

Ca2+ is a ubiquitous yet unusual second messenger in that it is not metabolized. Instead, Ca2+ levels in the cytosol are regulated by dynamic redistribution of Ca2+ across the plasma membrane and the membranes of organelles. In addition to the ER, acidic organelles such as lysosomes, endosomes, and secretory vesicles (in secretory cell types) store and release Ca2+. Endolysosomal Ca2+ homeostasis has several cellular functions; among them are apoptosis, trafficking, energy metabolism and fusion/fission events. The messenger NAADP appears particularly important in mobilizing acidic Ca2+ stores and in many cases NAADP-evoked signals are amplified by Ca2+ channels on the ER.1 The molecular basis for triggering of Ca2+ release from acidic organelles by NAADP however is unclear. Our recent evidence supports a central role for the endolysosomal 2-pore channels, TPC1 and TPC2.2
Figure 1 illustrates the main ion transporters and channels of the endolysosomal system. The driving force for most transporters is the endolysosomal H+ gradient. This is generated by the V-type H+ ATPase pump which together with the 2Cl-/H+ exchangers, CLC7 (in lysosomes) and CLC5 (in endosomes) load endolysosomes with HCl. The endolysosomes express 4 Na+/H+ exchangers (NHE6-NHE9), which likely participate in loading of Na+3. Ca2+ uptake by the endolysosomes requires the H+ gradient suggesting the presence of a H+/Ca2+ (and perhaps Na+/Ca2+) exchanger although this transporter(s) is unknown. The end result is that endolysosomes are rich in H+, Cl-, Na+ and Ca2+. In addition to TPCs, endolysosomes express a distinct family of Ca2+-permeable channels, the TRP mucolipins; TPC1 and TRPML3 are found mainly in endosomes and TPC2 and TRPML1 are expressed in lysosomes. The TRPMLs function as PI(3,5)P2-activated non-selective cation channels,4,5 whereas the TPCs function as Na+ permeable channel that can also conduct Ca2+ and are activated by PI(3,5)P22,3,6 and, as will be argued below, by NAADP.2
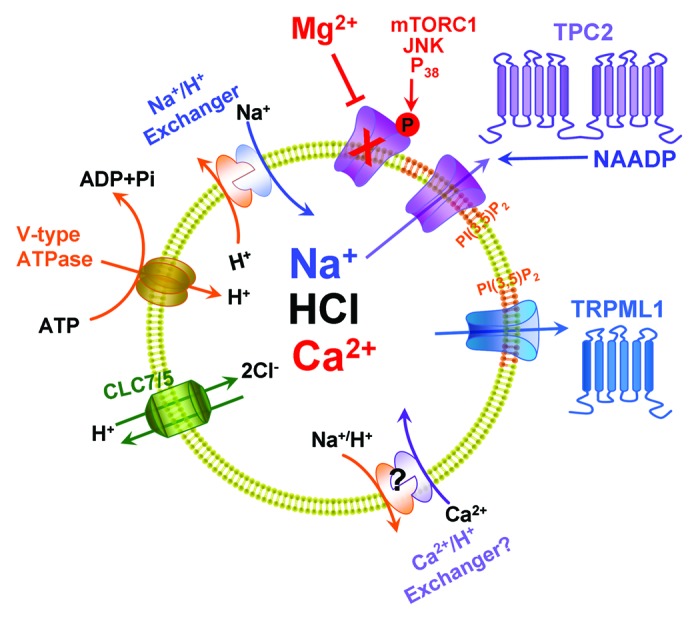
Figure 1. The major endolysosomal ion transporters. Ion transport by the endolysosomes is powered by the V-type H+ pump, which together with the 2Cl-/H+ exchangers CLC7 and CLC5 generate the endolysosomal H+ gradient. Na+/H+ exchangers and likely Ca2+/H+ exchangers utilize the H+ gradient to load the endolysosomes with Na+ and Ca2+. The 2 endolysosomal channels that have been associated with Ca2+ release are TRPMLs and TPCs. Both channels are activated by PI(3,5)P2 and TPC2 is activated by NAADP and is inhibited by cytoplasmic Mg2+ and by phosphorylation by multiple protein kinases.
While TRPML1 is dispensable for NAADP-mediated Ca2+ release,7 multiple observations indicate that the TPCs are essential for NAADP-mediated Ca2+ release. These include inhibition of NAADP responses by knockdown of TPCs, overexpression of dominant negative TPCs or knockout of TPC2 in mice. Conversely, NAADP-mediated Ca2+ release is enhanced by overexpression of TPCs.8 However, the role of the TPCs in NAADP-mediated Ca2+ release was questioned based on the findings that TPC currents in lysosomes are activated by PI(3,5)P2 and not by NAADP, that the currents are Na+-selective and the persistence of NAADP-mediated Ca2+ release in mouse line deleted of both TPC1 and TPC2.3 Further, it was suggested that the TPCs are inhibited when phosphorylated by the mTORC1 kinase to function as metabolic sensors by controlling the lysosomal membrane potential.6
In our recent study, we examined the response of TPCs to PI(3,5)P2 and NAADP, their regulation by protein kinases and their function as sensors of cell metabolic activity. We confirmed activation of TPC2 by PI(3,5)P2 and its permeability to Na+. Notably, the TPC2 current is regulated by cytoplasmic Mg2+ (Mg2+cyt).2 Mg2+cyt specifically inhibits the outward current (cations flowing from the cytosol into the lysosomes) with an apparent affinity within the physiological Mg2+ concentration. Hence, TPC2 functions as Mg2+cyt sensor, with Mg2+cyt determining the lysosomal membrane potential. Changes in Mg2+cyt that are observed with receptor stimulation and with changes in cytoplasmic ATP and cell energetics are thus transmitted to the cellular energetic hub, the lysosome, through rapid acute changes in Mg2+cyt. Our studies further showed that not only mTORC1, but multiple kinases, such as JNK and P38 kinases, are potent inhibitors of TPC2 (see Fig. 1). The JNK and P38 kinases are also important in cellular energetics and may provide a long-term response to changes in cellular energetics.
Another important finding of our study is that under controlled Mg2+ concentrations, TPC2 is readily activated by NAADP. Furthermore, the TPC2 current and NAADP-mediated Ca2+ release are identically regulated by Mg2+, PI(3,5)P2 and protein kinases.2 It is thus clear that (1) TPC2 is activated by NAADP and (2) the function of TPC2 is essential for NAADP-mediated Ca2+ release. Our data affirm a central role for TPCs in NAADP action and go partway in reconciling differences in experimental outcomes between labs. But questions remain. Although TPCs appear Ca2+-permeable under defined recording conditions, might Na+ be the main permeant ion under physiological conditions? If so, is it possible that changes in membrane potential through TPCs indirectly drive NAADP-evoked Ca2+ signals through an unidentified associated channel? Alternatively, might permeability of TPCs to Ca2+, even if limited, be sufficient to account for the Ca2+ mobilizing actions of NAADP in a cellular setting given amplification of NAADP responses by ER Ca2+ channels? Certainly more work is required in defining the biophysical properties of TPCs, TPC-interacting proteins and the cell biology underlying communication between acidic organelles and the ER. Further questions are what is the role TRPMLs in endolysosomal Ca2+ homeostasis and what is their physiological activator? What is the functional relationship between the TPCs and TRPMLs? These and other questions are likely to be addressed in the coming years.
Acknowledgments
We thank Drs Malini Ahuja and Sandip Patel for their valuable comments and suggestions.
References
- 1.Kiselyov KK, et al. . Channels (Austin) 2012; 6:344 - 51; http://dx.doi.org/ 10.4161/chan.21723; PMID: 22907062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jha A, et al. . EMBO J 2014; 33:501 - 11; http://dx.doi.org/ 10.1002/embj.201387035; PMID: 24502975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, et al. . Cell 2012; 151:372 - 83; http://dx.doi.org/ 10.1016/j.cell.2012.08.036; PMID: 23063126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong XP, et al. . Nat Commun 2010; 1:38; http://dx.doi.org/ 10.1038/ncomms1037; PMID: 20802798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HJ, et al. . J Biol Chem 2010; 285:16513 - 20; http://dx.doi.org/ 10.1074/jbc.M109.078204; PMID: 20378547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cang C, et al. . Cell 2013; 152:778 - 90; http://dx.doi.org/ 10.1016/j.cell.2013.01.023; PMID: 23394946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamaguchi S, et al. . J Biol Chem 2011; 286:22934 - 42; http://dx.doi.org/ 10.1074/jbc.M110.210930; PMID: 21540176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan AJ, et al. . Bioessays 2014; 36:173 - 83; http://dx.doi.org/ 10.1002/bies.201300118; PMID: 24277557 [DOI] [PubMed] [Google Scholar]