

Abstract
Viral infection during fetal or neonatal stages increases the risk of developing neuropsychiatric disorders such as schizophrenia and autism spectrum disorders. Although neurons express several key regulators of innate immunity, the role of neuronal innate immunity in psychiatric disorders is still unclear. Using cultured neurons and in vivo mouse brain studies, we show here that Toll‐like receptor 3 (TLR3) acts through myeloid differentiation primary response gene 88 (MYD88) to negatively control Disrupted in schizophrenia 1 (Disc1) expression, resulting in impairment of neuronal development. Cytokines are not involved in TLR3‐mediated inhibition of dendrite outgrowth. Instead, TLR3 signaling suppresses expression of several psychiatric disorder‐related genes, including Disc1. The impaired dendritic arborization caused by TLR3 activation is rescued by MYD88 deficiency or DISC1 overexpression. In addition, TLR3 activation at the neonatal stage increases dendritic spine density, but narrows spine heads at postnatal day 21 (P21), suggesting a long‐lasting effect of TLR3 activation on spinogenesis. Our study reveals a novel mechanism of TLR3 in regulation of dendritic morphology and provides an explanation for how environmental factors influence mental health.
Keywords: dendritic growth, dendritic spine formation, innate immunity, neonatal infection, neural development
Subject Categories: Immunology, Molecular Biology of Disease, Neuroscience
Introduction
Acute inflammation at early developmental stages has been suggested to cause brain dysfunction by interfering with neuronal development 1, 2, 3. Microbial infection, programmed cell death, tissue injury, and metabolic stress provide the sources for immune activation and trigger inflammatory responses 4. Innate pattern recognition receptors (PRRs) are the first line of defense molecules to recognize pathogen‐associated molecular patterns (PAMPs) derived from foreign pathogens and that detect endogenous stress signals through interactions with danger‐associated molecular patterns (DAMPs) 5. The major PRRs include Toll‐like receptors (TLRs), C‐type lectin receptors, retinoic acid‐inducible gene 1 (RIG‐I)‐like receptors, Nod‐like receptors (NLRs), and cytosolic DNA sensors 6, 7.
TLRs are the best‐characterized transmembrane PRRs that recognize various exogenous and endogenous molecular patterns 8, 9. TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11 are expressed on cell surfaces and recognize microbial membrane components, whereas TLR3, TLR7, TLR8, and TLR9 are expressed in endosomes to detect both microbial and endogenous nucleic acids 6, 8. Among them, TLR3 is widely expressed in multiple cell types and specifically recognizes double‐strand RNA (dsRNA) derived from viruses and dead cells 4, 8, 10, 11, 12. Unlike other TLRs that use MYD88 to transduce signaling, TLR3 only acts through TIR domain‐containing adapter‐inducing interferon‐β (TRIF) to trigger antiviral type I interferon and proinflammatory cytokine expression 7, 8.
TLR3 has been suggested to influence brain function in many aspects. The classical antiviral activity of TLR3 protects the brain from several neurotropic viral infections, for example, herpes simplex virus (HSV) and West Nile virus (WNV) 13. In addition to its canonical role in antiviral infection, TLR3 activation negatively regulates neural progenitor cell proliferation and axonal growth of dorsal root ganglia neurons 14, 15, suggesting roles for TLR3 in neurodevelopment. Indeed, TLR3 activation by virus invasion or administration of poly(I:C)—a synthetic dsRNA—at the early developmental stage increases the possibility of developing a psychiatric disorder such as autism or schizophrenia 1, 3, 16. Moreover, using a mouse genetic model, research has further indicated that, in the absence of an immune challenge, Tlr3 deletion enhances hippocampal‐dependent learning and memory, impairs amygdala‐related behavior, and increases anxiety 17. Therefore, TLR3 acts as a sensor to detect exogenous pathogens, as well as endogenous developmental or stress signals, to modulate neural development and function.
Peripheral immune responses induced by TLR3 activation have been suggested as being involved in the regulation of neuronal development and brain function 18, 19. However, accumulated studies indicate that TLR3 is expressed in neural progenitors, sensory neurons, and hippocampal and cortical neurons 14, 15, 20, 21, 22, with the evidence suggesting that poly(I:C) can directly activate TLR3 in neurons to control neuronal development, though the molecular mechanism is still unclear. Here, we used various mouse genetic models, combined with in vivo and in vitro poly(I:C) stimulation, to investigate the function and signaling of TLR3 activation in neuronal morphogenesis. Our results show that TLR3 uses a MYD88‐dependent pathway to regulate expression of a series of psychiatric disorder‐related genes, including Disc1, and subsequently influences neuronal development.
Results
TLR3 activation impairs neuronal morphogenesis
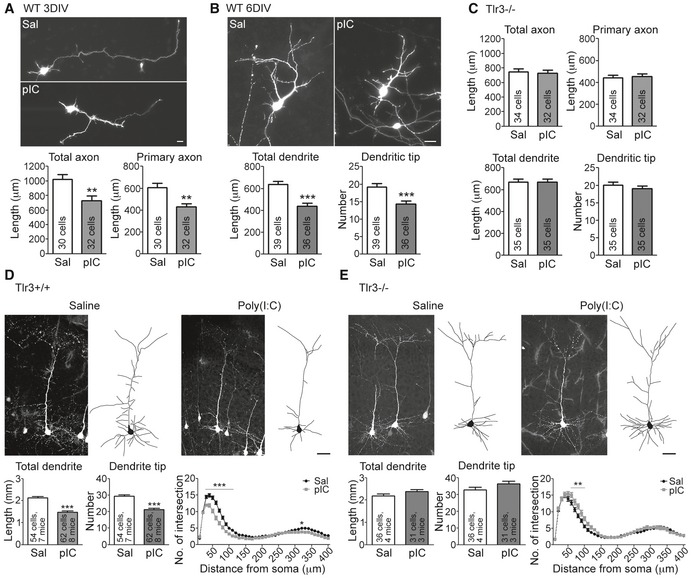
To study the regulation of neuronal morphology by TLR3, we used GFP‐transfected cortical and hippocampal mixed cultures. The GFP signals were used to outline neuronal morphology. Neurons were treated with poly(I:C) at 2 days in vitro (DIV) and 5 DIV for 24 h. Echoing a previous study 15, poly(I:C) treatment impaired axonal growth at 3 DIV in our system (Fig 1A). Moreover, we found that both dendritic length and tip number were reduced after poly(I:C) treatment (Fig 1B). The in vitro effect of poly(I:C) on axonal growth and dendritic arborization was mediated by TLR3, because Tlr3 −/− neurons did not respond to poly(I:C) (Fig 1C). We then investigated the effect of TLR3 activation on neuronal morphogenesis in vivo. Thy1‐Yfp transgenic mice received an intraperitoneal injection of saline or poly(I:C) at P4 and P5, and neuron morphology was monitored at P7. At P7, the Thy1‐Yfp transgene was only expressed in a few projection neurons in the retrosplenial granular cortex (RGC) and caudal hippocampal regions. Based on the YFP signals, we found that systemic administration of poly(I:C) impaired dendritic arborization of the RGC neurons, which was reflected in shorter total dendritic length, fewer branch tips, and a reduced intersection number in Sholl analysis (Fig 1D). The in vivo effect of poly(I:C) on dendritic arborization also depended on TLR3, since poly(I:C) did not inhibit dendritic growth of Tlr3 −/− mice (Fig 1E). These results suggest that TLR3 activation impairs neuronal morphology in vitro, as well as in vivo.
Figure 1. TLR3 activation negatively regulates neuronal morphogenesis in vivo and in vitro .
-
A, BPoly(I:C) treatment inhibits axonal growth at 3 DIV (A) and dendritic growth at 6 DIV (B) in WT neurons. Neurons were transfected with GFP at 1 or 4 DIV. One day later, neurons were treated with poly(I:C) for 24 h before harvest.
-
CAxon and dendrite morphology of Tlr3 −/− neurons after poly(I:C) treatment.
-
D, ENeuronal morphology in saline‐ and poly(I:C)‐treated Tlr3 +/+ ;Thy1‐Yfp (D) and Tlr3 −/−;Thy1‐Yfp (E) mouse brains. Total dendrite length, dendritic tip number, and Sholl analysis of dendritic processes were used to examine the dendrite phenotype.
MYD88 acts downstream of TLR3 in controlling dendritogenesis
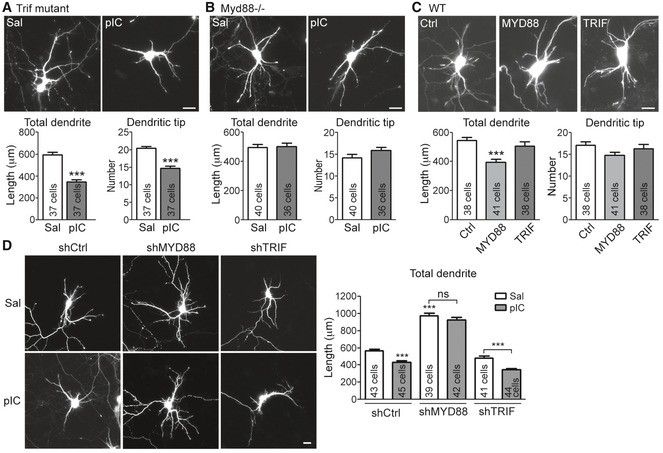
To investigate the downstream pathway of TLR3 in controlling dendritic arborization, Trif‐mutant 23 and Myd88 −/− 24 neurons were treated with poly(I:C). Unexpectedly, although Trif mutation impaired the cytokine production triggered by TLR3 activation (Fig EV1), Trif‐mutant neurons still had shorter dendrites and fewer dendritic tips in the presence of poly(I:C) (Fig 2A). For Myd88 −/− mice, the production of peripheral inflammatory cytokines is greatly reduced in response to TLR activation 24. Myd88 −/− neurons also lost the response to TLR7 activation 20, indicating that Myd88 is indeed functionally ablated in knockout mice. Here, we found that, similar to TLR7 activation, Myd88 −/− neurons also did not respond to TLR3 activation via poly(I:C) treatment (Fig 2B), suggesting that MYD88 is required for TLR3 to inhibit dendritic growth. To further confirm the roles of MYD88 and TRIF in dendritic growth, MYD88 and TRIF were overexpressed in cultured neurons. We found that only MYD88 overexpression was able to shorten the dendritic length (Fig 2C), suggesting that MYD88 overexpression triggers the downstream signaling to shorten dendritic length.
Figure EV1. Trif‐mutant neurons lose the response to poly(I:C) stimulation.
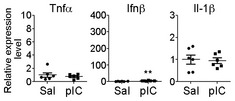
At 5 DIV, Trif‐mutant neurons were treated with poly(I:C) for 6 h, RNAs were extracted, and the expression levels of Tnfa, Ifnb, and Il1b were examined by using quantitative RT‐PCR. The scales of y‐axes are identical to those in Fig 4A for the responses of wild‐type neurons to poly(I:C) and saline control. For Ifnb, the expression levels in response to poly(I:C) decreased dramatically from 100‐fold to 4‐fold relative to Hprt transcripts when comparing wild‐type and Trif‐mutant neurons; although, compared with saline control, poly(I:C) still increased Ifnb expression in Trif‐mutant neurons. Data were analyzed by unpaired t‐test. N = 6. Mean values ± SEM are shown. **P < 0.001.
Figure 2. TLR3 acts through MYD88 to regulate neuronal morphology.
-
A, BQuantitation of dendrite morphology of Trif‐mutant neurons (A) and Myd88 −/− neurons (B) after poly(I:C) stimulation. Trif‐mutant or Myd88 −/− neurons were transfected with a GFP construct at 4 DIV and treated with poly(I:C) 1 day later. Neuron morphology was analyzed at 6 DIV according to the GFP signal. Mean values ± SEM of representatives of three independent experiments are shown. The numbers of analyzed neurons in the representative experiments are indicated in each column. Data were analyzed by unpaired t‐test.
-
CMYD88, but not TRIF, expression reduces dendrite outgrowth. HA‐tagged MYD88 and TRIF were cotransfected with a GFP construct into cultured wild‐type (WT) neurons at 4 DIV, and cell morphology was analyzed at 6 DIV. Mean values ± SEM of representatives of three independent experiments are shown. The numbers of analyzed neurons in the representative experiments are indicated in each column. Data were analyzed by one‐way ANOVA with Bonferroni's multiple comparison test.
-
DMYD88 knockdown, but not TRIF knockdown, robustly increases dendritic arbors and loses the response to poly(I:C). Three DIV neurons were transfected with control shRNA (shCtrl), MYD88 shRNA (shMYD88), or TRIF shRNA (shTRIF) and poly(I:C) was applied into the culture at 5 DIV for 24 h before harvest. Mean values ± SEM of representatives of three independent experiments are shown. The numbers of analyzed neurons in the representative experiments are indicated in each column. Data were analyzed by two‐way ANOVA with Bonferroni's multiple comparison test.
The effect of MYD88 on dendritic growth was also investigated by transfecting wild‐type neurons with the knockdown construct shMYD88. We found that MYD88 knockdown in cultured neurons was sufficient to promote dendritic growth (Fig 2D), suggesting the presence of intrinsic stimulants in the culture that downregulate dendritic growth via MYD88. Addition of poly(I:C) did not shorten the dendritic length of MYD88‐knockdown neurons, echoing the results from Myd88 −/− neurons showing that the effect of poly(I:C) on dendritic growth is mediated by MYD88. In contrast to MYD88, TRIF knockdown did not influence dendritic growth (Fig 2D) and poly(I:C) treatment still restricted dendritic growth of TRIF‐knockdown neurons (Fig 2D). The knockdown effects of MYD88 shRNA and TRIF shRNA on MYD and TRIF expression are shown in Fig EV2. Together, these results suggest that TLR3 activation by poly(I:C) uses a MYD88‐dependent pathway to inhibit dendritic growth.
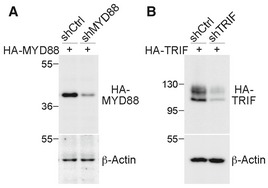
Figure EV2. The knockdown efficiency of MYD88‐ and TRIF‐knockdown constructs.
-
A, BNon‐silencing control shRNA (shCtrl), MYD88 shRNA (shMYD88), or TRIF shRNA (shTRIF) was cotransfected with HA‐tagged MYD88 or HA‐TRIF into HEK293T cells. Twenty‐four hours later, total cell extract was collected and subjected to immunoblot analysis. The effects of (A) shMYD88 and (B) shTRIF are shown. Immunoblotting was performed with HA tag and β‐actin antibodies. β‐Actin was used as a loading control. Molecular mass standards (kD) are indicated next to the gel blots.
The N‐terminal region of MYD88 is critical for TLR3 signaling
TLRs typically deliver the signals to their downstream TIR domain‐containing adaptor via a direct protein–protein interaction of the TIR domains 25. To further investigate the role of MYD88 in the TLR3 pathway, we examined the interaction between TLR3 and MYD88 using coimmunoprecipitation. In HEK293 cells, Myc‐tagged full‐length TLR3, but not the ectodomain of TLR3, was coprecipitated with HA tag antibody in the presence of HA‐tagged MYD88 (Fig 3A), suggesting a specific interaction between the cytoplasmic domain of TLR3 and MYD88. In addition, this interaction was enhanced in the presence of poly(I:C), as there was more TLR3 associated with the MYD88 immunocomplex upon poly(I:C) stimulation (Fig 3B). Although the TIR domain of MYD88 is the region that interacts with various TLRs 26, we found that the C‐terminal TIR domain of MYD88 was not coprecipitated with TLR3 (Fig 3C). Instead, the N‐terminal region of MYD88 that contains the death and intermediate domains precipitated with TLR3 (Fig 3C). These results suggest that MYD88 uses a special mechanism to interact with TLR3.
Figure 3. TLR3 interacts with MYD88.
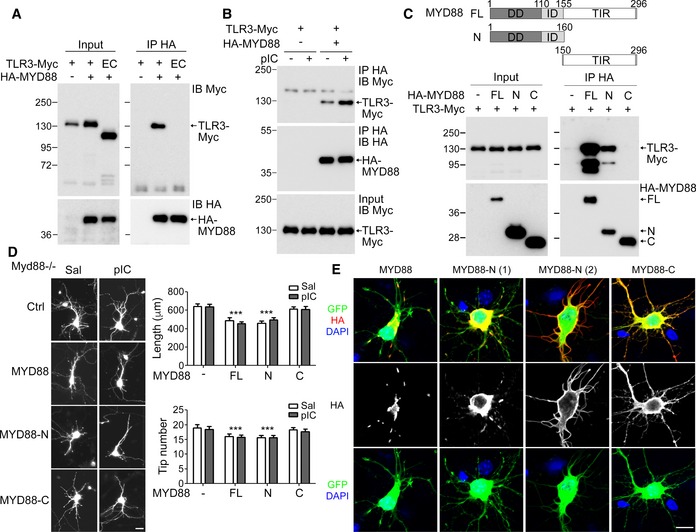
- Coimmunoprecipitation of TLR3 and MYD88. HEK293T cells were transfected with the indicated constructs. Twenty‐four hours later, immunoprecipitation (IP) was performed using HA antibody. The precipitates were immunoblotted (IB) with Myc and HA antibodies as indicated. EC, TLR3 ectodomain.
- Poly(I:C) treatment increases the interaction between TLR3 and MYD88. One day after transfection, cells were treated with saline or poly(I:C) for 30 min before harvesting.
- Upper, schematic of MYD88 constructs. DD, death domain; ID, intermediate domain; TIR, TIR domain. Lower, coimmunoprecipitation of TLR3 and the N‐terminal region of MYD88. For (A–C), representatives of three independent experiments are shown.
- Full‐length and N‐terminal MYD88 inhibit dendrite outgrowth. Myd88 −/− neurons were transfected with control vector (Ctrl), HA‐tagged MYD88 (MYD88), N‐terminal MYD88 (MYD88‐N), or C‐terminal MYD88 (MYD88‐C) with a GFP construct. Poly(I:C) was applied into the culture at 5 DIV and neuronal morphology was analyzed at 6 DIV. Mean values ± SEM of representatives of three independent experiments are shown. The numbers of analyzed neurons in the representative experiments are indicated in each column. Data were analyzed by two‐way ANOVA with Bonferroni's multiple comparison test. ***P < 0.0001. Scale bar, 20 μm.
- Expression patterns of full‐length and truncated fragments of MYD88 in Myd88 −/− neurons. Representative images are shown. Scale bar, 20 μm.
To further investigate the role of the MYD88 N‐terminal region in neurons, we transfected MYD88‐N, MYD88‐C, and full‐length constructs into Myd88 −/− neurons and monitored their effects on dendritic growth. We found that, similar to wild‐type neurons (Fig 2C), Myd88 overexpression in Myd88 −/− neurons reduced dendritic length (Fig 3D). Poly(I:C) treatment did not further shorten dendritic length, supporting that poly(I:C) and MYD88 act on the same pathway to regulate dendritic growth. MYD88‐N exhibited a similar pattern as full‐length MYD88 in regulating dendritic growth (Fig 3D), suggesting the essential and sufficient role of the N‐terminal region of MYD88 for dendritic regulation. In contrast to MYD88‐N and full‐length proteins, MYD88‐C did not have an effect on dendritic length, either in the presence or in the absence of poly(I:C) (Fig 3D). These data strengthen our evidence for the function of the N‐terminal region of MYD88 in controlling dendritic growth.
When we analyzed the Myd88 constructs in neurons, we noticed that full‐length MYD88 formed aggregates in neurons (Fig 3E). For MYD88‐N, ~70–80% of transfected cells also contained MYD88‐N aggregates (Fig 3E, MYD88‐N (1)). In the remaining 20–30% of transfected cells, MYD88‐N tended to concentrate at the edges of soma and entire dendritic processes (Fig 3E, MYD88‐N (2)). MYD88‐C was always evenly distributed in neurons (Fig 3E). Previous study has shown that MYD88 aggregation via its N‐terminal death domain is required for its activation and signal delivery to the downstream kinases 27, 28. Formation of protein aggregates suggests that overexpression of full‐length and the N‐terminal region of MYD88 in neurons is sufficient to activate the MYD88 downstream pathway, consistent with the aforementioned finding that overexpression of full‐length and the N‐terminal region of MYD88 restricted dendritic growth (Fig 3D).
Cytokines are not required for the effect of TLR3 on neurons
In general, TLR activation triggers expression of inflammatory and/or antiviral cytokines 5. A previous study showed that addition of these cytokines to cultured neurons impairs dendritic growth 29. We wondered whether cytokines are involved in the function of TLR3 in dendritic morphogenesis. Upon poly(I:C) stimulation, the RNA levels of Tnfa, Il‐1β, and Ifnb increased, but Il6 was not upregulated (Fig 4A). To investigate the roles of these cytokines, two sets of experiments were performed. The first one was to use poly(I:C) to stimulate Tnfa −/− neurons. Il6 −/− neurons were also included as a negative control, since TLR3 activation did not induce Il6 expression in cultured neurons. The results showed that poly(I:C) still effectively inhibited dendritic growth of these cytokine‐deficient neurons (Fig 4B and C), suggesting that IL‐6, as well as TNF‐α, is not critical for TLR3‐regulated dendritic growth. However, it is still possible that TLR3 activation induces expression of other cytokine(s) rather than TNF‐α to control dendritic growth. Therefore, we performed a second experiment using conditioned medium, which investigated whether neurons secrete cytokines into the culture medium to inhibit dendritic growth. Using conditioned medium, our previous study demonstrated that neuronal TLR7 activation promotes IL‐6 secretion and then inhibits dendritic growth 20. Here, the same experimental design was applied to explore whether TLR3 activation stimulates secretion of unknown factors into the medium to downregulate dendritic growth (Fig 4D, left panel). The conditioned medium of poly(I:C)‐treated neurons was collected and applied to naïve recipient neurons (Fig 4D, left panel). In WT recipient neurons, poly(I:C)‐treated conditioned medium inhibited dendritic growth, just like for the results of neurons treated directly with poly(I:C) (Fig 4D, right upper panel). However, we were concerned that the residual poly(I:C) in the conditioned medium was still able to activate the TLR3 of recipient neurons and result in dendritic shortening. To examine this possibility, we added the conditioned medium to Tlr3 −/− neurons and found that the poly(I:C)‐treated conditioned medium was unable to inhibit dendritic growth of Tlr3 −/− neurons (Fig 4D, right lower panel). These results suggest that TLR3 activation does not use secreted factor(s) to negatively control dendritic growth.
Figure 4. TLR3 activation does not use secreted factors to regulate neuronal morphology.

-
AQuantitative RT‐PCR analysis of proinflammatory cytokines Il6, Tnfa, Il1b and antiviral cytokine Ifnb in WT cultured cortical and hippocampal neurons. Data were analyzed by unpaired t‐test. Il6 and Tnfa, N = 8; Ifnb and Il1b, N = 6.
-
B, CPoly(I:C) treatment inhibits dendrite outgrowth of Il6 −/− neurons (B) and Tnfa −/− neurons (C).
-
DLeft, schematic of experiment using conditioned medium. Upper right, conditioned medium applied to WT neurons. Lower right, conditioned medium applied to Tlr3 −/− neurons. Dendrite morphology was analyzed 1 day after adding conditioned medium.
TLR3 activation cell‐autonomously regulates dendritogenesis
The aforementioned results suggest that a secreted factor is not involved in the effect of TLR3 on dendritic morphology. It seems possible that TLR3 activation acts cell‐autonomously to control neuronal morphology. To address the possibility, we applied TLR3 shRNA (shTLR3) to reduce TLR3 expression (Fig EV3). Since the knockdown construct coexpresses GFP, we can use GFP signal to identify the transfected neurons and outline neuronal morphology. Given that transfection efficiency in our neuronal cultures is < 1%, shTLR3‐transfected neurons are expected to be surrounded by many non‐transfected cells that still secrete cytokines in response to poly(I:C) stimulation. Therefore, TLR3‐knockdown neurons are still exposed to cytokines upon poly(I:C) treatment. For neurons transfected with non‐silencing control shCtrl 30, poly(I:C) treatment obviously restricted dendritic growth (Fig 5). For shTLR3‐expressing neurons, poly(I:C) treatment did not shorten dendritic length (Fig 5), supporting that secreted factors do not cause dendritic growth deficits under poly(I:C) treatment and that TLR3 activation acts cell‐autonomously to downregulate dendritic growth. Note that, similar to MYD88 knockdown (Fig 2D), TLR3 knockdown resulted in longer dendrites compared with control shCtrl (Fig 5), which also indicates the presence of an intrinsic ligand for TLR3 in the cultures. Because both mRNAs and the microtubule‐binding protein, stathmin, have been shown to serve as intrinsic ligands of TLR3 12, 31, dead cells in the cultures likely provide intrinsic stimulants to activate TLR3 and restrict dendritic growth. When TLR3 expression levels were reduced, knockdown neurons became unresponsive to the intrinsic ligands and grew better. Our previous study of TLR7 also indicated that neuronal TLR7 recognizes single‐stranded RNA in cultures and restricts dendritic growth 20. Thus, both TLR7 and TLR3 are able to use a cell‐autonomous mechanism to regulate dendritic growth.
Figure EV3. The effect of the TLR3‐knockdown construct on TLR3 expression.
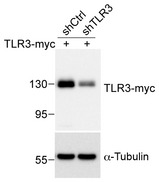
Control shRNA (shCtrl) or TLR3 shRNA (shTLR3) was cotransfected with Myc‐tagged TLR3 into HEK293T cells. Total cell extract was collected 24 h later and subjected to immunoblot analysis. Immunoblotting was performed with Myc tag and α‐tubulin antibodies. α‐Tubulin was used as a loading control.
Figure 5. TLR3 knockdown in neurons promotes dendrite outgrowth and is resistant to poly(I:C) stimulation.
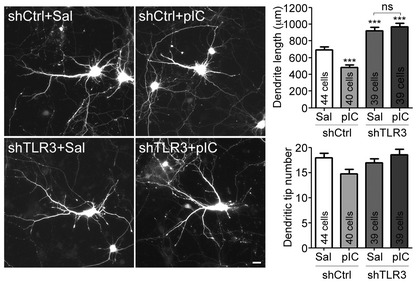
At 4 DIV, cultured cortical neurons were transfected with indicated plasmids. Three days later, neurons were treated with 10 μg/ml poly(I:C) for 24 h. Neuronal morphology was monitored by GFP signals at 8 DIV. Mean values ± SEM of representatives of three independent experiments are shown. The numbers of analyzed neurons in the representative experiments are indicated in each column. Data were analyzed by two‐way ANOVA with Bonferroni's multiple comparison test. Scale bar, 20 μm. ***P < 0.0001, ns: non‐significant. See also Fig EV3.
TLR3 regulates expression of psychiatric disease genes
TLR3 activation at early developmental stages induces autism‐ and schizophrenia‐like behavior 32. Our aforementioned results also suggest that TLR3 activation in neurons restricts dendritic growth. Therefore, we wondered whether TLR3 activation alters the expression of genes that regulate neuronal morphology and also are associated with schizophrenia and/or autism. Quantitative RT–PCR was used to determine the expression levels of several autism‐ or schizophrenia‐related genes in response to poly(I:C) stimulation, namely Auts2, Cdh8, Disc1, Fmr1, Mecp2, Nrg1, Nrxn1, Pten, Shank3, Tbr1, Tsc1, and Ube3a (Table EV1). We found that except for Cdh8, Mecp2, and Tsc1, all remaining nine genes in the list were downregulated in cultured cortical and hippocampal neurons upon poly(I:C) stimulation (Fig 6A). To confirm that the reduced expression of these genes is mediated by TLR3, the effect of poly(I:C) was examined using Tlr3 −/− neurons. The expression of Nrg1, Nrxn1, Shank3, and Tbr1 genes was still reduced in Tlr3 −/− neurons upon poly(I:C) treatment (Fig 6B), suggesting that TLR3 is not required for the effect of poly(I:C) on reducing Nrg1, Nrxn1, Shank3, and Tbr1 expression. It also indicates that only Auts2, Disc1, Fmr1, Pten, and Ube3a were specifically regulated by TLR3 activation (Fig 6B). In Myd88 −/− neurons, poly(I:C) treatment did not noticeably influence expressions of Auts2, Disc1, Fmr1, Pten, and Ube3a (Fig 6C), consistent with the role of MYD88 in the TLR3 pathway. The in vivo expression of these five genes was further investigated by injection of poly(I:C) into P5 mice. We found that Auts2, Disc1, Fmr1, and Ube3a still had lower RNA expression levels in poly(I:C)‐treated mouse brains (Fig 6D), indicating the role of TLR3 in controlling the expression of these autism‐ and schizophrenia‐related genes in brains.
Figure 6. TLR3 activation downregulates expression of neuropsychiatric disorder‐related genes.
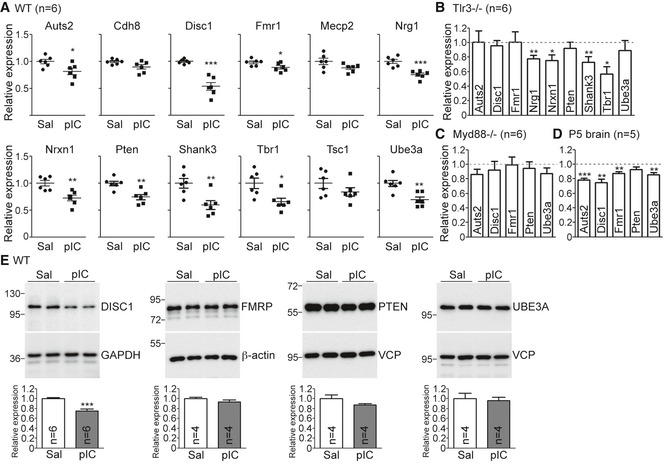
-
AExpression of 12 neuropsychiatric disorder‐related genes in saline‐ and poly(I:C)‐treated WT neurons.
-
BExpression of nine neuropsychiatric disorder‐related genes in poly(I:C)‐treated Tlr3 −/− neurons. Relative expression levels compared with vehicle control are shown. Dashed line indicates the level of saline control.
-
C, DExpression of Aust2, Disc1, Fmr1, Pten, and Ube3a in poly(I:C)‐treated Myd88 −/− neurons (C) and poly(I:C)‐treated P5 WT mouse brains (D). Similar to (B), relative levels compared with vehicle control are shown. Dashed lines indicate the level of saline control.
-
EDIV 5 WT neurons were treated with poly(I:C) for 24 h. The total neuronal lysates were subjected to immunoblotting with the indicated antibodies. VCP and β‐actin were used as loading controls. The protein levels were normalized with β‐actin.
We further examined the protein levels of the aforementioned genes 24 h after poly(I:C) treatment, that is, the time‐point for monitoring dendritic morphology. Only DISC1 protein levels were obviously lower upon poly(I:C) stimulation (Fig 6E). Moreover, the reduction in DISC1 protein levels was also dependent on TLR3 and MYD88, because poly(I:C) did not influence the DISC1 levels in Tlr3 −/− and Myd88 −/− neurons (Fig EV4). In addition, the results of immunostaining indicated that, similar to poly(I:C) treatment, MYD88 overexpression in neurons reduced DISC1 protein levels (Fig EV5). Together, these results suggest that TLR3 activation reduces Disc1 expression in neurons in a MYD88‐dependent manner.
Figure EV4. Poly(I:C) does not alter DISC1 protein levels in Tlr3 −/− neurons or Myd88 −/− neurons.
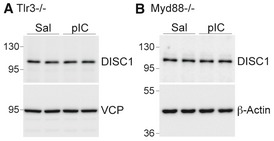
-
A, BAt 5 DIV, Tlr3 −/− (A) and Myd88 −/− (B) neurons were treated with poly(I:C) for 24 h. The total neuronal lysates were subjected to immunoblotting with the indicated antibodies. VCP and β‐actin were used as loading controls.
Figure EV5. Poly(I:C) treatment and MYD88 overexpression reduce the DISC1 protein levels in neurons.
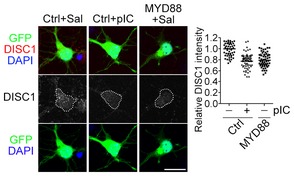
Neurons were transfected with control vector (Ctrl) or HA‐MYD88 (MYD88) with a GFP construct at 4 DIV. One day later, neurons were treated with saline or 10 μg/ml poly(I:C) for 24 h. Neurons were fixed and immunostained with DISC1 antibody at 6 DIV. Counterstaining with DAPI was performed to label the nuclei. DISC1 signal within the soma region (white dashed line as indicated) was quantified using ImageJ. The data were reproduced in two independent experiments. Only one set of representative data is shown. The dots indicate the relative DISC1 intensities of individual cells. The bars indicate mean of each group. Data were analyzed by one‐way ANOVA followed by Bonferroni's multiple comparison test. Ctrl − pIC versus Ctrl + pIC, P < 0.0001; Ctrl − pIC versus MYD88 − pIC, P < 0.0001. Scale bar, 20 μm.
DISC1 acts downstream of TLR3 in regulating dendritic growth
We then investigated the functional role of DISC1 in TLR3‐dependent neuronal morphogenesis by overexpression of DISC1 in poly(I:C)‐treated neurons. Neurons overexpressing DISC1 were resistant to poly(I:C) treatment, as the total dendrite lengths and total numbers of dendritic branch tips of poly(I:C)‐treated Disc1‐transfected neurons were comparable with those of neurons treated with vehicle controls with or without DISC1 overexpression (Fig 7A). The mouse DISC1 L604F mutant, which corresponds to the human DISC1 L607F mutant that is a common missense variant in schizophrenia patients 33, 34, was unable to rescue the dendritic defects caused by TLR3 activation (Fig 7A), further suggesting the specific effect of DISC1 on the TLR3 pathway. In addition to the in vitro‐cultured neurons, we also applied in utero electroporation to investigate the rescue effect of DISC1 overexpression in brains. Similar to the results of Thy1‐Yfp mice (Fig 1D), poly(I:C) treatment reduced the complexity of dendritic arbors in the layer 2/3 cortical neurons labeled with GFP by in utero electroporation (Fig 7B). With additional DISC1 expression, the neurons had comparable dendrite length, dendritic tip number, and dendritic complexity in the saline‐ and poly(I:C)‐treated mice (Fig 7C). These results demonstrate that DISC1 is critical for the effect of TLR3 activation on downregulation of dendritic morphogenesis.
Figure 7. DISC1 overexpression rescues the effects of TLR3 activation on dendritic arborization in vitro and in vivo .
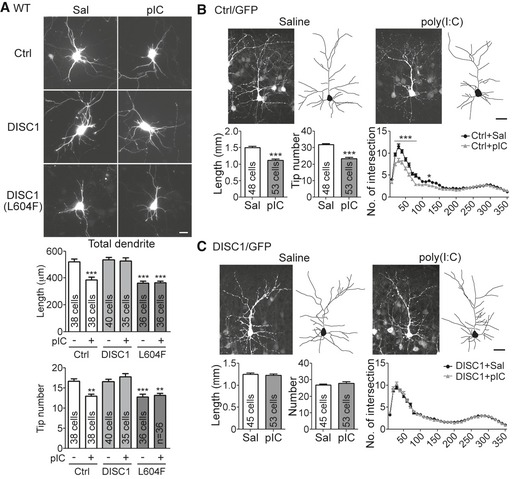
-
AOverexpression of WT DISC1, but not the DISC1 L604F mutant, in cultured neurons suppresses dendrite withdrawal induced by TLR3 activation. Mean values ± SEM of representatives of three independent experiments are shown. The numbers of analyzed neurons in the representative experiments are indicated in each column. Data were analyzed by two‐way ANOVA with Bonferroni's multiple comparison test. Scale bars, 20 μm. **P < 0.001, ***P < 0.0001.
-
B, COverexpression of DISC1 in cortical neurons of mouse brain is resistant to poly(I:C)‐triggered reduction in dendritic arborization. Vector control (Ctrl) (B) or Myc‐DISC1 (C) was coexpressed with GFP in cortical layer 2/3 neurons in mouse brain. After poly(I:C) injection at P4 and P5, the neuronal morphology was analyzed at P7 by tracing the GFP signals. The data of dendrite length and tip number were analyzed by unpaired t‐test. The Sholl data were analyzed by two‐way ANOVA with Bonferroni's multiple comparison test. The numbers of analyzed neurons collected from 3 to 4 mice of each group are indicated. Mean values ± SEM are shown. Scale bar, 30 μm. *P < 0.05, ***P < 0.0001.
In conclusion, these results support that TLR3 activation regulates expression of Disc1 to control dendritic arborization.
TLR3 activation impairs dendritic spinogenesis
We further investigated whether TLR3 activation also regulates dendritic spine formation. In cortical and hippocampal mixed cultures, poly(I:C) treatment increased dendritic spine density, but narrowed the spine heads at 18 DIV (Fig 8A), suggesting that TLR3 activation alters dendritic spine formation. To further confirm this effect in vivo, we intraperitoneally injected poly(I:C) or saline vehicle control into Thy1‐Yfp mice at P4 and P5 and characterized dendritic spines of layer 5 pyramidal neurons of the somatosensory cortex at P21 (Fig 8B). After being stimulated with poly(I:C), dendritic spine density was increased, whereas spine heads tended to be smaller (Fig 8C), which is similar to what we observed in cultured neurons (Fig 8A). The effect of poly(I:C) on dendritic spine density and size was TLR3‐specific, because dendritic spines of Tlr3 −/− mice were not altered by poly(I:C) treatment (Fig 8D). In conclusion, these analyses suggest that TLR3 activation during the neonatal stage has a long‐lasting effect on dendritic spine formation at later stages.
Figure 8. TLR3 activation alters spine morphology in vitro and in vivo .
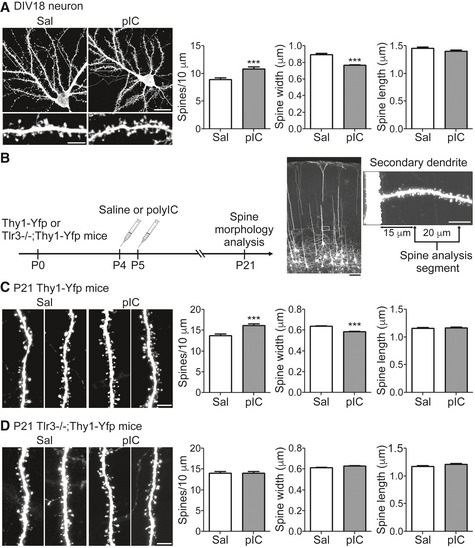
-
ACortical and hippocampal mixed neuronal cultures were transfected with a GFP construct at 12 DIV and treated with poly(I:C) (pIC) at 17 DIV for 24 h. Spine morphology was examined at 18 DIV. Three secondary dendrites of each neuron were selected to analyze the spine density, the width of the spine head, and the spine length. Mean values ± SEM of representatives of three independent experiments are shown. For the saline group, 15 neurons/45 dendrites were examined; for the poly(I:C) group, 16 neurons/48 dendrites were examined. Data were analyzed by unpaired t‐test. Scale bar, 20 μm in right upper panel and 5 μm in lower left panel. ***P < 0.0001.
-
BSchematic of the experimental procedure for in vivo spine morphology analysis in (C and D). Representative images of somatosensory cortical layer 5 neurons and a high magnification image of a secondary dendrite are shown. Scale bar, 100 μm in the brain section (black) and 10 μm in the dendrite segment (white).
-
C, DSpine morphology of P21 Thy1‐Yfp (C) and Tlr3 −/−; Thy1‐Yfp mice (D) after poly(I:C) stimulation at P4 and P5. One secondary dendrite of an apical dendrite of each somatosensory layer 5 cortical neuron was selected to examine the spine morphology. Three mice were analyzed for each group. Saline‐treated Thy1‐Yfp, 44 neurons; poly(I:C)‐treated Thy1‐Yfp, 43 neurons; saline‐treated Tlr3 −/− ;Thy1‐Yfp, 46 neurons; poly(I:C)‐treated Tlr3 −/− ;Thy1‐Yfp, 50 neurons. Mean values ± SEM are shown. Data were analyzed by unpaired t‐test. Scale bars, 5 μm. ***P < 0.0001.
Discussion
TLR3 is an endosomal TLR that recognizes viral and self dsRNA to trigger antiviral and inflammatory responses 35. In this report, we found three unexpected properties of TLR3 signaling. First, MYD88, but not TRIF, is required for TLR3 to control dendritic growth. Second, MYD88 uses its N‐terminal death and intermediate domains, but not the TIR domain, to interact with the TLR3 cytoplasmic region. Third, although TLR3 activation in neurons induces cytokine expression, cytokines are not required for TLR3 to negatively regulate dendritic growth.
TLR3 is known to utilize only TRIF to elicit an antiviral response 23, 36, and involvement of other adaptor molecules has not been suggested for TLR3‐dependent signaling. Here, in our cultured neurons, the data suggest that TRIF is required for poly(I:C) treatment to induce cytokine expression. However, TRIF‐mediated cytokine production is not required for regulation of dendritic growth. Instead, TLR3 uses MYD88 to control neuronal morphology, indicating that TLR3 uses both TRIF and MYD88 to deliver its downstream signaling. The TRIF‐dependent signaling is needed to regulate cytokine expression. The MYD88‐dependent pathway is crucial to controlling neuronal morphology. It is well known that TLR4 uses both MYD88 and TRIF to induce inflammatory responses 35. Recent studies have reported that TLR5 can also use TRIF to deliver signaling in intestinal epithelial cells 37, and TLR9 acts through TRIF as well as Sarm1 to promote a tolerogenic response in plasmacytoid dendritic cells and induce apoptosis in neurons, respectively 38, 39. Therefore, canonical TLR signal pathways may not be applied universally. Distinct cell types and various physiological conditions may alter TLR signal pathways.
The death and TIR domains of MYD88 have been identified to interact with multiple proteins. Fas‐associated protein with death domain (FADD), interleukin‐1 receptor‐associated kinases (IRAKs), and interferon regulatory factor 7 (IRF7) bind to the death domain of MYD88 40, 41, 42, 43. The TIR domain of MYD88 associates with IL‐1R, MYD88‐adapter‐like (MAL, #32), IRF3, and TLRs (except TLR3) 26, 44, 45. Unlike other TLR–MYD88 interactions, the TLR3 and MYD88 interaction is a TIR domain to death domain association. This kind of TLR3–MYD88 interaction may form a different protein complex to trigger distinct cell responses. The downstream signaling of this TLR3–MYD88 complex needs to be further characterized.
Previous reports indicate that maternal cytokines, such as IL‐6 and IL‐17a, from pregnant mice infected with virus or receiving poly(I:C) stimulation cause abnormal fetal brain development and develop autism‐ and schizophrenic‐like behavior in adult offspring 18, 19, 32. Some viruses are neurotropic and may cross the placenta to infect the fetus, for example, West Nile virus and Zika virus 46, 47, and these viruses have been suggested to trigger TLR3 signaling 48, 49. Therefore, both developing neurons and microglia may encounter the invasive virus directly and activate their TLR3 pathway to defend themselves from the foreign pathogens. Indeed, our data suggest that poly(I:C) treatment induces Tnfa, Il1b, and Ifnb expression in cultured neurons, which may be involved in defense against pathogenic infection. However, cytokines produced by neurons upon poly(I:C) stimulation are either inefficient or insufficient to modulate neuronal morphology. Instead, our data suggest that neuronal TLR3 controls expression of a series of autism‐ and schizophrenia‐related genes to regulate neuronal morphology. It is reasonable to speculate that in addition to peripheral and microglial immune responses 21, 32, 50, neuronal TLR3 activation may also contribute to altering neuronal and brain function. It will be important to further verify the effect of neuronal TLR3 on brain development and function using neuron‐specific Tlr3‐knockout mice.
In this report, we provide evidence that the protein levels of DISC1 were noticeably reduced after poly(I:C) treatment. Overexpression of wild‐type DISC1, but not a mutant identified from schizophrenia patients, effectively rescues the dendritic defects caused by TLR3 activation, suggesting a critical role of DISC1 in TLR3‐regulated neurodevelopment. DISC1 is associated with schizophrenia, autism, bipolar disorders, and depression 51, 52, 53, 54. It is a versatile molecule involved in neurogenesis, postmitotic neural migration, dendritic arborization, dendritic spine formation, and synaptic plasticity 55, 56, 57, 58, 59. DISC1 associates with microtubule‐associated proteins, motor molecules, signaling proteins, and RNA binding proteins to influence cytoskeleton function, protein and RNA trafficking, and neuronal development and plasticity 59, 60, 61, 62, 63, 64. Reduction in DISC1 expression by TLR3 activation may then influence the downstream pathways of DISC1 and result in neurodevelopmental impairment. It would certainly be interesting to further determine whether a specific downstream effector of DISC1 is particularly sensitive to TLR3 activation.
Several studies have suggested that environment–genetic interactions play an important role in neurodevelopmental disorders. For instance, maternal immune activation and Tsc2 haploinsufficiency work together to disturb social behavior in offspring 65. Expression of the Disc1 mutant in mice exacerbates the behavioral defects induced by poly(I:C) injections at prenatal stages 66, 67, 68. Our study indicates regulation of DISC1 expression by TLR3 activation, implying that environmental factors, such as viral infection, regulate expression of the genes associated with psychiatric disorders and influence neurodevelopment. Our findings further support that genetic polymorphism plus environmental stimulations may contribute to the diversity of psychiatric disorders.
Our research shows that, compared with the saline control group, poly(I:C)‐treated mice had a higher dendritic spine density at P21, which resembles one of the common features of autism spectrum disorders, namely impaired synapse elimination during maturation 69, 70. Since TLR3 activation at P4 and P5 influences dendritic spine density and morphology at P21, this result indicates a long‐term effect of TLR3 activation on neurodevelopment. Recently, epigenetic reprogramming has been shown to control long‐lasting effects of innate immune responses in peripheral tissues 71. It is very likely that neuronal innate immunity uses a similar mechanism to control gene expression over a long‐lasting period and thus impact on neural plasticity and cognition, which presents a very intriguing subject for future study.
In the current study, the RNA levels of Auts2, Disc1, Fmr1, and Ube3a were noticeably reduced upon TLR3 activation. However, only DISC1 proteins were obviously decreased 24 h after TLR3 activation. It is unclear why only DISC1 protein levels were altered. Perhaps, the other proteins are more stable and thus take a longer time to reflect the effect of RNA reduction. If TLR3 does alter the protein levels of these genes, these proteins may impact on other neuronal phenotypes or responses. It would be intriguing to examine these possibilities in the future.
A previous study showed that poly(I:C) treatment did not influence Disc1 expression in mice 72, which conflicts with the findings of our study. There are at least two possibilities to explain the discrepancy. First, an outbred strain of ICR mice was used in the previous paper, whereas we used mice in the C57BL/6 background in our study. Different genetic backgrounds may result in different responses to poly(I:C) stimulation. In particular, ICR mice, but not C57BL/6, have been shown to carry a 25‐bp deletion in the Disc1 gene and cannot express full‐length DISC1 proteins 73. Second, the experimental procedures between the previous work and our study differ. In our study, a single shot of poly(I:C) was intraperitoneally injected into neonatal mice at P5. Six hours later, cortex and hippocampus were harvested for quantitative RT‐PCR. In the previous work, poly(I:C) was injected once per day from P2 to P6. Only the hippocampus was harvested for quantitative RT‐PCR 2 and 24 h after the last injection. These differences may also contribute to the differences between our results and those of the previous study.
In conclusion, our data suggest that TLR3 uses a non‐canonical MYD88‐dependent pathway to control Disc1 expression and cell‐autonomously regulate neuronal morphology. Neuronal innate immunity may regulate neuronal morphology in response to exogenous immune challenge as well as intrinsic developmental signals or stress responses.
Materials and Methods
Animals and treatments
Thy1‐Yfp 74, Tlr3 −/− 75, Trif‐mutant 23, Myd88 −/− 24, Il6 −/− 76, and Tnfa −/− 77 mice in a C57BL/6 background were purchased from Jackson Laboratory. Animals were housed in the animal facility of the Institute of Molecular Biology, Academia Sinica, with a 12‐h light/12‐h dark cycle and controlled temperature and humidity. All animal experiments were performed with the approval of the Academia Sinica Institutional Animal Care and Utilization Committee and in strict accordance with its guidelines and those of the Council of Agriculture Guidebook for the Care and Use of Laboratory Animals. For cortical and hippocampal mixed neuronal cultures, E16‐17 mouse embryos of both genders were used. For in vivo neuronal morphology analysis, Thy1‐Yfp and Tlr3 −/− ;Thy1‐Yfp mice of both genders received an intraperitoneal injection of saline or 5 mg/kg poly(I:C) (HMW; InvivoGen) daily at P4 and P5. The brains were harvested at P7 for dendritic arbor analysis and at P21 for examining spine morphology. Neuronal morphology was monitored with YFP signal.
Cell culture, transfection, drug treatment, immunoprecipitation, and immunoblotting
Transfections of HEK293T cells were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions 30. Mouse cortical and hippocampal mixed neurons were cultured in Neurobasal medium/DMEM (1:1) with B27 supplement and transfected using calcium phosphate precipitation methods as described 20, 30. Cotransfected or coexpressed GFP was used to outline neuronal morphology. Neurons were treated with 10 μg/ml poly(I:C) for 24 h before being harvested. Immunoprecipitation and immunoblotting were performed as described 30, except that the protein signals were visualized and quantified using ImageQuant LAS 4000 with the software ImageQuant LAS 4000 Biomolecular Imager (GE Healthcare).
Immunocytochemistry and immunohistochemistry
Immunostaining of cultured neurons and 100‐μm‐thick brain sections was performed as described 30. Immunofluorescent images of cultured neurons were visualized at room temperature with a fluorescence microscope (Axioimage M2; Zeiss) equipped with a 20×/NA 0.80 (Plan‐Apochromat) objective lens and acquired using a cooled charge‐coupled device camera (Rolera EM‐C2; QImaging) with Zen software (Zeiss). For brain sections, neuronal images were captured at room temperature with a confocal microscope (LSM 700, Zeiss) equipped with a 20×/NA 0.80 (Plan‐Apochromat) objective lens and Zen acquisition and analysis software (Zeiss). The images were processed using Photoshop (Adobe) with minimal adjustment of brightness or contrast applied to the entire images. The camera lucida drawings were performed with ImageJ.
In utero electroporation
In utero electroporation was performed as previously described with slight modifications 22. Briefly, pregnant wild‐type B6 mice were exposed to 1.8% isoflurane in oxygen during surgery and the embryos at embryonic day 15 were used for in vivo electroporation of the cortex. Control plasmid pCAGEN (Addgene) or pCAG‐Myc‐DISC1 was mixed with pCAG‐GFP (Addgene) in a 3:1 ratio and injected into one of the lateral ventricles of embryo brain using a glass micropipette. The embryonic brain then received five pulses (30 V for 50 ms) of electric shock with 950‐ms intervals using an ECM830 square wave pulse generator (BTX, Harvard Apparatus). Poly(I:C) injection, perfusion, brain fixation/sectioning, and neuronal image acquisition were performed as described in the previous sections.
Neuronal morphometry
For axons, the lengths of both the primary axon (i.e. the longest axon) and total axons were measured. For dendrites, the total dendrite length was measured, including primary dendrites and all dendritic branches, and the number of dendritic tips was counted. For neuronal morphology in brains, assessment of the intersection number in Sholl analysis 78 was also undertaken. For in vivo spine analysis, secondary dendrites of apical dendrites of layer 5 cortical neurons from the somatosensory cortex were chosen to examine spine density, the width of the spine head, and spine length. To minimize the effects of bias, the critical experiments were performed blind by relabeling the samples with the assistance of other laboratory members. For cultured neurons, all experiments were repeated at least three times. For each repeat, at least 30 neurons were randomly picked from each group for analysis. The conclusions of all results shown in this manuscript can be repeated in three independent experiments. For in vivo analysis, at least 30 neurons collected from three or more mice were used for analysis. All measurements were carried out using the software ImageJ.
Quantitative RT‐PCR
Wild‐type (WT), Tlr3 −/− , and Myd88 −/− cultured neurons were treated with 10 μg/ml poly(I:C) for 6 h before harvesting. To quantify gene expression in brains, WT mice received an intraperitoneal injection of saline or 5 mg/kg poly(I:C) at P5. Six hours after injection, mouse cortex and hippocampus were harvested for total RNA extraction using Trizol reagent according to the manufacturer's instructions (Invitrogen) followed by DNase I digestion (New England BioLabs). Reverse transcription and quantitative RT‐PCR analysis was performed using the Transcriptor First Strand cDNA Synthesis Kit (Roche) with an oligo(dT)18 primer and the Universal ProbeLibrary probes (UPL; Roche) system, respectively 20, 22. Primer sets for cytokines were as described 20. The primer sets for neurodevelopmental disease genes and internal control Hprt for quantitative RT‐PCR analysis are listed in Table EV1. Levels of target mRNA were normalized to levels of Hprt mRNA measured at the same time on the same reaction plate.
Antibodies and plasmids
Detailed information on the antibodies and DNA constructs used in this report is summarized in Tables EV2 and EV3, respectively.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software. For animal studies, all available mice were subjected to analysis and no randomization was performed. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications 20, 30, 79, 80. No sample was excluded from the analyses. The majority of data met the assumption (normal distribution) of statistical tests, except Fig 2A Tip/pIC, 4A Tnfa/Sal and Ifnb/Sal, 4B Tip/Sal, 4C Tip/pIC, 7C Tip/pIC, 8A width and length, 8C width and length, and 8D width and length. Experiments were performed blind by relabeling the samples with the assistance of other laboratory members. For two‐group experiments, the unpaired t‐test was used. For experiments with more than two groups, one‐way ANOVA with post hoc Bonferroni correction was applied. For the Sholl analysis, two‐way ANOVA with post hoc Bonferroni correction was used. Data are presented as the mean ± SEM.
Author contributions
C‐YC and Y‐PH were involved in conceptualization; C‐YC and H‐YL made investigation (H‐YL, initial analysis of Fig 4A); C‐YC, H‐YL and Y‐PH were involved in writing; C‐YC and Y‐PH performed visualization; Y‐PH was involved in supervision, project administration and funding acquisition.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File
Acknowledgements
We thank Dr. Kozo Kaibuchi (Nagoya University, Japan) for the kind gifts of the Disc1 construct and antibody; Dr. Constance L. Cepko for the pCAGEN (Addgene plasmid # 11160) and pCAG‐GFP (Addgene plasmid # 11150); the Genomic Core Facility of the Institute of Molecular Biology, Academia Sinica, for technical support; Dr. John O'Brien for English editing; and the members of Yi‐Ping Hsueh's laboratory for relabeling samples for the blinded experiments. This work was supported by grants from the Simons Foundation (SFARI# 388449), Academia Sinica (AS‐103‐TP‐B05), and the Ministry of Science and Technology (MOST 104‐2321‐B‐001‐050 and 105‐B‐001‐031) to Y.‐P. Hsueh. C.‐Y. Chen is also supported by the Ministry of Science and Technology (MOST 104‐2811‐B‐001‐048).
EMBO Reports (2017) 18: 169–183
References
- 1. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, Toovey S, Prinssen EP (2014) Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 10: 643–660 [DOI] [PubMed] [Google Scholar]
- 2. Estes ML, McAllister AK (2015) Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Rev Neurosci 16: 469–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB (2015) Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry 2: 258–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin C, Flavell RA (2013) Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol 132: 287–294 [DOI] [PubMed] [Google Scholar]
- 5. Kawai T, Akira S (2010) The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 11: 373–384 [DOI] [PubMed] [Google Scholar]
- 6. Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820 [DOI] [PubMed] [Google Scholar]
- 7. Zohaib A, Sarfraz A, Kaleem QM, Ye J, Mughal MN, Navid MT, Khan FA, Duan X, Zhu B, Wan S et al (2016) The Yin and Yang of antiviral innate immunity in central nervous system. Curr Pharm Des 22: 648–655 [DOI] [PubMed] [Google Scholar]
- 8. Kawai T, Akira S (2011) Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650 [DOI] [PubMed] [Google Scholar]
- 9. Czirr E, Wyss‐Coray T (2012) The immunology of neurodegeneration. J Clin Invest 122: 1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL (2008) TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med 205: 2609–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Green NM, Moody KS, Debatis M, Marshak‐Rothstein A (2012) Activation of autoreactive B cells by endogenous TLR7 and TLR3 RNA ligands. J Biol Chem 287: 39789–39799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kariko K, Ni H, Capodici J, Lamphier M, Weissman D (2004) mRNA is an endogenous ligand for Toll‐like receptor 3. J Biol Chem 279: 12542–12550 [DOI] [PubMed] [Google Scholar]
- 13. Carty M, Reinert L, Paludan SR, Bowie AG (2014) Innate antiviral signalling in the central nervous system. Trends Immunol 35: 79–87 [DOI] [PubMed] [Google Scholar]
- 14. Lathia JD, Okun E, Tang SC, Griffioen K, Cheng A, Mughal MR, Laryea G, Selvaraj PK, ffrench‐Constant C, Magnus T et al (2008) Toll‐like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. J Neurosci 28: 13978–13984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cameron JS, Alexopoulou L, Sloane JA, DiBernardo AB, Ma Y, Kosaras B, Flavell R, Strittmatter SM, Volpe J, Sidman R et al (2007) Toll‐like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci 27: 13033–13041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Majidi J, Kosari‐Nasab M, Salari AA (2016) Developmental minocycline treatment reverses the effects of neonatal immune activation on anxiety‐ and depression‐like behaviors, hippocampal inflammation, and HPA axis activity in adult mice. Brain Res Bull 120: 1–13 [DOI] [PubMed] [Google Scholar]
- 17. Okun E, Griffioen K, Barak B, Roberts NJ, Castro K, Pita MA, Cheng A, Mughal MR, Wan R, Ashery U et al (2010) Toll‐like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc Natl Acad Sci USA 107: 15625–15630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR (2016) The maternal interleukin‐17a pathway in mice promotes autism‐like phenotypes in offspring. Science 351: 933–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH (2007) Maternal immune activation alters fetal brain development through interleukin‐6. J Neurosci 27: 10695–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu HY, Hong YF, Huang CM, Chen CY, Huang TN, Hsueh YP (2013) TLR7 negatively regulates dendrite outgrowth through the Myd88‐c‐Fos‐IL‐6 pathway. J Neurosci 33: 11479–11493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu HY, Chen CY, Hsueh YP (2014) Innate immune responses regulate morphogenesis and degeneration: roles of Toll‐like receptors and Sarm1 in neurons. Neurosci Bull 30: 645–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu HY, Huang CM, Hung YF, Hsueh YP (2015) The microRNAs Let7c and miR21 are recognized by neuronal Toll‐like receptor 7 to restrict dendritic growth of neurons. Exp Neurol 269: 202–212 [DOI] [PubMed] [Google Scholar]
- 23. Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S et al (2003) Identification of Lps2 as a key transducer of MyD88‐independent TIR signalling. Nature 424: 743–748 [DOI] [PubMed] [Google Scholar]
- 24. Hou B, Reizis B, DeFranco AL (2008) Toll‐like receptors activate innate and adaptive immunity by using dendritic cell‐intrinsic and ‐extrinsic mechanisms. Immunity 29: 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kawai T, Akira S (2006) TLR signaling. Cell Death Differ 13: 816–825 [DOI] [PubMed] [Google Scholar]
- 26. Ohnishi H, Tochio H, Kato Z, Orii KE, Li A, Kimura T, Hiroaki H, Kondo N, Shirakawa M (2009) Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc Natl Acad Sci USA 106: 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin SC, Lo YC, Wu H (2010) Helical assembly in the MyD88‐IRAK4‐IRAK2 complex in TLR/IL‐1R signalling. Nature 465: 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Into T, Inomata M, Niida S, Murakami Y, Shibata K (2010) Regulation of MyD88 aggregation and the MyD88‐dependent signaling pathway by sequestosome 1 and histone deacetylase 6. J Biol Chem 285: 35759–35769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gilmore JH, Fredrik Jarskog L, Vadlamudi S, Lauder JM (2004) Prenatal infection and risk for schizophrenia: IL‐1beta, IL‐6, and TNFalpha inhibit cortical neuron dendrite development. Neuropsychopharmacology 29: 1221–1229 [DOI] [PubMed] [Google Scholar]
- 30. Chen CY, Lin CW, Chang CY, Jiang ST, Hsueh YP (2011) Sarm1, a negative regulator of innate immunity, interacts with syndecan‐2 and regulates neuronal morphology. J Cell Biol 193: 769–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bsibsi M, Bajramovic JJ, Vogt MH, van Duijvenvoorden E, Baghat A, Persoon‐Deen C, Tielen F, Verbeek R, Huitinga I, Ryffel B et al (2010) The microtubule regulator stathmin is an endogenous protein agonist for TLR3. J Immunol 184: 6929–6937 [DOI] [PubMed] [Google Scholar]
- 32. Patterson PH (2011) Maternal infection and immune involvement in autism. Trends Mol Med 17: 389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh KK, De Rienzo G, Drane L, Mao Y, Flood Z, Madison J, Ferreira M, Bergen S, King C, Sklar P et al (2011) Common DISC1 polymorphisms disrupt Wnt/GSK3beta signaling and brain development. Neuron 72: 545–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Malavasi EL, Ogawa F, Porteous DJ, Millar JK (2012) DISC1 variants 37W and 607F disrupt its nuclear targeting and regulatory role in ATF4‐mediated transcription. Hum Mol Genet 21: 2779–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O'Neill LA, Golenbock D, Bowie AG (2013) The history of Toll‐like receptors – redefining innate immunity. Nat Rev Immunol 13: 453–460 [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K et al (2003) Role of adaptor TRIF in the MyD88‐independent toll‐like receptor signaling pathway. Science 301: 640–643 [DOI] [PubMed] [Google Scholar]
- 37. Choi YJ, Im E, Chung HK, Pothoulakis C, Rhee SH (2010) TRIF mediates Toll‐like receptor 5‐induced signaling in intestinal epithelial cells. J Biol Chem 285: 37570–37578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Volpi C, Fallarino F, Pallotta MT, Bianchi R, Vacca C, Belladonna ML, Orabona C, De Luca A, Boon L, Romani L et al (2013) High doses of CpG oligodeoxynucleotides stimulate a tolerogenic TLR9‐TRIF pathway. Nat Commun 4: 1852 [DOI] [PubMed] [Google Scholar]
- 39. Mukherjee P, Winkler CW, Taylor KG, Woods TA, Nair V, Khan BA, Peterson KE (2015) SARM1, not MyD88, mediates TLR7/TLR9‐induced apoptosis in neurons. J Immunol 195: 4913–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhande R, Dauphinee SM, Thomas JA, Yamamoto M, Akira S, Karsan A (2007) FADD negatively regulates lipopolysaccharide signaling by impairing interleukin‐1 receptor‐associated kinase 1‐MyD88 interaction. Mol Cell Biol 27: 7394–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muzio M, Ni J, Feng P, Dixit VM (1997) IRAK (Pelle) family member IRAK‐2 and MyD88 as proximal mediators of IL‐1 signaling. Science 278: 1612–1615 [DOI] [PubMed] [Google Scholar]
- 42. George J, Motshwene PG, Wang H, Kubarenko AV, Rautanen A, Mills TC, Hill AV, Gay NJ, Weber AN (2011) Two human MYD88 variants, S34Y and R98C, interfere with MyD88‐IRAK4‐myddosome assembly. J Biol Chem 286: 1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S et al (2004) Interferon‐alpha induction through Toll‐like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5: 1061–1068 [DOI] [PubMed] [Google Scholar]
- 44. Gay NJ, Symmons MF, Gangloff M, Bryant CE (2014) Assembly and localization of Toll‐like receptor signalling complexes. Nat Rev Immunol 14: 546–558 [DOI] [PubMed] [Google Scholar]
- 45. Iliev DB, Sobhkhez M, Fremmerlid K, Jorgensen JB (2011) MyD88 interacts with interferon regulatory factor (IRF) 3 and IRF7 in Atlantic salmon (Salmo salar): transgenic SsMyD88 modulates the IRF‐induced type I interferon response and accumulates in aggresomes. J Biol Chem 286: 42715–42724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. O'Leary DR, Kuhn S, Kniss KL, Hinckley AF, Rasmussen SA, Pape WJ, Kightlinger LK, Beecham BD, Miller TK, Neitzel DF et al (2006) Birth outcomes following West Nile Virus infection of pregnant women in the United States: 2003–2004. Pediatrics 117: e537–e545 [DOI] [PubMed] [Google Scholar]
- 47. Mlakar J, Korva M, Tul N, Popovic M, Poljsak‐Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V et al (2016) Zika virus associated with microcephaly. New Engl J Med 374: 951–958 [DOI] [PubMed] [Google Scholar]
- 48. Daffis S, Samuel MA, Suthar MS, Gale M Jr, Diamond MS (2008) Toll‐like receptor 3 has a protective role against West Nile virus infection. J Virol 82: 10349–10358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera‐Lecoin M, Surasombatpattana P, Talignani L, Thomas F et al (2015) Biology of Zika virus infection in human skin cells. J Virol 89: 8880–8896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA (2006) Microglia recognize double‐stranded RNA via TLR3. J Immunol 176: 3804–3812 [DOI] [PubMed] [Google Scholar]
- 51. Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ (2001) Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain‐expressed genes: clinical and P300 findings in a family. Am J Hum Genet 69: 428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Millar JK, Wilson‐Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, St Clair DM, Muir WJ, Blackwood DH et al (2000) Disruption of two novel genes by a translocation co‐segregating with schizophrenia. Hum Mol Genet 9: 1415–1423 [DOI] [PubMed] [Google Scholar]
- 53. Kilpinen H, Ylisaukko‐Oja T, Hennah W, Palo OM, Varilo T, Vanhala R, Nieminen‐von Wendt T, von Wendt L, Paunio T, Peltonen L (2008) Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry 13: 187–196 [DOI] [PubMed] [Google Scholar]
- 54. Zheng F, Wang L, Jia M, Yue W, Ruan Y, Lu T, Liu J, Li J, Zhang D (2011) Evidence for association between Disrupted‐in‐Schizophrenia 1 (DISC1) gene polymorphisms and autism in Chinese Han population: a family‐based association study. Behav Brain Funct 7: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK (2008) The DISC locus in psychiatric illness. Mol Psychiatry 13: 36–64 [DOI] [PubMed] [Google Scholar]
- 56. Brandon NJ, Sawa A (2011) Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci 12: 707–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hattori T, Shimizu S, Koyama Y, Yamada K, Kuwahara R, Kumamoto N, Matsuzaki S, Ito A, Katayama T, Tohyama M (2010) DISC1 regulates cell‐cell adhesion, cell‐matrix adhesion and neurite outgrowth. Mol Psychiatry 15: 778 798–809 [DOI] [PubMed] [Google Scholar]
- 58. Lee FH, Fadel MP, Preston‐Maher K, Cordes SP, Clapcote SJ, Price DJ, Roder JC, Wong AH (2011) Disc1 point mutations in mice affect development of the cerebral cortex. J Neurosci 31: 3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tsuboi D, Kuroda K, Tanaka M, Namba T, Iizuka Y, Taya S, Shinoda T, Hikita T, Muraoka S, Iizuka M et al (2015) Disrupted‐in‐schizophrenia 1 regulates transport of ITPR1 mRNA for synaptic plasticity. Nat Neurosci 18: 698–707 [DOI] [PubMed] [Google Scholar]
- 60. Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, Bonnert TP, Whiting PJ, Brandon NJ (2007) Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry 12: 74–86 [DOI] [PubMed] [Google Scholar]
- 61. Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH (2004) Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron 44: 263–277 [DOI] [PubMed] [Google Scholar]
- 62. Morris JA, Kandpal G, Ma L, Austin CP (2003) DISC1 (Disrupted‐In‐Schizophrenia 1) is a centrosome‐associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Hum Mol Genet 12: 1591–1608 [DOI] [PubMed] [Google Scholar]
- 63. Taya S, Shinoda T, Tsuboi D, Asaki J, Nagai K, Hikita T, Kuroda S, Kuroda K, Shimizu M, Hirotsune S et al (2007) DISC1 regulates the transport of the NUDEL/LIS1/14‐3‐3epsilon complex through kinesin‐1. J Neurosci 27: 15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shinoda T, Taya S, Tsuboi D, Hikita T, Matsuzawa R, Kuroda S, Iwamatsu A, Kaibuchi K (2007) DISC1 regulates neurotrophin‐induced axon elongation via interaction with Grb2. J Neurosci 27: 4–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ehninger D, Sano Y, de Vries PJ, Dies K, Franz D, Geschwind DH, Kaur M, Lee YS, Li W, Lowe JK et al (2012) Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol Psychiatry 17: 62–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, Pogorelov V, Ladenheim B, Yang C, Krasnova IN et al (2010) Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry 68: 1172–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nagai T, Kitahara Y, Ibi D, Nabeshima T, Sawa A, Yamada K (2011) Effects of antipsychotics on the behavioral deficits in human dominant‐negative DISC1 transgenic mice with neonatal polyI: C treatment. Behav Brain Res 225: 305–310 [DOI] [PubMed] [Google Scholar]
- 68. Lipina TV, Zai C, Hlousek D, Roder JC, Wong AH (2013) Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia‐related behaviors in mice. J Neurosci 33: 7654–7666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14: 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR, Brynczka C, Rush R, Thomas A, Paylor R et al (2012) Reversal of disease‐related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med 4: 152ra128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O'Neill LA, Xavier RJ (2016) Trained immunity: a program of innate immune memory in health and disease. Science 352: aaf1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ibi D, Nagai T, Kitahara Y, Mizoguchi H, Koike H, Shiraki A, Takuma K, Kamei H, Noda Y, Nitta A et al (2009) Neonatal polyI: C treatment in mice results in schizophrenia‐like behavioral and neurochemical abnormalities in adulthood. Neurosci Res 64: 297–305 [DOI] [PubMed] [Google Scholar]
- 73. Kuroda K, Yamada S, Tanaka M, Iizuka M, Yano H, Mori D, Tsuboi D, Nishioka T, Namba T, Iizuka Y et al (2011) Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum Mol Genet 20: 4666–4683 [DOI] [PubMed] [Google Scholar]
- 74. Feng G, Mellor RH, Bernstein M, Keller‐Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28: 41–51 [DOI] [PubMed] [Google Scholar]
- 75. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double‐stranded RNA and activation of NF‐kappaB by Toll‐like receptor 3. Nature 413: 732–738 [DOI] [PubMed] [Google Scholar]
- 76. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G (1994) Impaired immune and acute‐phase responses in interleukin‐6‐deficient mice. Nature 368: 339–342 [DOI] [PubMed] [Google Scholar]
- 77. Pasparakis M, Alexopoulou L, Episkopou V, Kollias G (1996) Immune and inflammatory responses in TNF alpha‐deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med 184: 1397–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sholl DA (1953) Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 87: 387–406 [PMC free article] [PubMed] [Google Scholar]
- 79. Huang TN, Chuang HC, Chou WH, Chen CY, Wang HF, Chou SJ, Hsueh YP (2014) Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat Neurosci 17: 240–247 [DOI] [PubMed] [Google Scholar]
- 80. Chuang HC, Huang TN, Hsueh YP (2014) Neuronal excitation upregulates Tbr1, a high‐confidence risk gene of autism, mediating Grin2b expression in the adult brain. Front Cell Neurosci 8: 280 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File