

Like all tumors, human gliomas are remarkably tolerant of hypoxia. Conversely, hypoxia rapidly activates cell death pathways in healthy brain cells. Therapies aimed at reversing the hypoxia-tolerance of cancerous cells offer considerable promise in the treatment of brain tumors; however, the underlying cellular mechanisms that permit tumor cells to tolerate prolonged hypoxia are poorly understood. Key to normal cellular responses to hypoxia are a number of ion channels whose expression or activity are modified by changes in oxygen. Such changes in ion channel function alter cellular ion gradients that control downstream signaling cascades, which in turn mediate a wide variety of intra- and inter-cellular 2nd messenger systems that are critical to cellular viability, growth, and proliferation. Therefore, the ability of ion channels to respond to hypoxia and beneficially regulate these downstream cellular pathways plays a key role in determining cellular tolerance to low oxygen stress. As such, differences in the oxygen-sensitivity of ion channels between healthy vs. cancerous cells are excellent candidates to contribute to the hypoxia-tolerance of tumor cells. Of particular interest in the search for treatments of human glioma is one particular family of ion channels, the Ca2+-activated and voltage-dependent K+ (BK) channels, which are found in both the plasmalemmal and mitochondrial inner membranes.
BK channels are critical signaling intermediates that detect changes in local oxygen availability and coordinate cellular responses to hypoxia in healthy brain cells.1,2 The function of BK channels in cancerous cells is less well understood; however, in biopsies from human gliomas, BK channel expression is upregulated relative to in healthy tissue and this change correlates with the malignancy grade of tumors.3 Perhaps more importantly, pharmacologically activating plasma membrane BK (plasmaBK) channels activates apoptotic cell death pathways in a human glioma cell line.4 Together, these studies suggest a strong correlation between plasmaBK-channel activity and glioma cell viability and tumor proliferation. In combination with their role as cellular oxygen sensors in healthy brain tissue, this putative connection between BK channel function and glioma viability suggests that there may be a link between the function of plasmaBK channels in glioma cells and their tolerance to hypoxia. It is therefore reasonable to hypothesize that differences in the expression or function of plasmaBK channels between healthy brain cells and glioma contribute to the divergent hypoxia-tolerance of these 2 cell types. Experiments designed to elucidate functional differences between plasmaBK channels in healthy vs. glioma cells may thus inform the development of treatments for hypoxia-tolerant tumors.
In a recent study, we employed electrophysiological approaches to examine the oxygen-sensitivity and channel kinetics of mitochondrial and plasmalemmal BK channels in a human glioma cell line (LN229 cells).5 We found that unlike in healthy neurons and glia, plasmaBK channels in human glioma are not sensitive to hypoxia. Furthermore, we found that activating plasmaBK channels (but not mitochondrial BK channels) increases glioma cell death in both normoxia and hypoxia (1% O2), and that cell viability decreases by > 50% following 24 h of hypoxia when plasmaBK channels are activated. Conversely, untreated LN229 cells tolerate 24 h of hypoxia without significant mortality. The exact mode via which plasmaBK channel activation killed glioma cells in our study is unclear but another recent study from Sontheimer’s group demonstrated that plasmaBK channel activation is central to the execution of volume-dependent apoptosis in a different human glioma cell line.6 Although this study did not investigate hypoxia, these authors reported that TNF-α-induced apoptosis is dependent upon the activation of plasmaBK channels via a Ca2+-dependent mechanism. Specifically, Ca2+ release from endoplasmic reticulum (ER) stores leads to the activation of plasmaBK channels, which permits the large-scale K+ efflux that is the key mediator of volume-dependent apoptosis (Fig. 1).
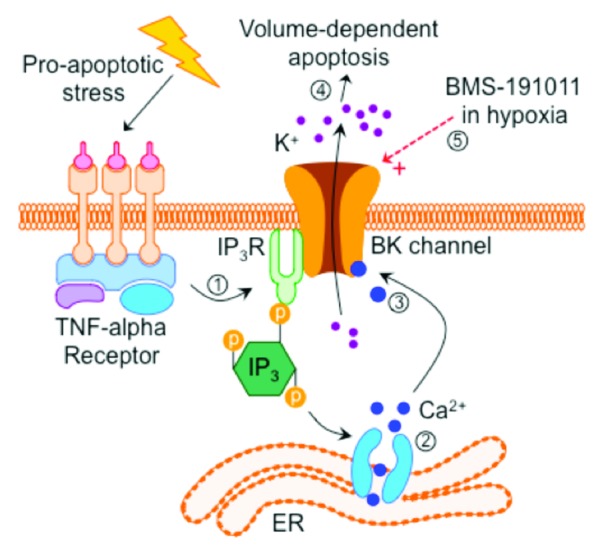
Figure 1. Model of plasmaBK channel-mediated cell death in human glioma cells. (1) Pharmacological activation of apoptosis induces cytosolic Ca2+ accumulation via (2) inositol triphosphate (Ins(1,4,5)P3)-mediated stimulation of ER Ca2+ release. (3) Elevated cytoplasmic [Ca2+] chronically activates plasmaBK channels, which permit K+ efflux, leading to (4) volume-dependent apoptosis. (5) During hypoxia, pharmacological activation of plasmaBK channels may induce glioma cell death via a similar mechanism.
Plasmalemmal BK channels are particularly well suited to a role in this mechanism as they (1) are unusually sensitive to local changes in cytosolic [Ca2+] relative to BK channels in healthy glia,7 and (2) are located in close proximity to the intracellular point of Ca2+ release from the ER: plasmaBK channels co-localize with, and are connected via lipid rafts to inosital-triphosphate receptors, which are the primary receptors that mediate intracellular Ca2+ release from ER stores.8 It remains to be determined whether a similar mechanism is involved in the loss of cellular viability in hypoxic glioma cells in which plasmaBK channels are pharmacologically activated; however, based on our findings and previous work in the field, we propose that the muted or absent hypoxic response of glioma plasmaBK channels might represent a strategic adaptation to hypoxia that permits tumor cells to survive in otherwise deleterious hypoxic environments by preventing the execution of volume-dependent apoptosis. Tumor cells are generally known to tolerate hypoxic environments but the mechanism of their hypoxic resistance is unknown. Enhancing plasmaBK channel activity or “re-activating” the sensitivity of these channels to hypoxia may offer therapeutic potential in the treatment of cancers; however, the data currently available is largely correlative and further studies that directly investigate the putative link between altered plasmaBK channel function and the hypoxia-tolerance of glioma cells are required to determine if there is a causative link between these 2 phenomenon.
References
- 1.Liu H, Moczydlowski E, et al. . J Clin Invest 1999; 104:577 - 88; http://dx.doi.org/ 10.1172/JCI7291; PMID: 10487772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng Y, et al. . Cell Physiol Biochem 2008; 22:127 - 36; http://dx.doi.org/ 10.1159/000149790; PMID: 18769039 [DOI] [PubMed] [Google Scholar]
- 3.Sontheimer H. . Exp Biol Med (Maywood) 2008; 233:779 - 91; http://dx.doi.org/ 10.3181/0711-MR-308; PMID: 18445774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Debska-Vielhaber G, et al. . J Physiol Pharmacol 2009; 60:27 - 36; PMID: 20065494 [PubMed] [Google Scholar]
- 5.Gu XQ, et al. . Glia 2014; 62:504 - 13; http://dx.doi.org/ 10.1002/glia.22620; PMID: 24446243 [DOI] [PubMed] [Google Scholar]
- 6.McFerrin MB, et al. . Am J Physiol Cell Physiol 2012; 303:C1070 - 8; http://dx.doi.org/ 10.1152/ajpcell.00040.2012; PMID: 22992678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ransom CB, et al. . Glia 2002; 38:281 - 91; http://dx.doi.org/ 10.1002/glia.10064; PMID: 12007141 [DOI] [PubMed] [Google Scholar]
- 8.Weaver AK, et al. . J Biol Chem 2007; 282:31558 - 68; http://dx.doi.org/ 10.1074/jbc.M702866200; PMID: 17711864 [DOI] [PMC free article] [PubMed] [Google Scholar]