperates with Mps3 to promote fully DSB interaction to the SPBs (Fig 4D). As previously reported, elimination of the N‐terminus of Mps3 does not sensitize cells to MMS (Oza et al, 2009; Horigome et al, 2014; Fig EV4C). However, in agreement with the defects observed in DSB‐SPB tethering, cell viability in mps3Δ75–150 mutants is severely affected in the presence of phleomycin (Fig EV4C). These data support a model whereby a DSB‐SPB interaction through the N‐terminus of Mps3 might be required to maintain viability under conditions that generate DSBs.
Figure EV4. DSBs are preferentially recruited to Mps3 loaded onto the SPBs.
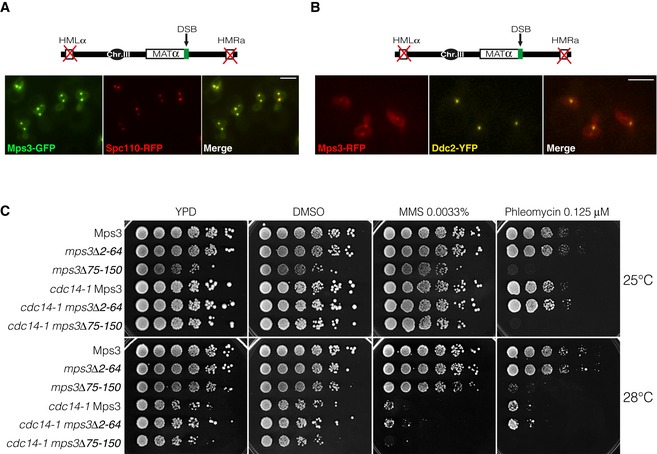
- Mps3 localizes to the nuclear membrane and SPBs in response to a single DSB. A non‐reparable DSB was induced in cells expressing Mps3‐GFP. Spc110‐RFP was used as a SPB reporter. Scale bar: 3 μm.
- DSBs are preferentially recruited to Mps3 bound to the SPBs. A non‐reparable DSB was induced in cells harbouring Mps3‐RFP. Ddc2‐YFP was used as a DSB reporter. Scale bar: 3 μm.
- Elimination of the amino acids 75–150 of the N‐terminus domain of Mps3 sensitizes cells to phleomycin. Tenfold serial dilutions from overnight cultures of cells expressing truncated versions of the N‐terminus domain of Mps3 in wild‐type and cdc14‐1 mutants. Cells were dropped and grown on solid rich media or media containing DMSO (mock), MMS or Phleomycin at 25 or 28°C.
Next, we sought to investigate whether DSB‐SPB tethering depends on a competent DNA damage checkpoint. We measured D1, D2 and D3 distances (monitored by labelling Ddc2 with the GFP and Spc110 with RFP) in cells lacking a proficient DNA damage checkpoint. It has been reported that the lethality of the essential DNA damage protein Rad53 is suppressed by the removal of the RNR inhibitor Sml1. A double sml1Δ rad53Δ mutant, while viably, is drastically affected in DNA damage checkpoint activation (Zhao et al, 1998). We observed that sml1Δ rad53Δ mutants were dramatically affected in promoting DSB‐SPB binding when compared to a wild‐type strain or an sml1Δ single mutant (Fig 4E). This demonstrates that an intact DNA damage checkpoint is necessary for stimulating DSB‐SPB tethering.
The spindle pole body component Spc110 is a target of Cdc14 during the DNA damage response
We have previously shown that Cdc14 is released from the nucleolus in response to DNA damage, implying that the phosphatase is activated during the DDR execution. In order to identify potential targets of Cdc14 specifically in response to a DNA lesion that could attain for DSB‐SPB tethering, we performed quantitative phospho‐proteomics by mass spectrometry of wild‐type and cdc14‐1 samples before and after expression of the HO endonuclease (Fig 5A). Using this approach, we screened for proteins containing quantitatively low levels of phosphorylated residues after induction of the HO endonuclease that occurs only when Cdc14 was active. The identified targets were grouped into different GO categories depending on the protein's molecular function (Fig 5A). We identified a large number of new potential Cdc14 targets previously described for their role in the DNA damage response. Within this category, quantitative comparison between the wild‐type and cdc14‐1 before and after the induction of the DSB was applied to classify the discovered targets depending on their relative phosphorylation across the experimental conditions. Consequently, the higher the ratio, the more phosphorylation of a particular protein was found in cdc14‐1 mutants when compared to wild‐type cells specifically during the DNA damage response.
Figure 5. The spindle pole body component Spc110 is a target of Cdc14 during the DNA damage response.
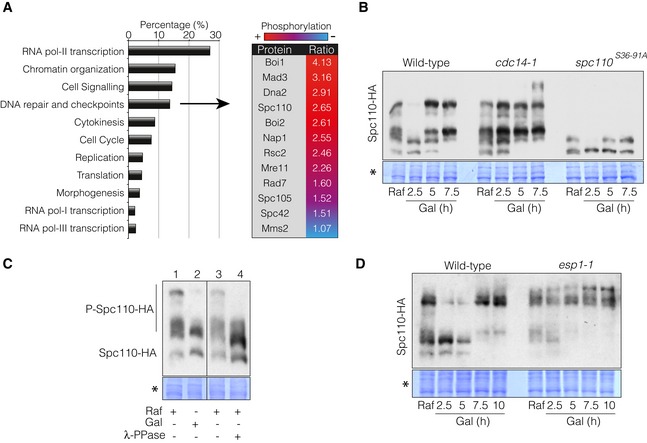
- Identification of Cdc14 phospho‐targets during the DNA damage response by mass spectrometry analysis. Wild‐type and cdc14‐1 cells were grown overnight and blocked in G2/M by using nocodazole to avoid cell cycle‐dependent changes in protein phosphorylation between both strains. After the block was attained, cells were transferred to 37°C for 45 min to deplete Cdc14 activity prior to HO induction by galactose addition for 4 h. Differential phosphorylation of phospho‐peptides detected between the wild‐type and cdc14‐1 were grouped into broad categories depending on the molecular function of the proteins. The table includes the DNA damage and checkpoint‐related proteins with Cdc14‐dependent hyper‐phosphorylated status and the relative ratio between the wild‐type and cdc14‐1 during the DNA damage response. Red and blue indicate relative amount of the residue phosphorylation between both strains (red, high; blue, low).
- Cdc14 dephosphorylates Spc110 in response to a DSB. Spc110 was tagged with the 6HA epitope in wild‐type, cdc14‐1 and spc110 S36‐91A backgrounds. Cells were grown overnight in raffinose‐containing media and supplemented with galactose to induce HO expression. Samples were collected at each time point indicated. Proteins were TCA extracted, separated on Phos‐Tag‐containing gels and blotted. Coomassie staining is shown as a loading control.
- Lambda‐phosphatase treatment of protein extracts in the absence of DNA damage reduces Spc110 phosphorylation levels to the same extent as samples taken during the DNA damage response. Cells were grown overnight in raffinose‐containing media (line 1) and incubated with galactose for 2.5 h to induce HO expression (line 2). Extracts from raffinose‐containing cultures were treated with mock (line 3) or λ‐PPase (line 4). All samples were separated in a Phos‐Tag‐containing gel and blotted. Coomassie staining is shown as a loading control.
- The canonical FEAR pathway is required for Spc110 dephosphorylation during the DNA damage response. Spc110 was labelled with the 3HA tag in both wild‐type and esp1‐1 background strains. Cells were grown overnight in raffinose‐containing media and supplemented with galactose to induce the HO expression. Samples were collected at each time point indicated. Proteins were TCA extracted, separated on Phos‐Tag‐containing gels and blotted. Coomassie staining is shown as a control for loading.
Correlating with a putative function of Cdc14 in promoting DSB‐SPB interaction during the induction of a DSB, several SPB‐associated proteins were identified. Among these targets, we focused our attention on the spindle pole body protein Spc110, the intra‐nuclear receptor for the γ‐tubulin complex. It has previously been described that both Cdc14 and Cdc28‐Clb5 regulate the cell cycle phosphorylation status of Spc110, contributing to spindle integrity (Huisman et al, 2007). To verify the phosphatase activity of Cdc14 over Spc110 upon DNA damage, we used Phos‐Tag gels to follow Cdc14‐dependent changes in Spc110 phosphorylation after inducing a DSB. Prior to HO induction in wild‐type and cdc14‐1 mutant cells, Spc110 migrated with a characteristic pattern of multiple bands suggesting different phosphorylated species. Interestingly, 2.5 h after adding galactose to the media the higher molecular weight bands of Spc110 transiently disappeared in a Cdc14‐dependent manner (Fig 5B) to reappear at later time points. A similar distribution of bands was observed in samples collected 2.5 h from the HO induction and undamaged cells whose protein extract was treated with lambda‐phosphatase (Fig 5C). This result indicates that the slower mobility bands of Spc110 observed previously were indeed phosphorylated isoforms of the protein. Phosphorylation of Spc110 in response to DNA damage is mainly due to the effect imposed by the Cdk activity, since mutations cancelling two Cdk consensus sites at position 36 and 91 (spc110 S36‐91A) largely abolished the phosphorylation of the protein (Fig 5B). Remarkably, cdc14‐1 mutant cells accumulated higher Spc110‐phosphorylated forms along the HO induction, demonstrating that the phosphatase is actively dephosphorylating Spc110 during the DNA damage response (Fig 5B). Supporting these observations, inactivation of the FEAR pathway component Esp1 during the induction of the DNA break showed the same defects in the kinetics of Spc110 phosphorylation than those observed in cdc14‐1 mutant cells (Fig 5D). We conclude that in response to DNA damage, the canonical FEAR pathway is responsible for Cdc14 activation, which in turn promotes Spc110 dephosphorylation.
Cdc14 activity over Spc110 is necessary for stabilization of the metaphase spindle during the response to DNA damage
Spindle polarity is attained by the asymmetric anchoring of the dynein motor Dyn1 specifically to astral microtubules emanating from the daughter SPB. This mechanism generates the force required to drag the spindle to the neck, and thereby align it along the cell axis (Markus et al, 2012). Our results point to a function of Cdc14 in DNA repair by modulating the phosphorylation state of the SPB component Spc110. Interestingly, Spc110 phosphorylation by Cdk contributes to spindle integrity by controlling the spindle polarity at the level of astral microtubule organization (Huisman et al, 2007). Elimination of both Cdk consensus sites of Spc110 renders cells to a symmetrical localization pattern of Dyn1 during SPB separation and thus to defects in spindle polarity (Huisman et al, 2007). To check whether Cdc14 was required for spindle orientation upon DNA damage, we induced the endonuclease HO in wild‐type and cdc14‐1 cells harbouring both Spc110 and Tub1 tagged with RFP and GFP, respectively. Inactivation of Cdc14 during the induction of the DNA lesion increased oscillatory SPB movements rendering to a continuous misalignment of the metaphase spindle (Fig 6A and B, and Movie EV3). These defects are not due to a differential expression of the Tub1‐GFP since both strains contain equal amounts of tubulin–GFP by Western blot (Fig 6A). A similar phenotype was observed when using the phospho‐Cdk mutant spc110 S36‐91A (Fig 6B) indicating that the control of Spc110 phosphorylation is important to maintain spindle polarity during the DNA damage response. Accordingly, Dyn1 asymmetric distribution on astral microtubules (MTs) at the daughter SPB was affected in both cdc14‐1 and spc110 S36‐91A mutants (Fig 6C). We also detected a significant increase in cells with Dyn1 being loaded on astral MTs emerging from both mother and daughter SPBs (Fig 6C). These results clearly indicate that the steady state of Spc110 phosphorylation is fundamental in promoting stabilization and orientation of the metaphase spindle at the level of astral microtubule organization during the DDR execution.
Figure 6. Cdc14 activity is needed for stabilization of the metaphase spindle in response to DNA damage.
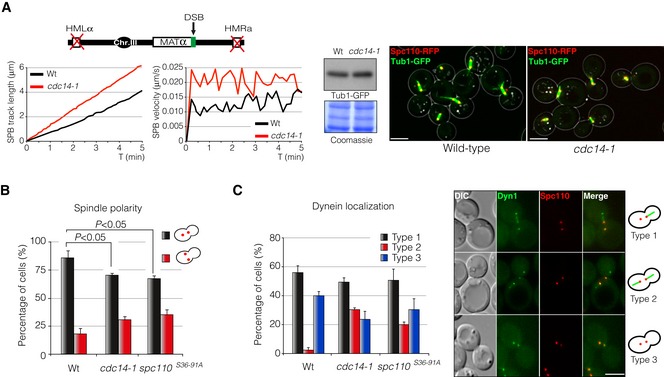
- Time‐lapse experiments of living cells harbouring a non‐reparable DSB were carried out after expressing the HO endonuclease in wild‐type and cdc14‐1 mutant cells. Spc110‐RFP was used as a SPB reporter and Tub1‐GFP was used as a tubulin marker. Three z‐planes images every 5‐s intervals over a period of 5 min were captured and used to quantify the average SPB track length and velocity using Spc110‐RFP as SPB marker. At least 100 cells were scored. A Western blot is shown to confirm the equal amount of Tub1‐GFP in both wild‐type and cdc14‐1 backgrounds. Coomassie blue staining is shown as a loading control. A representative maximum projection image is depicted. Scale bar: 3 μm.
- The steady‐state phosphorylation of Spc110 is required to maintain spindle polarity in response to DNA damage. Wild‐type, cdc14‐1 and spc110 S36‐91A cells expressing Spc110‐RFP were scored for their ability to maintain spindle polarity at the axial plane of the cell upon induction of a single DSB induced by the HO endonuclease. A maximum projection of nine z‐planes was used to score the cells. At least 100 cells from three independent experiments were scored. Graph represents the percentage ± SD of cells with oriented/mis‐oriented SPBs with respect to the axial plane of the cell. P‐value was calculated using a two‐tailed unpaired Student's t‐test.
- The asymmetric loading of dynein into astral MTs emanating from the daughter SPB is affected in cdc14‐1 and spc110 S36‐91A cells during the DNA damage response. Dynein distribution on astral MTs emerging from each SPB was scored by using Dyn1‐GFP in strains containing Spc110‐RFP. Graph represents the percentage ± SD of cells grouped into three categories depending on the pattern of dynein distribution on astral MTs: Type 1, dynein is exclusively loaded on astral MTs emerging from the daughter SPB; Type 2, dynein is equally loaded on astral MTs emanating from both SPBs; Type 3, dynein is absent from astral MTs. At least 100 cells from three independent experiments were scored. A representative picture showing the three patterns is included. Scale bar: 3 μm.
Microtubule organization is essential to stimulate DSB‐SPB interaction and DSB repair by homologous recombination
The experiments described in the previous section suggest a function of Cdc14 in spindle stabilization and DSB‐SPB recruitment upon DNA damage. If the role of Cdc14 in DNA repair is to regulate spindle orientation and dynamics, then disruption of the spindle integrity should phenocopy the defects observed in cdc14‐1 cells upon DNA damage, including DNA repair. To test this prediction, we investigated whether addition of the microtubule‐depolymerizing drug nocodazole affected both DSB‐SPB interaction and DNA repair. We expressed the HO endonuclease in wild‐type and cdc14‐1 cells in the presence or absence of nocodazole and measured D1, D2 and D3 distances. A strain with deleted HM loci was used to avoid repair of the DSB. As predicted, nocodazole treatment in wild‐type cells increased D2 values when compared to a DMSO negative control (from 0.38 to 0.60 μm). On the contrary, D3 distance was reduced (from 1.00 to 0.89 μm), indicating a lack of DSB‐SPB interaction when the drug was present (Fig 7A). It should be noted that the inter‐SPB distance D1 was drastically reduced in nocodazole‐treated cells (from 1.19 to 0.78 μm), probably due to a collapse of the metaphase spindle. As shown before, cdc14‐1 mutant cells presented a higher D2 distance in mock DMSO (0.55 μm) when compared to wild‐type cells. However, nocodazole addition did not increase the high D2 distance observed in cdc14‐1 cells in the drug's absence (0.56 μm; Fig 7A). These data show that microtubule stability and spindle integrity are required for the maintenance of DSB‐SPB attachment during the DNA damage response.
Figure 7. Microtubule destabilization affects DSB‐SPB interaction and DSB repair by homologous recombination.
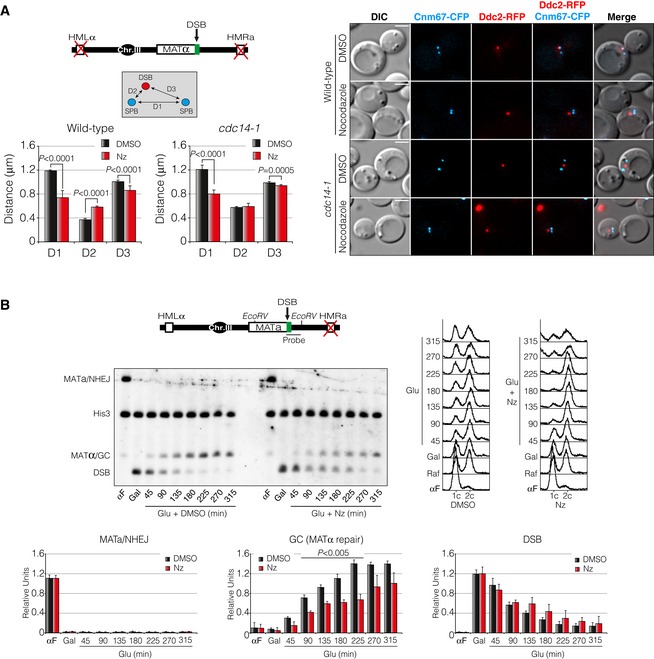
- Cells treated with nocodazole are affected in DSB‐SPB interaction. Cnm67‐CFP and Ddc2‐RFP were used as SPB and DSB markers, respectively. Diagram depicts the genomic background used. D1: inter‐SPB distance; D2: DSB distance to proximal SPB; D3: DSB distance to distal SPB. D1, D2 and D3 distances after inducing a DSB in the presence of nocodazole or mock dimethyl sulphoxide (DMSO) were scored and plotted. Graph represents the mean ± SD of D1, D2 and D3 distances of three independent experiments. At least 100 cells per experiment were scored using the maximum projection of three z‐planes. P‐values were calculated using a two‐tailed unpaired Student's t‐test. A representative picture is shown. Scale bar: 3 μm.
- Physical analysis of wild‐type cells containing the repair pathway choice assay described in Fig 2B. The diagram with the genomic information, the restriction enzymes used and the location of the probe are shown. Cells were synchronized in G1 by using the α‐factor pheromone and released into fresh media for 1 h. Induction of HO expression was attained by adding galactose for 2 h. After formation of the DSB, glucose was added to repress the HO in the presence of nocodazole or mock DMSO and samples were taken to analyse the kinetics of the repair. DNA was extracted, digested with EcoRV, separated on agarose gels and blotted. Both probes for the MATa‐distal sequence and the HIS3 gene (as a loading control) were used. FACS analyses are shown. Graphs show the quantification of MATa restoration (NHEJ), mating‐type switching by gene conversion (HR), and DSB kinetics formation. All data were normalized with the HIS3 gene. Graphs show the mean ± SD from three independent experiments. P‐values were calculated using a two‐tailed unpaired Student's t‐test.
To correlate this observation with a defect in DNA repair, we used a MATa strain with an HMLα as the sole template for the repair of the DSB. Repair of a HO‐induced break by HR is associated with mating‐type switching from MATa to MATα, while NHEJ will conserve the mating type as MATa (Fig 7B). We blocked the culture in G1 using the α‐factor pheromone and released the cells in fresh raffinose‐containing media. After expressing the HO endonuclease by supplementing the media with galactose, glucose was added to repress HO expression and the fate of the broken DNA was followed by Southern blot in the presence or absence of nocodazole. Remarkably, DSB repair kinetics was delayed when cells were treated with nocodazole during the repair process compared to cells treated with DMSO only (Fig 7B). Together, these results imply that microtubule and/or spindle stabilization upon DNA damage is required for DNA repair by homologous recombination, possibly by modulating the DSB‐SPB interaction.
Inactivation of Spc110 impairs SPB dynamics, DSB‐SPB interaction and DSB repair by homologous recombination
Our previous results showed that Spc110 is a target of Cdc14 at SPBs in response to DNA damage. If the control that Cdc14 exerts over Spc110 is required for SPB integrity, DSB‐SPB interaction and DNA repair, then the lack of Spc110 activity should present similar defects than those observed in cdc14‐1 cells during DSB induction. To check for this hypothesis, we constructed the conditional allele spc110‐220, a thermosensitive mutant that can be inactivated by shifting the temperature to 32°C (Sundberg et al, 1996). We introduced the spc110‐220 allele in a non‐reparable HO system where the SPB reporter Cnm67 was labelled with CFP and Ddc2 with RFP. We monitored DSB‐SPB interaction by measuring D1, D2 and D3 distances in both wild‐type and spc110‐220 mutant after induction of a single DSB. Phenocopying a cdc14‐1 background, spc110‐220 cells showed a consistent increase in D2 distance (from 0.39 to 0.65 μm) indicating a failure to promote DSB‐SPB interaction (Fig EV5A). It should be remarked that, as previously reported, we scored cells with a high variation in inter‐SPB distance (D1), probably due to the essential role of Spc110 in SPB assembly and, in turn, spindle formation. Together with a lack of DSB‐SPB interaction, we also noted an increase in SPB dynamics when Spc110 was inactivated. Time‐lapse experiments of living cells showed that spc110‐220 cells presented an increase in SPB accumulated track distance and higher average velocity than wild‐type control cells (Fig EV5B).
Figure EV5. Low Spc110 activity results in high SPB dynamics, loss of DSB‐SPB interaction and inefficient DSB repair by homologous recombination.
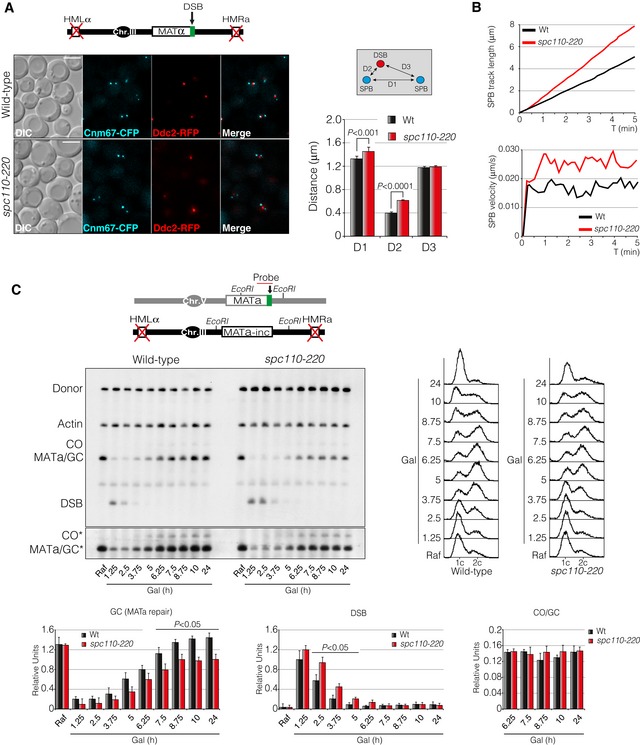
- D1, D2 and D3 distances measured in wild‐type and spc110‐220 cells after inducing a non‐reparable DSB at the MAT locus. Cnm67‐CFP and Ddc2‐RFP were used as SPB and DSB markers, respectively. Graph represents the mean ± SD of D1, D2 and D3 distances of three independent experiments. At least 100 cells per experiment were scored using the maximum projection of three z‐planes. P‐values were calculated using a two‐tailed unpaired Student's t‐test. A diagram with the genome information and a representative picture are shown. Scale bar: 3 μm.
- Time‐lapse experiments in living cells were carried out after expressing the HO endonuclease in wild‐type and spc110‐220 cells. Three z‐planes every 10‐s intervals over a period of 5 min were captured and used to quantify the average SPB track length and velocity using Spc110‐RFP as SPB marker. At least 100 cells were scored.
- Physical analysis of wild‐type and spc110‐220 mutant strains carrying the inter‐chromosomal gene conversion assay depicted (top diagram). Cells were grown overnight and exposed to HO expression by adding galactose at 32°C, thus producing a DSB on chromosome V. Samples were taken at different time points, genomic DNA was extracted, digested with EcoRI and analysed by Southern blot. Blots were hybridized with a MATa‐only and ACT1 (loading control) DNA sequences. FACS profiles for DNA content are included. Graphs represent quantification of gene conversion, DSB induction and crossover versus non‐crossover ratio. All data were normalized with the actin control. Graphs show the mean ± SD from three independent experiments. P‐values were calculated using a two‐tailed unpaired Student's t‐test. Asterisk denotes an overexposed film to visualize crossover formation. Data information: DSB, double‐strand break; SPB, spindle pole body; Raf, raffinose; Gal, galactose; GC, gene conversion; CO, crossover.
Source data are available online for this figure.
In order to assess whether these defects could lead to an impaired DNA damage repair, we analysed the efficiency to restore a DSB generated by the HO endonuclease by Southern blot using the inter‐chromosomal repair system. We carried out measurements to determine the repair efficiency by HR with or without crossover association. Resembling the DNA repair deficiency observed in cdc14‐1 mutants, spc110‐220 cells showed a reduction in DSB repair by gene conversion with no significant difference in the ratio between crossover and non‐crossover products (Fig EV5C). These experiments demonstrate that SPBs and/or metaphase spindle integrity upon DNA damage is a vital feature for promoting recruitment of the DNA lesion to the SPBs proximity and in turn for recombinational DNA repair.
Loss of Cdk‐dependent Spc110 phospho‐regulation affects spindle stability, DSB‐SPB interaction and DNA repair by homologous recombination
We have previously shown that Spc110 phosphorylation during the activation of the DDR is largely abolished in a phospho‐deficient spc110 S36‐91A mutant (Fig 5B), indicating that Cdk is actively phosphorylating Spc110 during the DDR. To determine the importance of the Cdk‐dependent Spc110 phosphorylation in the DDR, we checked for the effect of expressing the spc110 S36‐91A allele during the induction of a DNA lesion. Phenotypic analysis of this mutant revealed growth sensitivity when exposed to MMS or phleomycin to a similar extent as in cdc14‐1 mutant cells (Fig 8A). To test whether this sensitivity to genotoxic drugs was due to defects in DSB‐SPB interaction, we examined the distribution of D1, D2 and D3 distances in both wild‐type and spc110 S36‐91A strains upon induction of a single DSB. Accordingly, spc110 S36‐91A cells exhibited an increased D2 distance (from 0.42 to 0.64 μm) demonstrating that Spc110 phosphorylation by Cdk is essential to promote DSB‐SPB interaction (Fig 8B). Concurrently, we observed an increase in SPB accumulated track distance and higher average velocity when the two Cdk consensus sites of Spc110 were mutated to alanine (Fig 8C). In order to address whether these defects impair DSB repair by homologous recombination, we analysed the efficiency to restore a DNA break generated by the HO endonuclease by Southern blot using an inter‐chromosomal repair system. We also calculated the ratio of gene conversion versus crossover associated with the repair between the wild‐type strain and the spc110 S36‐91A mutant. As predicted, DSB repair kinetics in a spc110 S36‐91A mutant was delayed when compared with a wild‐type strain. However, no significant differences were observed in the ratio between crossover and non‐crossovers repair outcomes (Fig 8D). Reintroduction of a wild‐type Spc110 version of the protein complemented both the spc110 S36‐91A sensitivity to phleomycin (Appendix Fig S1A) and the defects in DNA repair by homologous recombination (Appendix Fig S1B), confirming the importance of Spc110 phosphorylation in the DDR. We conclude that lack of Spc110 phosphorylation during the induction of a DNA break leads to high SPB dynamics, loss of DSB‐SPB recruitment and inefficient DNA repair.
Figure 8. Cdk‐dependent phosphorylation of Spc110 is essential to stabilize the metaphase spindle, stimulate DSB‐SPB interaction and promote DNA repair by homologous recombination.
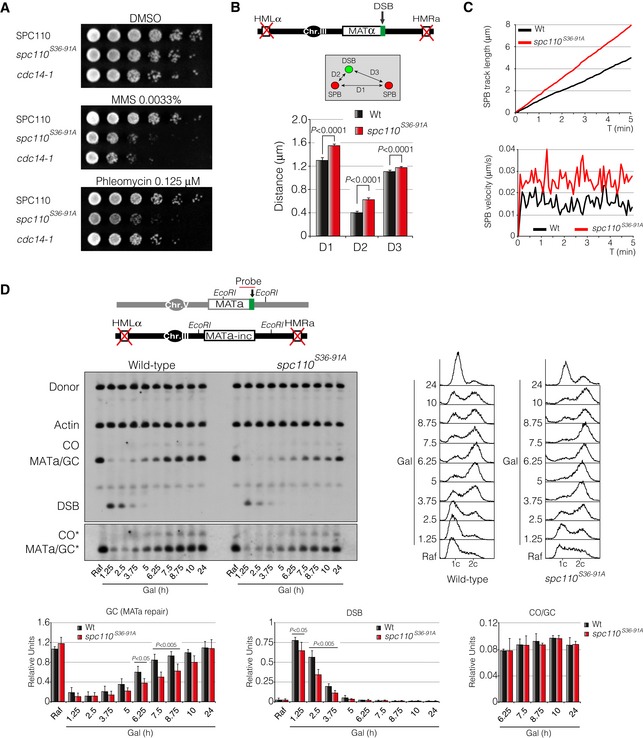
- Growth sensitivity to MMS and phleomycin of a Cdk phospho‐deficient mutant of Spc110. Tenfold serial dilutions from overnight cultures of wild‐type, spc110 S36‐91A and a cdc14‐1 cells grown on solid rich media containing mock DMSO, MMS or phleomycin at 30°C.
- Diagram showing the genomic background and the approximation used to measure DSB‐SPB interaction. Spc110 and spc110 S36‐91A were labelled with the RFP in a background containing Ddc2‐GFP as DSB reporter. D1: inter‐SPB distance; D2: DSB distance to proximal SPB; D3: DSB distance to distal SPB. At least 100 cells from three independent experiments were scored using the maximum projection of nine z‐planes. Graph shows the mean ± SD. P‐values were calculated using a two‐tailed unpaired Student's t‐test.
- Time‐lapse experiments of living cells expressing the HO endonuclease in wild‐type and spc110 S36‐91A mutant cells. Five z‐planes every 5‐s intervals over a period of 5 min were captured. Quantifications were made using the average SPB track length and velocity of at least 100 cells. Spc110 and spc110 S36‐91A were labelled with the RFP and used as a SPB reporter.
- Southern blot of wild‐type and spc110 S36‐91A mutant harbouring the inter‐chromosomal gene conversion assay portrayed. An overnight culture was induced by adding galactose at 32°C to express the HO endonuclease. Samples were collected at different time points, genomic DNA was extracted, digested with EcoRI and analysed by Southern blot. Blots were probed with a MATa‐only and actin (for loading control) DNA sequences. FACS profiles for DNA content are included. Graphs show quantification of gene conversion, DSB induction and crossover versus non‐crossover ratio. All data were normalized using the actin signal. Graphs represent the mean ± SD from three independent experiments. P‐values were calculated using a two‐tailed unpaired Student's t‐test. Asterisk denotes an overexposed film to visualize the formation of crossover products.
The fact that in the absence of Cdc14 function we observed similar phenotypes than those described previously in a spc110 S36‐91A mutant background suggests that the fine‐tune balance of Spc110 phosphorylation during the DDR might be tightly regulated by Cdk/Cdc14 to promote spindle integrity and, in turn, DSB‐SPB recruitment and DNA repair. To confirm the physiological importance of the dual Cdk/Cdc14‐dependent Spc110 phosphorylation during the DNA damage response, we generated a phospho‐mimic version of Spc110 by mutating the two Cdk consensus sites to aspartic acid residues (spc110 S36‐91D). In agreement with a dual Cdk/Cdc14 regulation over Spc110 during the response to DNA damage, cells expressing spc110 S36‐91D showed similar phenotypes than those observed in a phospho‐deficient spc110 S36‐91A. Regarding the sensitivity to genotoxic drugs, cells expressing spc110 S36‐91D exhibited a comparable sensitivity to phleomycin than cells bearing the phospho‐deficient spc110 S36‐91A when compared with a wild‐type strain (Fig 9A). However, in contrast to the pronounce sensitivity to MMS of an spc110 S36‐91A mutant, spc110 S36‐91D cells presented similar growth levels when compared with DMSO control plates. This experiment indicates that dephosphorylation of Spc110 in response to DNA damage is especially important when cells are exclusively exposed to phleomycin‐mediated DSBs. It is worth mentioning that the presence of a phospho‐mimic version of Spc110 developed growth defects when growing in media without genotoxic drugs (Fig 9A), arguing for an essential function of S36‐91 dephosphorylation during an unperturbed cell cycle. In agreement with a defect in cell viability under phleomycin‐containing media, spc110 S36‐91D cells presented an increased D2 distance (from 0.37 to 0.56 μm) when measuring DSB‐SPB interaction (Fig 9B) and an elevated SPB track length and velocity when compared with a wild‐type strain (Fig 9C). Finally, to fully confirm the importance of Spc110 dephosphorylation during the DNA damage response, we analysed the efficiency of spc110 S36‐91D to repair a DSB by homologous recombination using an inter‐chromosomal repair system (Fig 9D). As predicted, cells expressing the phospho‐mimic version of Spc110 repaired the HO‐induced DSB with a slower kinetics rate than a wild‐type background without affecting the levels of crossover formation (Fig 9D).
Figure 9. Constitutive Cdk‐dependent Spc110 phosphorylation affects metaphase spindle stabilization, DSB‐SPB tethering and DNA repair by homologous recombination.
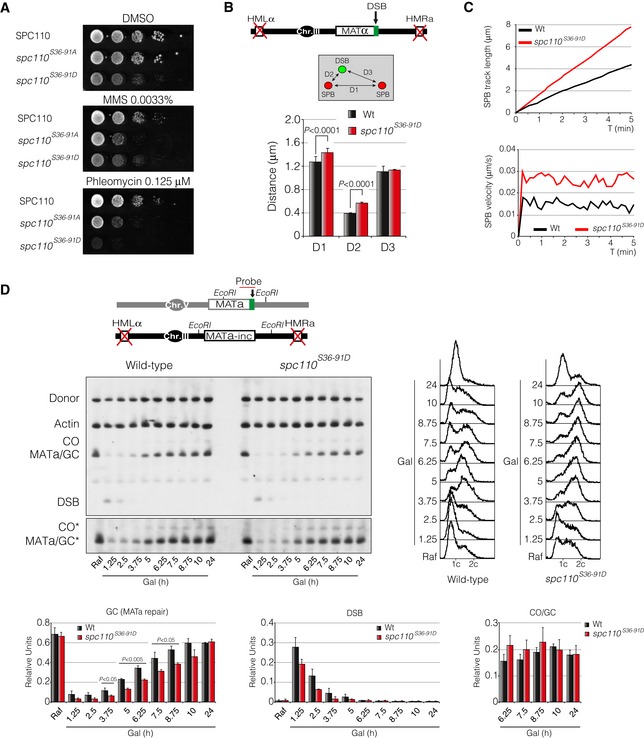
- DNA damage sensitivity to MMS and phleomycin of a Cdk phospho‐mimic mutant of Spc110. Tenfold serial dilutions from mid‐log phase cultures of wild‐type, spc110 S36‐91A and spc110 S36‐91D cells grown on solid rich media containing mock DMSO, MMS or phleomycin at 30°C.
- Schematic representation of the genomic background and the approximation used to measure DSB‐SPB tethering. Wild‐type Spc110 and spc110 S36‐91D were labelled with the RFP. Ddc2 was tagged with the GFP to be used as a DSB reporter. D1: inter‐SPB distance; D2: DSB distance to proximal SPB; D3: DSB distance to distal SPB. At least 100 cells from three independent experiments were scored using the maximum projection of nine z‐planes. Graph shows the mean ± SD. P‐values were calculated using a two‐tailed unpaired Student's t‐test.
- Time‐lapse experiments of living cells after induction of the HO endonuclease in wild‐type and spc110 S36‐91D cells. Three z‐planes every 10‐s intervals over a period of 5 min were captured to measure the average SPBs track length and velocity. Spc110 and spc110 S36‐91D were labelled with the RFP and used to measure SPBs kinetics. At least 100 cells from three independent experiments were scored.
- Southern blot of wild‐type and spc110 S36‐91D mutant bearing the inter‐chromosomal gene conversion assay depicted. Mid‐log phase cells were HO‐induced at 32°C by adding galactose to the media. Samples were collected at different time points and the genomic DNA was extracted. Preps were digested with EcoRI and analysed by Southern blot. Blots were hybridized with MATa‐only and actin probes. FACS profiles for DNA content are displayed. Graphs show quantification of gene conversion, DSB induction and crossover versus non‐crossover ratio. All data were normalized using the actin signal. Graphs represent the mean ± SD from three independent experiments. P‐values were calculated using a two‐tailed unpaired Student's t‐test. Asterisk denotes an overexposed film to visualize the formation of crossover products.
These results demonstrate that metaphase spindle stability during DDR activation is attained by a tightly regulated activity of Cdk/Cdc14 acting over Spc110. Importantly, the steady state of Spc110 phosphorylation is critical not only to promote spindle stability but also to stimulate DSB‐SPB interaction and in turn recombinational DNA repair to minimize genome instability.
Discussion
Phosphorylation after DNA damage has been thoroughly studied for the last years, focusing mostly in the activation of the signalling pathways and the kinases involved in the response to a DNA lesion. Even though it is generally accepted that dephosphorylation is crucial for restoring the effects imposed by kinases, there have only been sporadic studies about the role of protein dephosphorylation during the DDR execution. In this study, we have evaluated the importance of the Cdc14 phosphatase in the repair of a DNA lesion in the budding yeast S. cerevisiae. Our data show that Cdc14 is required for cell viability under different DNA damage conditions. Cdc14 exerts this function by stimulating intra‐ and inter‐chromosomal DNA repair. Remarkably, in response to a DNA lesion, Cdc14 is transiently released from the nucleolus into the nucleoplasm where it dephosphorylates multiple DDR‐related proteins. One of these targets is the intra‐nuclear receptor for the γ‐tubulin complex Spc110. Spc110 dephosphorylation by Cdc14 might contribute to the stabilization of the metaphase spindle at the bud axis during the DNA damage response. This observation suggests that SPB integrity is a requisite for DNA repair. Accordingly, disruption of spindle stability by treating cells with nocodazole, reducing Spc110 activity or replacing the Cdk consensus sites of the protein impairs DNA repair by homologous recombination, demonstrating that stabilization of the metaphase spindle is indeed an important feature of the repair process. How is SPB stabilization achieved during the DDR? It has been reported that during mitosis, Spc110 phosphorylation by the Cdk‐Clb5 is required for spindle polarity at the level of astral microtubule organization (Huisman et al, 2007). Surprisingly, cells lacking Cdc14 activity during the DDR activation exhibit similar SPB polarity defects. These observations suggest that the balance of Spc110 phosphorylation by Cdk/Cdc14 during the DNA damage response must be tightly regulated to elicit spindle orientation and stabilization. In full agreement with this observation, both a phospho‐deficient spc110 S36‐91A and a phospho‐mimic spc110 S36‐91D presented similar phenotypes in DSB‐SPB tethering, spindle orientation and DNA repair. Supporting these data, it was recently established that both spc110 S36‐91A and spc110 S36‐91D presented slow growth at 37°C in the absence of the SAC gene MAD2 (Lin et al, 2014). This suggests that lack of phospho‐Spc110 regulation can be compensated by a cell cycle delay promoted by the SAC and implies that both Cdk and Cdc14 might work in tandem over Spc110 to ensure a timely execution of mitosis. Additionally, it has been reported that in mitosis, the simultaneous substitution of both Cdk and Mps1 consensus sites of Spc110 (S36‐S91 and S60‐T64‐T68, respectively) to alanine or aspartic acid exhibited a marked reduction in microtubule nucleation at SPBs when compared with a wild‐type control (Lin et al, 2014). This observation supports a model whereby both Cdk and Cdc14 could stimulate microtubule nucleation through the regulation of Spc110 phosphorylation along the cell cycle, and explains, at least in part, the similar defects observed in both spc110 S36‐91A and spc110 S36‐91D mutants during the response to a DNA break. Still, due to the high redundancy in the phenotypes observed in both mutants, we cannot certainly attribute these mutations as phospho‐mimetic or phospho‐deficient. However, the fact that both cdc14‐1 and spc110 S36‐91D cells present similar phenotypes in response to DNA damage, the distinct sensitivity to MMS in both spc110 S36‐91A and spc110 S36‐91D, and the observation that these mutations do not alter the overall structure of the protein (Lin et al, 2014) argue against this possibility. It is worth mentioning that recently, an additional potential Cdk site (T18) has been identified at the N‐terminus of Spc110. While in vitro assays have demonstrated that T18 phosphorylation by Cdk inactivates oligomerization by the γ‐tubulin complex, in in vivo experiments both a phospho‐mimetic and a phospho‐mutant version of T18 showed identical growth defects due to a failure in promoting microtubule nucleation (Lin et al, 2014). Surprisingly, the fact that T18 is phosphorylated only by Cdk‐Clb2 in mitosis indicates that this potential site is probably regulated in a different manner than the previously described S36‐91. Whether phosphorylation/dephosphorylation of this new phospho‐acceptor site by Cdk/Cdc14 has a specific role during the DDR execution to control spindle stabilization is an attractive question for the future. Additionally, we cannot discard that other kinases could be participating in the Spc110 regulation during the DDR. As previously commented, Spc110 contains three Mps1 phospho‐acceptor sites closely surrounded by the two S36‐91 Cdk consensus sequences mentioned before. Because of their relative proximity, it is tempting to speculate that the phosphorylation of Cdk/Mps1 sites could intrinsically affect (positively or negatively) one another. On the other hand, as Mps1 phospho‐sites do not contain a canonical Cdk target sequence, it is very unlikely that Cdc14 could have a role in dephosphorylating these residues. This infers that other phosphatases could be cooperating with Cdc14 specifically at Mps1 phospho‐sites in the regulation of Spc110 phosphorylation. In this respect, the involvement of these new players could exponentially increase the level of complexity of Spc110 phospho‐regulation during the DNA damage response.
A fundamental question that can be extracted from these observations is how the phosphorylation state of SPB components at the inner side of the SPBs could influence the processes involved in nuclear positioning outside the nucleus. Nuclear movements are generated at the outer SPB plaque by the recruitment of the γ‐tubulin complex to the Spc72 protein (Knop & Schiebel, 1998). Interestingly, Spc72 and Spc110 are bound to Spc42 to form a bridge that connects the nucleoplasm with the cytoplasm. In this scenario, signals at the inner side of the SPB in response to DNA damage could be transmitted to the outer side by physical changes in protein conformation of the Spc110‐Spc42‐Spc72 complex to modulate the recruitment of γ‐tubulin at the outer side of the SPB. Encouragingly, a spc72Δ mutant was identified in a large‐scale survey as hypersensitive to genotoxic compounds such as caffeine (Parsons et al, 2004) and hydroxyurea (Hartman & Tippery, 2004), evidencing the importance of this process in DNA repair. A similar mechanism has been proposed for the LINC complex in S. pombe, where the SUN protein Sad1 and the KASH protein Kms1 (functional orthologues of Mps3 and Csm4, respectively, in S. cerevisiae) collaborate with DNA repair by connecting DSB at the inner side of the nuclear envelope with the microtubule cytoskeleton at the cytoplasm (Swartz et al, 2014). We have demonstrated that dynein‐specific localization at the astral MTs emanating from the daughter SPB is affected in cells lacking Cdc14 activity or in the spc110 S36‐91A mutant, suggesting that a signal at the inner SPB side can influence the activity of the dynein motor at the outer SPB side to enhance spindle stability and polarity. Unexpectedly, both hyper‐phosphorylated Spc110 (existing in cdc14‐1 mutant cells) and hypo‐phosphorylated Spc110 (spc110 S36‐91A mutant) showed similar defects in spindle polarity and dynein loading. This observation, together with the fact that Cdc14 is only transiently released from the nucleolus, suggests that a temporal fine‐tune phosphorylation of Spc110 in response to DNA damage must be particularly important to achieve spindle orientation. This steady‐state phosphorylation of Spc110 is determinant to stimulate DSB interaction with the SPBs in order to execute an efficient DNA repair pathway, suggesting that a spatial regulation is also required to achieve an appropriate DDR execution. Nevertheless, we cannot disregard that dephosphorylation of other SPB targets by Cdc14 could cooperate in the maintenance of spindle polarity. Supporting this notion, in the screening looking for substrates of Cdc14 in response to DNA damage, the SPB component Spc42 presented elevated phosphorylation levels in the absence of Cdc14 activity during the induction of a DSB.
Many nuclear events have been described to be specifically located to nuclear sub‐compartments. It has been postulated that organization of DSBs into repair centres may provide several advantages to the cell by coordinating cell cycle progression with the repair status or by facilitating a local concentration of proteins at the DNA lesion that are needed for certain steps of the DNA repair process (Lisby et al, 2003). A DSB with a functional donor for HR shifts at least transiently to the nuclear periphery where it binds either the nuclear membrane protein Mps3 or the nuclear pore sub‐complex Nup84 (Oza & Peterson, 2010; Horigome et al, 2014). Recently, Gasser and colleagues have proposed that while Mps3‐DSB binding specifically occurs during S/G2 and requires the chromatin remodeler INO80 and the recombinase activity of Rad51, Nup84‐DSB interaction occurs independently of the cell cycle phase and without the need of either INO80 or Rad51 (Horigome et al, 2014). These observations strongly suggest that DSBs re‐localization is, at least in part, a cell cycle‐regulated process implying that DNA lesions produced in S/G2 are preferentially engaged to the nuclear rim. Taking into account that homologous recombination occurs specifically at the S/G2 phases of the cell cycle, it is tempting to speculate that DSB‐SPB tethering is a prerequisite specific for homology‐dependent repair. Mps3 was firstly identified in a screen searching for mutants defective in SPB assembly (Jaspersen et al, 2002). Currently, several roles have been assigned to this protein in SPB duplication and insertion in the nuclear membrane, nuclear organization and meiotic telomere bouquet formation. Regarding its role in DNA repair, it is believed that Mps3 could attain for most of DSBs to nuclear envelope tethering since deletion of the amino‐terminal domain of the protein abolishes the peripheral localization of DNA lesions (Oza et al, 2009). Interestingly, Mps3 is an essential SPB protein required for its assembly and stability (Jaspersen et al, 2006). Even though it has been reported that Mps3 localizes throughout the inner nuclear membrane, we have shown that most of the protein is restricted to the SPBs in response to DNA damage. This specific localization of Mps3 could account for our observation that DSBs generated by the expression of the HO endonuclease preferentially re‐localize from the nucleoplasm to the SPBs. Accordingly, elimination of the N‐terminus domain of Mps3 disrupts DSB‐SPB interaction indicating that DNA lesions are preferentially targeted to the Mps3 located at the SPBs. It is important to remark that these mutants are deeply sensitized to phleomycin confirming that recruitment of DNA lesions to the SPBs is indeed necessary for DNA repair and cell viability under genotoxic stress. Alternative to the role of Mps3 in tethering DSBs to the nuclear membrane adjacent to the SPBs, several studies have revealed that the nuclear pore can also bind DSBs through the Nup84 component. It has been proposed that a DSB recruited to the nuclear pore stimulates its repair by gene conversion in a Nup84‐dependent manner (Nagai et al, 2008). To date, it is believed that interaction of DSBs with the nuclear pore is a safeguard mechanism that compensates for a deficient Mps3‐DSB tethering (Horigome et al, 2014). In this scenario, both Mps3 and Nup84 could cooperate in the recruitment of a DNA lesion to stimulate repair by homologous recombination. This hypothesis fits with previous observation showing that mutations that abrogate both Mps3‐DSB and Nup84‐DSB pathways have additive repair defects, confirming that both perinuclear anchoring mechanisms collaborate in the repair of DNA lesions (Horigome et al, 2014).
We have shown that metaphase spindle stabilization and orientation is a requirement to promote fully DSB interaction with putative repair centres located at the SPB. It is interesting that microtubule behaviour is indispensable for DSB‐SPB recruitment and DNA repair. One possibility to explain this prerequisite is that the mechanical forces operating at the cytoplasmic side of the SPBs could be transmitted inside the nucleus to increase chromosomal dynamics that stimulate DSB interaction with the SPBs. This theory fits perfectly with previous observations demonstrating that yeast chromosomal movements are indeed increased in response to DNA damage (Dion & Gasser, 2013). It is currently unclear whether a similar mechanism exists in mammalian cells. Analyses of the dynamics of single DSBs in living mammalian cells have demonstrated that broken ends are positionally stable (Soutoglou et al, 2007). One possibility to explain DSB mobility restriction in mammalian cells might come from the predominant use of NHEJ. In fact, inactivation of NHEJ by depletion of the DNA‐end binding protein Ku80 leads to a significant increase in the ability of a DSB generated by the I‐SceI endonuclease to locally diffuse (Soutoglou et al, 2007). Supporting this observation, U2OS cells exposed to γ‐radiation show enhanced mobility for DSBs (Krawczyk et al, 2012). Alongside, DSBs induced at the nuclear lamina restrict HR and allow NHEJ, revealing another layer of regulation of DNA repair pathway choice imposed by nuclear compartmentalization (Lemaitre et al, 2014). Whether the nuclear periphery or centrosomes of mammalian cells contain a permissive environment to support DNA repair by homologous recombination remains to be directly investigated.
Independently of Cdc14's role in spindle stabilization and DSB‐SPB recruitment, we cannot rule out additional roles of the phosphatase in DNA repair by directly modulating the phosphorylation levels of DNA repair components. Supporting this idea, our mass spectrometry data have revealed that several proteins involved in different steps of the DNA repair process are also hyper‐phosphorylated in the absence of Cdc14 activity, including the MRX subunit Mre11 and the nuclease Dna2. Both proteins have recently been described to be direct targets of the Cdk in response to DNA damage (Cannavo & Cejka, 2014; Simoneau et al, 2014). Cdk regulation of Mre11 facilitates HR repair of DSBs by limiting competition from the NHEJ machinery. Interestingly, several lines of evidence suggest that this mechanism could be evolutionarily conserved, since the human Mre11 interacts directly with Cdk2 (Buis et al, 2012). On the other hand, Dna2‐dependent Cdk phosphorylation stimulates its recruitment to DSBs, resection and subsequent Mec1‐dependent phosphorylation. Whether Cdc14 is capable to counterbalance the Cdk activity over these components of the DDR during the repair pathway is a question that remains to be directly addressed.
In summary, our data fit into a model whereby the spatiotemporal localization and activation of Cdc14 could provide an additional control in the phosphorylation status of diverse repair factors. In this scenario, Cdc14 could be counterbalancing the effect imposed by the Cdk over these substrates to reset each step of the repair process to the original state once they have been accomplished. Up to date, several works have identified numerous Cdk substrates during the activation of the different stages of the DDR, including resection, homologous recombination and the DNA damage checkpoint (Ira et al, 2004). Yet, less is known about the role of Cdc14 in counteracting the phosphorylation state of these targets. Here we have shown that in response to DNA damage, Cdc14 is released from the nucleolus to dephosphorylate several Cdk substrates. This observation is in line with previous results in S. pombe demonstrating that Cdc14 orthologue Clp1 is also detached from the nucleolus in a Chk1‐dependent manner in response to several sources of DNA damage (Diaz‐Cuervo & Bueno, 2008; Broadus & Gould, 2012). We have shown that the function of the canonical checkpoint component Rad53 is essential to stimulate DSB‐SPB recruitment, suggesting that a fully DNA damage checkpoint activation is also essential in S. cerevisiae to promote Cdc14 activation. Interestingly, the partial and transient Cdc14 nucleolar release observed in response to DNA damage is confined in the nucleoplasm, a specific feature of the FEAR‐activated Cdc14. It is believed that the amount of Cdc14 liberated by the FEAR pathway is insufficient to promote Cdk deactivation and mitotic exit, explaining why cells are maintained at the G2/M arrest even when Cdc14 has been released and activated. Strikingly, given that mammalian Cdc14B is also mobilized from the nucleolus in response to DNA damage (Bassermann et al, 2008; Mocciaro et al, 2010), our results may provide insight towards a conserved function of the phosphatase during the DDR execution.
We are just starting to understand what are the specific functions of Cdc14 in DNA repair, how its activity is regulated and what the targets in the process are. More work is required to comprehend the exact role of Cdc14 during the repair of a DNA lesion. Finally, the regulatory mechanisms proposed herein are likely to be relevant in higher eukaryotes, since both human orthologues Cdc14A and Cdc14B positively regulate DNA double‐strand break repair (Mocciaro et al, 2010). In this regard, it is important to determine whether these phosphatases contribute to the DNA damage sensitivity and poor prognostic associated with human diseases caused by mutated or epigenetically inactivated DDR components.
Materials and Methods
Yeast strains, growing conditions and plasmids
All yeast strains used in this study are listed in Appendix Table S1. Epitope tagging of endogenous genes was performed by gene targeting using polymerase chain reaction (PCR) products as described in Janke et al (2004). Cells were grown in YEP containing 2% raffinose. The induction of the HO expression was obtained by galactose addition to a final concentration of 2%. Samples were collected for FACS, DNA and protein analyses just prior to and at different time points following addition of galactose. For repair experiments, DSB repair and cell recovery was promoted by repression of the HO by adding 2% glucose to the medium. When using phleomycin as the source of DNA damage, cells were grown in YEPD prior to addition of the drug.
The pAC5 plasmid containing the cdc14‐1‐GFP‐KanMX4 cassette was created by PCR amplification of the cdc14‐1‐GFP‐KanMX4 locus from strain AC40 and cloned into a TOPO® vector (Life Technologies). The vector was linearized with KpnI prior to transformation. The temperature‐sensitive allele spc110‐220 was generated by introducing a C911R amino acid change in the calmodulin‐binding site. The C‐terminal domain of the gene was amplified by PCR and cloned into a pRS306 plasmid digested with XbaI and SalI. Point mutagenesis was attained by PCR amplification using oligonucleotides containing a TGT to CGT mutation in the 5′ end, followed by DpnI digestion and ligation. The pAC42 plasmid generated containing the C911R substitution was confirmed by sequencing, linearized with AflII and transformed. The correct integration of the plasmid at the URA3 locus was tested by PCR. Positive colonies were isolated in 5‐fluoroorotic acid media, and clones carrying the mutation were identified by sequencing. The Spc110 variant lacking CDK consensus phosphorylation sites was generated by gene synthesis of a SacI‐StyI fragment containing the promoter and 5′ region of the gene and cloned into a pUC57 plasmid (pAC44). The SacI‐StyI fragment of pAC44 was subcloned into a pRS304 carrying a SacI‐EcoRI fragment of an extended 5′ region of Spc110 (pAC47), generating the plasmid pAC48. pAC48 was AfeI‐linearized and transformed. Same strategy was used to generate the Spc110 variant containing the CDK phospho‐mimic consensus sites. A SacI‐StyI fragment holding the residues S36 and S91 changed to aspartic acid was synthetized and subcloned into pAC47, generating the plasmid pAC66. The integration of the construct was directed by linearizing pAC66 with AfeI prior to transformation. For SPC110 complementation assays, a BamHI/SalI PCR fragment containing the promoter and the ORF of the gene was cloned into pRS306, generating the plasmid pAC63. pAC63 was targeted to the URA3 locus by linearizing the plasmid with StuI previous transformation. pRS316 plasmids expressing the wild‐type Mps3 (PSS269) and the N‐terminus truncated versions mps3Δ2–64 (PSS326) and mps3Δ75–150 (PSS327) fused to the RFP were a gift from Professor P. San Segundo Sequences and details are available upon request.
Southern blots
Cell lysis was performed by incubating the samples with 40 units of lyticase in DNA preparation buffer (1% SDS, 100 mM NaCl, 50 mM Tris–HCl, 10 mM EDTA) for 10 min. DNA was isolated by phenol:chloroform:isoamylalcohol (25:24:1) extraction, precipitated with ethanol and resuspended in TE buffer. Isolated DNA was digested with the appropriate restriction enzyme and separated on 1% agarose gels and subjected to Southern blotting. Probing was performed by labelling with a mix of nucleotides containing fluorescein‐12‐dUTP (Fluorescein‐High Prime; Roche). The oligonucleotides used to generate the probes are listed in Appendix Table S2. Detection was achieved by using an anti‐fluorescein antibody conjugated with alkaline phosphatase (Anti‐Fluorescein‐AP Fab fragments; Roche).
Western blots
Samples were prepared by trichloroacetic acid (TCA) extraction. Cells were collected by centrifugation and fixed with 20% TCA. Cell breakage was performed with glass beads in a FastPrep machine (MPBio) (three 20‐s cycles at power setting 6.5). Protein precipitation was attained by centrifugation at 2,500 g for 5 min at 4°C. After precipitation, the pellet was solubilized in 1 M Tris–HCl pH8 and SDS‐PAGE loading buffer and boiled for 10 min. Insoluble material was removed by centrifugation, and the supernatant was loaded in a 6% acrylamide gel. When separating Spc110 phospho‐bands, gels were supplemented with 30 μM of Phos‐Tag and 60 μM of MnCl2. For Net1 phosphorylation analysis, 10 μM of Phos‐Tag and 20 μM of MnCl2 were used. Proteins were transferred into a PVDF membrane (Hybond‐P; GE Healthcare) and blocked with 5% milk in PBS–Tween (0.1%). Anti‐Rad53 antibody was used at a 1:1,000 dilution (Santa Cruz), and the secondary anti‐goat antibody was used at a concentration of 1:5,000 (Santa Cruz). Anti‐HA antibody was used at a 1:2,500 dilution (Roche), and the secondary anti‐mouse was used at a concentration of 1:25,000 (GE Healthcare). After several washes in PBS–Tween, membranes were incubated with SuperSignal® West Femto (Thermo Scientific), followed by exposure to ECL Hyperfilm (GE Healthcare).
Microscopy
In vivo fluorescence microscopy of green fluorescent protein (GFP), yellow fluorescent protein (YFP), cyan fluoresce protein (CFP), red fluoresce protein (RFP) and 4′‐6‐diamidino‐2‐phenylindole (DAPI) staining was performed in a Deltavision microscope (PersonalDV; Imsol) equipped with a Cool Snap HQ CCD camera (Photometrics). Images were analysed by using the softWoRx Suite visualization and analysis software. For time‐lapse experiments, images were acquired by using a motorized Olympus IX81 microscope equipped with a Confocal Yokogawa Spinning Disc illumination system (Roper Scientific) and an Electron‐Multiplier‐CCD Evolve camera (Photometrics). When using the Spinning Disc microscope, images were collected and analysed with a MetaMorph image analysis software. For image processing and analysis, ImageJ software and FIJI software were used. Track length and velocity were measured by using the FIJI MTrackJ plug‐in.
Mass spectrometry
Protein extracts from 107 cells were prepared by extracting with urea buffer. Cells were collected by centrifugation and washed in 8 M urea buffer (8 M Urea, 0.1 N NaOH, 0.05 M EDTA, 2% 2‐mercaptoethanol). Two volumes of glass beads and urea buffer were added to the pellet prior to breaking the cells in a FastPrep machine (MPBio) (three 20‐s cycles at power setting 5.5). Solutions were made up to 0.5 ml with fresh urea buffer and centrifuged at 13,000 g for 10 min at 4°C. Supernatant was transferred to a fresh tube and SDS‐PAGE loading buffer was added, and it was boiled for 10 min. Proteins were separated in an 8% acrylamide gel. Gel lanes were excised into six equal regions and destained with 50 mM ammonium bicarbonate in 10% acetonitrile. Protein disulphide bonds were reduced using 10 mM dithiothreitol (incubated at 56°C for 30 min) and then alkylated with 25 mM iodoacetamide (incubated at room temperature in the dark for 20 min). 500 ng of Promega Gold trypsin was added to each sample and incubated overnight at 37°C. Peptides were extracted using 10% acetonitrile containing 5% formic acid, and the solution was dried in a SpeedVac centrifuge. Dried peptide samples were solubilized in 1% acetonitrile and 0.1% trifluoroacetic acid (TFA) and desalted using Oasis HLB (Waters Corporation). Desalted peptides were eluted using 1 M glycolic acid in 80% acetonitrile and 5% TFA.
Phospho‐proteomic analysis was performed essentially as described before (Wilkes et al, 2015). Briefly, phospho‐peptides were enriched using TiO2 chromatography and dried up. Phospho‐peptides were solubilized in 10 μl of 0.1% TFA prior to loading onto an Ultimate 3,000 nanoflow HPLC. Peptides were on a 90‐min gradient, 4–45% mobile phase B (80% acetonitrile, 0.1% formic acid), and electrosprayed directly into an LTQ XL Orbitrap mass spectrometer. The mass spectrometer was operated in positive ionization mode and in data‐dependent acquisition (DDA) mode. The top seven most abundant ions were subjected to multistage activation (MSA) collision‐induced dissociation fragmentation.
Mascot was used for peptide identification from the MS/MS data by searching against the SwissProt S. cerevisiae database. Fixed modification of serine, threonine and tyrosine was used in Mascot Daemon. MS1 tolerance of 10 ppm and MS2 fragmentation tolerance of 600 mmu were used. A decoy database was also searched so as to report results with a false discovery rate of 1%. Pescal software was used for quantification of the phospho‐peptides using the peak heights of the extracted ion chromatograms. Replicates were averaged and statistical significance of differences across conditions assessed by unpaired t‐test of log‐transformed data. P‐values were adjusted using the Benjamini–Hochberg FDR method.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository database (http://www.ebi.ac.uk/pride) with the data set identifier PXD004966.
Author contributions
AC‐B made the original observation that Cdc14 was implicated in DNA repair by homologous recombination in S. cerevisiae. MTV was responsible for the development of the experiments showing the correlation between metaphase spindle instability and DNA repair. FR collaborated in the demonstration that Spc110 was a target of Cdc14 in response to DNA damage. ED provided technical support for all experiments shown in this work. PF and PRC were responsible for performing the mass spectrometry experiments and the analysis of the data. AC‐B. wrote the manuscript, and all authors analysed the data, discussed the results and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9
Acknowledgements
We would like to thank L. Aragon, P. San Segundo, F. Prado, JF. Diffley, R. Rothstein and J. Haber for providing valuable strains. We particularly thank P. San Segundo for providing reagents, strains and plasmids. We thank L. Aragon, P. San Segundo, CR. Vazquez de Aldana and J. Correa for helpful comments, discussions and reading of the manuscript. We especially thank CR Vazquez de Aldana for his technical support and advice at the microscopy facility. This work was funded by grants from the Ministerio de Economía y Competitividad (BFU2013‐41216‐P) and the Ramón y Cajal Programme (RYC‐201210363) conceded to A.C‐B. F.R. was recipient of a fellowship from Ministerio de Economía y Competitividad.
The EMBO Journal (2017) 36: 79–101
References
- Aylon Y, Liefshitz B, Kupiec M (2004) The CDK regulates repair of double‐strand breaks by homologous recombination during the cell cycle. EMBO J 23: 4868–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam R, Chen SL, Shou W, Mah AS, Alexandru G, Nasmyth K, Annan RS, Carr SA, Deshaies RJ (2004) Phosphorylation by cyclin B‐Cdk underlies release of mitotic exit activator Cdc14 from the nucleolus. Science 305: 516–519 [DOI] [PubMed] [Google Scholar]
- Barlow JH, Rothstein R (2009) Rad52 recruitment is DNA replication independent and regulated by Cdc28 and the Mec1 kinase. EMBO J 28: 1121–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M (2008) The Cdc14B‐Cdh1‐Plk1 axis controls the G2 DNA‐damage‐response checkpoint. Cell 134: 256–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco MG, Matos J, West SC (2014) Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol Cell 54: 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M (2008) Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 9: 297–308 [DOI] [PubMed] [Google Scholar]
- Broadus MR, Gould KL (2012) Multiple protein kinases influence the redistribution of fission yeast Clp1/Cdc14 phosphatase upon genotoxic stress. Mol Biol Cell 23: 4118–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buis J, Stoneham T, Spehalski E, Ferguson DO (2012) Mre11 regulates CtIP‐dependent double‐strand break repair by interaction with CDK2. Nat Struct Mol Biol 19: 246–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bupp JM, Martin AE, Stensrud ES, Jaspersen SL (2007) Telomere anchoring at the nuclear periphery requires the budding yeast Sad1‐UNC‐84 domain protein Mps3. J Cell Biol 179: 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen BP, Chen DJ (2006) Role of non‐homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 5: 1042–1048 [DOI] [PubMed] [Google Scholar]
- Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11‐Rad50‐Xrs2 to resect DNA breaks. Nature 514: 122–125 [DOI] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D'Andrea AD (2016) Repair pathway choices and consequences at the double‐strand break. Trends Cell Biol 26: 52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Niu H, Chung WH, Zhu Z, Papusha A, Shim EY, Lee SE, Sung P, Ira G (2011) Cell cycle regulation of DNA double‐strand break end resection by Cdk1‐dependent Dna2 phosphorylation. Nat Struct Mol Biol 18: 1015–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement A, Solnica‐Krezel L, Gould KL (2011) The Cdc14B phosphatase contributes to ciliogenesis in zebrafish. Development 138: 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente‐Blanco A, Mayan‐Santos M, Schneider DA, Machin F, Jarmuz A, Tschochner H, Aragon L (2009) Cdc14 inhibits transcription by RNA polymerase I during anaphase. Nature 458: 219–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente‐Blanco A, Sen N, Mayan‐Santos M, Sacristan MP, Graham B, Jarmuz A, Giess A, Webb E, Game L, Eick D, Bueno A, Merkenschlager M, Aragon L (2011) Cdc14 phosphatase promotes segregation of telomeres through repression of RNA polymerase II transcription. Nat Cell Biol 13: 1450–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad MN, Lee CY, Wilkerson JL, Dresser ME (2007) MPS3 mediates meiotic bouquet formation in Saccharomyces cerevisiae . Proc Natl Acad Sci USA 104: 8863–8868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Cuervo H, Bueno A (2008) Cds1 controls the release of Cdc14‐like phosphatase Flp1 from the nucleolus to drive full activation of the checkpoint response to replication stress in fission yeast. Mol Biol Cell 19: 2488–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V, Gasser SM (2013) Chromatin movement in the maintenance of genome stability. Cell 152: 1355–1364 [DOI] [PubMed] [Google Scholar]
- Eissler CL, Mazon G, Powers BL, Savinov SN, Symington LS, Hall MC (2014) The Cdk/cDc14 module controls activation of the Yen1 Holliday junction resolvase to promote genome stability. Mol Cell 54: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Luis J, Clemente‐Blanco A, Aragon L, Machin F (2014) Cdc14 targets the Holliday junction resolvase Yen1 to the nucleus in early anaphase. Cell Cycle 13: 1392–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamot M, Manchado E, Chiesa M, Gomez‐Lopez G, Pisano DG, Sacristan MP, Malumbres M (2011) Cdc14b regulates mammalian RNA polymerase II and represses cell cycle transcription. Sci Rep 1: 189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE (2012) Mating‐type genes and MAT switching in Saccharomyces cerevisiae . Genetics 191: 33–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman J, Tippery NP (2004) Systematic quantification of gene interactions by phenotypic array analysis. Genome Biol 5: R49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM (2014) SWR1 and INO80 chromatin remodelers contribute to DNA double‐strand break perinuclear anchorage site choice. Mol Cell 55: 626–639 [DOI] [PubMed] [Google Scholar]
- Huisman SM, Smeets MF, Segal M (2007) Phosphorylation of Spc110p by Cdc28p‐Clb5p kinase contributes to correct spindle morphogenesis in S. cerevisiae . J Cell Sci 120: 435–446 [DOI] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1‐Top3 suppress crossovers during double‐strand break repair in yeast. Cell 115: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, Haber JE, Foiani M (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431: 1011–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno‐Borchart A, Doenges G, Schwob E, Schiebel E, Knop M (2004) A versatile toolbox for PCR‐based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962 [DOI] [PubMed] [Google Scholar]
- Jaspersen SL, Giddings TH Jr, Winey M (2002) Mps3p is a novel component of the yeast spindle pole body that interacts with the yeast centrin homologue Cdc31p. J Cell Biol 159: 945–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Martin AE, Glazko G, Giddings TH Jr, Morgan G, Mushegian A, Winey M (2006) The Sad1‐UNC‐84 homology domain in Mps3 interacts with Mps2 to connect the spindle pole body with the nuclear envelope. J Cell Biol 174: 665–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Hicks WM, Li J, Tay SY, Haber JE (2011) Protein phosphatases pph3, ptc2, and ptc3 play redundant roles in DNA double‐strand break repair by homologous recombination. Mol Cell Biol 31: 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Schiebel E (1998) Receptors determine the cellular localization of a gamma‐tubulin complex and thereby the site of microtubule formation. EMBO J 17: 3952–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk PM, Borovski T, Stap J, Cijsouw T, ten Cate R, Medema JP, Kanaar R, Franken NA, Aten JA (2012) Chromatin mobility is increased at sites of DNA double‐strand breaks. J Cell Sci 125: 2127–2133 [DOI] [PubMed] [Google Scholar]
- Lemaitre C, Grabarz A, Tsouroula K, Andronov L, Furst A, Pankotai T, Heyer V, Rogier M, Attwood KM, Kessler P, Dellaire G, Klaholz B, Reina‐San‐Martin B, Soutoglou E (2014) Nuclear position dictates DNA repair pathway choice. Genes Dev 28: 2450–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TC, Neuner A, Schlosser YT, Scharf AN, Weber L, Schiebel E (2014) Cell‐cycle dependent phosphorylation of yeast pericentrin regulates gamma‐TuSC‐mediated microtubule nucleation. eLife 3: e02208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R (2003) Colocalization of multiple DNA double‐strand breaks at a single Rad52 repair centre. Nat Cell Biol 5: 572–577 [DOI] [PubMed] [Google Scholar]
- Markus SM, Kalutkiewicz KA, Lee WL (2012) Astral microtubule asymmetry provides directional cues for spindle positioning in budding yeast. Exp Cell Res 318: 1400–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiasen DP, Lisby M (2014) Cell cycle regulation of homologous recombination in Saccharomyces cerevisiae . FEMS Microbiol Rev 38: 172–184 [DOI] [PubMed] [Google Scholar]
- Mazon G, Symington LS (2013) Mph1 and Mus81‐Mms4 prevent aberrant processing of mitotic recombination intermediates. Mol Cell 52: 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Petrova B, Lombardi IM, Bertazzi DT, Hub B, Zentgraf H, Pereira G (2010) Targeted localization of Inn1, Cyk3 and Chs2 by the mitotic‐exit network regulates cytokinesis in budding yeast. J Cell Sci 123: 1851–1861 [DOI] [PubMed] [Google Scholar]
- Meitinger F, Palani S, Pereira G (2012) The power of MEN in cytokinesis. Cell Cycle 11: 219–228 [DOI] [PubMed] [Google Scholar]
- Mocciaro A, Schiebel E (2010) Cdc14: a highly conserved family of phosphatases with non‐conserved functions? J Cell Sci 123: 2867–2876 [DOI] [PubMed] [Google Scholar]
- Mocciaro A, Berdougo E, Zeng K, Black E, Vagnarelli P, Earnshaw W, Gillespie D, Jallepalli P, Schiebel E (2010) Vertebrate cells genetically deficient for Cdc14A or Cdc14B retain DNA damage checkpoint proficiency but are impaired in DNA repair. J Cell Biol 189: 631–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai S, Dubrana K, Tsai‐Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ (2008) Functional targeting of DNA damage to a nuclear pore‐associated SUMO‐dependent ubiquitin ligase. Science 322: 597–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL (2009) Mechanisms that regulate localization of a DNA double‐strand break to the nuclear periphery. Genes Dev 23: 912–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza P, Peterson CL (2010) Opening the DNA repair toolbox: localization of DNA double strand breaks to the nuclear periphery. Cell Cycle 9: 43–49 [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE (1999) Multiple pathways of recombination induced by double‐strand breaks in Saccharomyces cerevisiae . Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C (2004) Integration of chemical‐genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol 22: 62–69 [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE (2001) Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage‐induced G2/M arrest. Mol Cell 7: 293–300 [DOI] [PubMed] [Google Scholar]
- Saponaro M, Callahan D, Zheng X, Krejci L, Haber JE, Klein HL, Liberi G (2010) Cdk1 targets Srs2 to complete synthesis‐dependent strand annealing and to promote recombinational repair. PLoS Genet 6: e1000858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoneau A, Robellet X, Ladouceur AM, D'Amours D (2014) Cdk1‐dependent regulation of the Mre11 complex couples DNA repair pathways to cell cycle progression. Cell Cycle 13: 1078–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T (2007) Positional stability of single double‐strand breaks in mammalian cells. Nat Cell Biol 9: 675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Visintin R, Amon A (2002) Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell 108: 207–220 [DOI] [PubMed] [Google Scholar]
- Sundberg HA, Goetsch L, Byers B, Davis TN (1996) Role of calmodulin and Spc110p interaction in the proper assembly of spindle pole body compenents. J Cell Biol 133: 111–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz RK, Rodriguez EC, King MC (2014) A role for nuclear envelope‐bridging complexes in homology‐directed repair. Mol Biol Cell 25: 2461–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A (1998) The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk‐dependent phosphorylation. Mol Cell 2: 709–718 [DOI] [PubMed] [Google Scholar]
- Wei Z, Peddibhotla S, Lin H, Fang X, Li M, Rosen JM, Zhang P (2011) Early‐onset aging and defective DNA damage response in Cdc14b‐deficient mice. Mol Cell Biol 31: 1470–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes EH, Terfve C, Gribben JG, Saez‐Rodriguez J, Cutillas PR (2015) Empirical inference of circuitry and plasticity in a kinase signaling network. Proc Natl Acad Sci USA 112: 7719–7724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shim EY, Davis M, Lee SE (2009) Regulation of repair choice: Cdk1 suppresses recruitment of end joining factors at DNA breaks. DNA Repair 8: 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9