

1. Introduction
The 199th ENMC workshop on FHL1-related myopathies brought together 20 clinicians and basic scientists from 10 different countries (Australia, Austria, Canada, Egypt, France, Germany, Italy, Japan, United Kingdom, USA) in Naarden from the 7th to the 9th of June 2013. FHL1-related myopathies are a clinically and pathologically heterogeneous group of conditions caused by mutations in the FHL1 gene, which encodes various alternatively spliced isoforms. Topics addressed included clinical, molecular genetic and pathophysiological aspects in patients as well as in animal and cellular models, in particular as they pertain to the formation of cytoplasmic aggregates referred to as reducing bodies (RB) and the analysis of FHL1 binding partners. Intensive discussions focused on proper disease classification, such as the subdivision of FHL1-related myopathies in two major subgroups, depending on the presence of RB in patient’s biopsy.
2. FHL1 and FHL1-myopathies: One gene, three isoforms and a spectrum of disorders
Christina Mitchell commenced the workshop by providing an overview of FHL1 protein structure and function. The known FHL1 protein isoforms, FHL1A, FHL1B and FHL1C, are all encoded by the human FHL1 gene which has 8 exons (Fig. 1a) and is located on chromosome Xq26. mRNA splicing of all protein coding exons (3–8) except exon 7, generates FHL1A (commonly called FHL1), the predominant isoform in skeletal and cardiac muscles, which comprises a N-terminal half LIM domain followed by four complete LIM domains. FHL1B and FHL1C, minor isoforms, share the same N-terminal two and a half LIM domains as FHL1A, however alternative splicing results in unique C-termini: FHL1B contains nuclear import and export sequences and a RBP-J-binding domain, while FHL1C C-terminus only consists of a RBP-J-binding domain [1]. The structure of the protein-binding LIM domains was discussed in detail, specifically the importance of the highly conserved cysteine and histidine residues which bind Zn2+ and are required for fold and stability of each LIM domain structure.
Fig. 1.
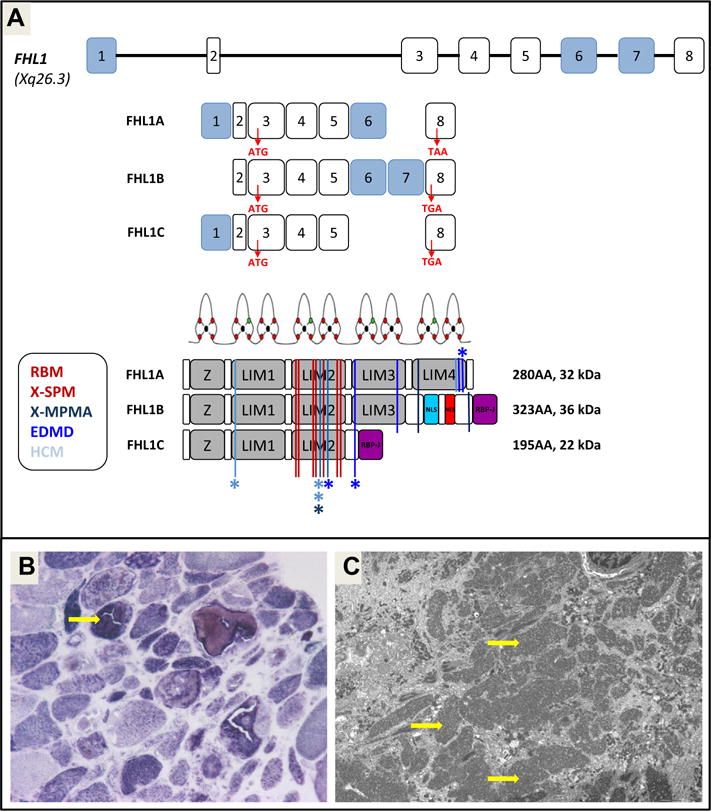
(a) Scheme presenting the FHL1 gene, mRNA and protein isoforms and the localization of the different FHL1 mutations giving ride to different myopathies. (b) Typical structure of reducing bodies revealed by menadione–NBT staining; and (c) by electronic microscopy.
Christina Mitchell also discussed key findings related to the normal function of FHL1 protein in skeletal muscle, which have largely resulted from studies in her laboratory. In rodent skeletal muscle myoblasts, FHL1A localizes to the nucleus and to focal adhesions and is able to shuttle between these compartments. In mature muscle FHL1A localizes to the I-band and the M-line [2]. FHL1A has established roles in promoting fusion of myoblasts involving binding and activation of the transcription factor NFATc1. These results were confirmed in vivo by generation of skeletal-muscle specific FHL1A transgenic mice which established a role for FHL1A in promoting muscle growth through activation of the calcineurin/NFATc1 pathway [3]. There was group discussion about a role for FHL1A in promoting a calcineurin/NFATc1-dependent shift in muscle fiber types from glycolytic to oxidative and how this relates to the clinical features of FHL1 myopathies. Thus, the current published literature has begun to dissect a role for FHL1A in skeletal muscle; however this still remains to be fully understood. Furthermore, a need for better understanding of the functions of the FHL1B and FHL1C isoforms was highlighted.
The discussion continued with Carsten Bönnemann who presented the various hitherto recognized disease phenotypes and types of mutations underlying them. Mutations in FHL1 were first reported in 2008 with the description of FHL1 mutations in three neuromuscular phenotypes: X-linked myopathy with postural muscle atrophy and generalized hypertrophy (XMPMA), X-linked dominant scapuloperoneal myopathy (X-SM), and classical reducing body myopathy (RBM) [4–6]. Whereas in the first two syndromes the FHL1 gene was found using linkage analysis in families, in RBM it was implicated using laser capture of the inclusions followed by proteomic analysis in two sporadic patients. Subsequently many reports have described additional mutations that delineates the phenotypic spectrum further, and added the phenotypes of Emery-Dreifuss muscular dystrophy (EDMD) and hypertrophic cardiomyopathy (HCM) with no or minimal musculo-skeletal involvement to the clinical manifestations of FHL1 mutations [7,8].
A literature review of reported FHL1 mutations and phenotypes was presented at the beginning of the meeting and helped to delineate the status quo and define the open questions that were discussed further during the meeting. Up to the beginning of 2013, 34 different mutations in FHL1 associated with disease have been reported in 24 publications and were supplemented with additional observations throughout the meeting. A distinct group of mutations corresponds to missense mutations confined to the 2nd LIM (LIM2) domain (Fig. 1a) and appeared to be associated with the occurrence of reducing bodies (RB) in the muscle biopsy as initially reported in classic RBM (15 different mutations). These mutations follow an X-linked dominant mechanism, i.e. females were affected, but males carrying the same mutation were affected more severely and showed symptoms earlier. The second group of FHL1 mutations included nonsense mutations, deletions and splice site mutations, as well as missense mutations in LIM3 or LIM4 (Fig. 1a). These mutations were associated with the phenotypes of XMPMA, EDMD and HCM with minimal extracardiac features. These phenotypes typically did not show reducing bodies in the biopsy. These non-2nd LIM domain/non-RBM mutations behave in a more X-linked recessive fashion, but not purely though as female carriers may show mild signs of disease.
Gisèle Bonne then presented the UMD-FHL1 databases, which gathers all FHL1 mutations and associated phenotypes reported so far in the literature. This mutation database has been developed using the Universal mutation database tools (UMD) developed by the team of Christophe Béroud (Inserm U910, Marseille; www.umd.be). The main objectives of such mutation database are to (i) list exhaustively all reported mutations associated to their clinical phenotypes, (ii) provide tools for the exploration of phenotype/genotype relations, and (iii) explore the pathomechanisms of the FHL1-mutations. Numerous similar initiatives of mutation databases are currently developed for various neuromuscular disorders [9]. Currently the UMD-FHL1 database contains 173 entries corresponding to 173 subjects from 69 families, carrying 44 different FHL1 mutations including 10 unpublished cases (G. Bonne, P. Richard, et al., personal com.). Among the reported FHL1 mutations, four mutations affect only FHL1A isoform and are associated with EDMD or HCM [7,8], and one mutation located in exon 7, modifies only FHL1B isoform and is associated with XMPMA [10]. This UMD-FHL1 database will be available on the web site of the UMD database (www.umd.be). Gisèle Bonne also suggested to move data from the UMD-FHL1 database where clinical information is collected only at one time point, usually the time of the molecular diagnosis, to a patient registry for FHL1-related diseases with the option to collect prospective and longitudinal clinical data. Such a tool would be particularly helpful to carefully monitor the natural history of these various forms of FHL1-related disease in order to clearly define phenotypic overlap and distinctions, intra- and interfamilial variability, progression of the diseases and the frequency and time course of cardiac and respiratory involvement in particular. Such patient registries are currently set up for other neuromuscular indications under the initiatives of Treat-NMD alliance (http://curecmd.org/studies/family-registry/) or Cure-CMD (http://www.cmdir.org). In the context of FHL1-related diseases, such development would necessitate the support of the international FHL1-myopathy consortium.
3. Myopathies with reducing bodies
Carsten Bönnemann and Joachim Schessl recounted the characteristics of the first series of 11 patients (9 families) with RBM due to histidine and cysteine zinc coordinating missense mutations in the second LIM domain [11]. These 11 patients included the two original patients in whom FHL1 was identified by Schessl et al. in the Bönnemann laboratory as the most prominent component of the reducing bodies using laser microdissection/capture followed by proteomic analysis, leading to the identification of mutations in FHL1 [4] and two mother and son familial cases in whom the sons were affected earlier and more severely compared to the mothers. The mothers had progressive disease with increasing problems ambulating in adulthood, whereas the sons had lost ambulation as teenagers. The 7 sporadic cases (6 female and 1 male) in the series were due to de novo mutations with severe childhood onset disease, ranging from 13 months to 7 years of age, with the earliest onset and most rapid progression in the sporadic male case (onset between 13 and 17 months of age, loss of ambulation at 31 months of age), whereas the most severely affected female patient showed onset before 2 years of age and lost ambulation at 3 years of age. Other noteworthy clinical features included frequent and early restrictive respiratory disease necessitating respiratory support in 5 of the childhood cases, as well as sudden cardiac arrest at 3 years of age in one childhood case. Muscle weakness was diffuse or had a scapulo-pelvic-peroneal distribution. The most commonly mutated residue was histidine 123 (3 different missense mutations in 6 unrelated patients), associated with severe and early disease in sporadic female patients and even more progressive in the sporadic male patient mentioned earlier. Histomorphologically all patients had typical reducing bodies, often adjacent to the nucleus, which was also surrounded by a band of electron-dense material of similar ultrastructure compared to the reducing bodies. Notably, there were also coexisting cytoplasmic bodies in all cases examined.
Joachim Schessl then reported on 3 additional German families with various FHL1 mutations. In one family, two brothers with a late-onset X-linked scapulo-axio-peroneal myopathy presented with remarkable intrafamilial clinical variability. The p.C224W mutation in the 4th LIM domain of FHL1 was identified, the same mutation that had previously been reported in a family with XMPMA [5]. Unexpectedly, there was clear evidence with light microscopy and, after extensive searching, with electron microscopy for the presence of reducing bodies in the muscle biopsies from both patients, thus reporting this histological phenomenon for the first time in patients with mutations in the fourth LIM-domain of FHL1 [12]. Another large family with reducing body myopathy due to a FHL1 p.C150S mutation was presented with a severely affected female index patient, a less severe affected mother, but very severe affected brothers with death in their teens [13]. Finally, consistent with earlier observations in patients with RBM, in a family originally described as a mixed myopathy with reducing bodies and cytoplasmic bodies in which the more severely affected male proband presented with a significant rigid spine and contractures [14], a second LIM domain missense mutation in a zinc coordinating cysteine (p.C150R) was subsequently identified [15], clarifying this case as primary FHL1 related reducing body myopathy.
Michio Hirano described a large Italian–American family with X-SM initially diagnosed in 1973 by Lewis P. Rowland due to a p.W122S mutation in the second LIM domain of FHL1 identified in his laboratory based on linkage data and candidate gene analysis [6]. Among the 16 mutation carriers, 6 men and 8 women were clinically affected while 2 women (ages 32 and 50) were asymptomatic. Age-at-onset ranged from 20 to 58 years-old and was younger in men (26.1 years ± 4.7 years, mean ± standard deviation) than in women (35.4 ± 11.9). Affected individuals had often asymmetric scapuloperoneal distribution defined by early foot drop, proximal arm weakness preceding hand weakness, and scapular winging, with more significant severity and progression in men, who became wheelchair-bound more frequently and earlier (6–8 years after onset; 35.8 years ± 4.0) than women (4 of 8; 54.8 ± 19.3). However, there was significant clinical variability with one man with onset at age 35 and still ambulating with a cane when he died at age 69, while one woman had early onset at age 20 died of pneumonia in the setting of respiratory insufficiency at age 54. Finger and wrist flexors and ankle plantar flexors were relatively spared even late in the disease while facial weakness was noted in two individuals. Contractures occurred late in the ankles, elbows and forearm supinators. Respiratory muscle weakness was prominent; two men were ventilator-dependent before age 40, with respiratory failure in the setting of pneumonia being the cause of death earlier in men (50 ± 9.7 years) than in women (71.6 ± 11.3). Cardiac involvement included “acute heart failure” in two men, and right bundle branch block in another.
Serum CK was elevated up to 10 times the upper limit of normal, muscle biopsies in five patients revealed myopathic changes while detailed analyses of three muscle biopsies revealed eosinophilic cytoplasmic inclusions in up to 6% of muscle fibers that stained positively for desmin and with Menadione NBT without substrate in one sample characteristic of reducing bodies. Autopsies of 5 patients revealed diffuse fibroadipose replacement of skeletal muscles including the diaphragm in one patient examined, but small muscles of the hands and feet were relatively spared. The heart of one patient showed biventricular hypertrophy and evidence of acute and chronic myocardial infarction, but hearts of two other patients were grossly unremarkable.
Roberta Battini presented the progression and the outcome of 2 Italian patients, a boy (21 year old) and his mother (38 year old) from the onset over the time. These patients were previously reported in the first paper of the FHL1 gene identification in RBM [4] and in the subsequent clinical and histological characterization report [11] and were found to carry the p.C153R mutation affecting the second LIM domain.
After a normal pregnancy and delivery and normal psychomotor development, disease onset in the proband was reported at 7 years with evidence of a rigid spine, asymmetrical contractures of the elbows and mild scoliosis. Blood CK level was about 1000 U/L. At 10 years, the boy showed increased spinal rigidity involving neck and progressive elbow and wrists contractures (more on the right) with consequent loss of function. Blood CK level was about 1500 U/L. In addition, he showed pronounced axial and proximal muscle weakness, generalized muscle hypotrophy except a relative hypertrophy of vastus lateralis and tricepes brachii. At 13 years, joint contractures in the neck, elbows (more on the left), wrist (more on the right), knee and lower limbs made progress and the child lost autonomous gait to become wheelchair dependent at age of 14 years. Blood CK level were 1770. At age of diagnosis, he presented moderate respiratory difficulties with occasional BiPAP use and no cardiac involvement. Blood CK level was 275 U/L. Muscle MRI studies from pelvis, thighs and legs performed at diagnosis showed a selective sparing of the glutei, which appeared hypertrophic (a feature that is not typically observed in other muscle disorders); at thigh level there was predominant involvement of adductor magnus and of postero-medial muscles and at calf level there was some involvement of soleus. A similar pattern was observed in his mother (30 years) who showed a milder but similar muscle involvement at thigh and calf level, with also glutei hypertrophy [16]. In that period the mother was pregnant and prenatal diagnosis was performed and a healthy boy was born.
The proband, now 21 years, on re-examination showed generalized muscle hypotrophy and a relatively mild hypertrophy of vastus lateralis and tricepes brachii. Ventilatory support is now daily: he uses continuous nocturnal BiPAP, plus additionally every 3–4 h during the day. He shows mild dysphagia in particular with liquids. His mother (38 years) shows long standing muscle fatigability, and now progressively more noticeable proximal weakness with asymmetric involvement of the upper limbs, difficulties with climbing stairs and a minimal Gowers’ sign but has no contractures. No cardiac and respiratory involvements are present.
Yukiko K. Hayashi and Sherine Shalaby reported 15 patients in 10 families with FHL1 myopathy identified in Japan [17–19]. All patients have a mutation in the LIM2 domain and have histological evidence for RB in their muscles. Clinically, all patients were quite normal at birth and until muscle weakness appeared. Male patients showed earlier onset of the disease with a more severe clinical phenotype compared with female patients in the same family. However, in this series, three sporadic female patients also showed early onset before 3 years of age followed by rapidly progressive weakness including neck control, and 2 died from respiratory failure within a few years. No cardiac involvement was noticed. Three male patients had rigid spine and joint contractures during school age, followed by rapidly progressive muscle weakness. Others (1 male and 8 female) showed adult onset of a milder form. Shoulder-girdle muscle involvement was commonly seen together with scapular winging. Six of 15 patients showed limb-girdle type of muscle involvement, whereas 6 others had a more scapulo-peroneal form, and often showed drop foot. Asymmetrical muscle involvement was commonly seen. On muscle images, paraspinal muscles were involved from early stages. Conduction block was seen in 3, but no patient showed overt cardiomyopathy. All biopsied muscles contained RB, but their number and distribution were quite variable. Rimmed vacuoles and cytoplasmic bodies were also commonly seen. In some muscle specimens markedly atrophic fibers with RB accumulated, while other parts of muscles appeared to be nearly normal. Cytoplasmic inclusions were immunoreactive for FHL1, desmin, ubiquitin, and several proteins associated with unfolded protein response [20]. Immunoblotting analysis showed variable amounts of FHL1. Electron microscopic observations revealed electron-dense characteristic materials especially around myonuclei. Data of this series confirm that FHL1 myopathy with LIM2 domain mutations showed a wide variety of clinical and pathological features. Sherine Shalaby then emphasized that FHL1 myopathy is a rare disease and it is likely that some patients may be overlooked due to the clinical variability, the selectivity of muscle involvement, the presence of inclusions seen in other protein aggregate myopathies and the presence of neurogenic changes.
Based on the series of Japanese patients they defined features that could be used to allow for early clinical suspicion of the disease. These include (in addition to the inclusions in the biopsy): (1) family history with no male to male transmission and in which males are more severely affected than females, (2) sporadic presentation of early childhood onset of severe myopathy, rapidly progressive in boys but also girls (in severe de novo mutation in the LIM2 domain), and (3) patients presenting with early axial muscle involvement and asymmetry. To allow for early histological recognition of inclusions as reducing bodies, menadione NBT staining should be performed more routinely.
Now, since she is back to Egypt, Dr. Shalaby and her colleagues use the above clinical and pathological insight about FHL1/RBM myopathy in attempt to identify this disease in Egyptian patients. They currently suspect 2 cases to have RBM clinically. They hope they can identify FHL1 myopathy in Egyptian patients aiming at shedding more light on this rare disease.
4. FHL1 in depth: RB negative subgroup
Christian Windpassinger presented on the data reported in 2008 by his group, where they identified FHL1 as the causative gene for a hitherto unknown X-chromosomal recessive muscular disease in a large multigenerational Austrian family referred to as XMPMA [5]. The first signs of the disease are usually noticed by the patients around the age of 30.
Patients initially showed contractures of tendons in upper and lower limbs which lead to gait problems and a restriction of movement of the neck. Additionally, male patients displayed a characteristic “athletic” appearance due to hypertrophy of muscles composed by a high percentage of fast twitch fibers, whereas muscles with a high percentage of slow twitch fibers show atrophy. As initial degenerative processes were restricted to phasic muscles maintaining the posture, the group referred to the disease as X-linked myopathy with postural muscle atrophy (XMPMA). The FHL1 mutation p.C224W was found to cosegregate with the disease in this family. The mutation affects one of the crucial cysteine residue of the 4th LIM domain of FHL1A, FHL1B but not the shorter isoform FHL1C. Western blot analysis showed that FHL1 protein expression was markedly reduced in muscle biopsies of the patients. No inclusions were identified in the biopsies.
Further systematic clinical evaluation of the cardiac phenotype by this group revealed a novel variant of hypertrophic cardiomyopathy, which is mainly characterized by spongy structures found within the left ventricular myocardium [21]. Moreover, all patients showed mild left ventricular hypertrophy accentuated at the midventricular level, late post gadolinium enhancement as a marker of fibrosis, and impaired diastolic and systolic function. Subsequent functional analysis of the preserved isoform FHL1C revealed that this isoform is a binding partner of the potassium channel Kv1.5 (KCNA5), a protein known to be associated with sudden cardiac death and atrial fibrillation. Furthermore, they could show that Kv1.5 was markedly reduced in skeletal muscle biopsies of XMPMA patients [22].
In summary, XMPMA represents as a mild form in the spectrum of FHL1 related muscle disorders, perhaps related to the fact, that the isoform FHL1C remains unaffected and thereby some of the multiple functions of FHL1 might be preserved.
Gisèle Bonne briefly presented the clinical data first reported by her group in 2009, when they identified FHL1 as the causative gene for X-linked EDMD in a cohort of patients for whom mutations in the two main genes responsible for EDMD, LMNA or EMD, were excluded [7,23]. Clinically, patients with FHL1 mutations presented with EDMD-typical clinical features, i.e. myopathy with scapulo-peroneal and/or axial distribution, joint contractures and cardiomyopathy associated with arrhythmias and conduction defects. The associated cardiac disease is peculiar in that it is characterized by hypertrophic cardiomyopathy. Heterozygous female carriers were either asymptomatic or had cardiac disease and/or mild myopathy. Importantly, some of the FHL1-mutated male relatives had isolated cardiac disease, with possibly clinically overt hypertrophic cardiomyopathy. Notably, some patients presented also with vocal cord palsy and/or dysphasia. Dr. Bonne then reported more recent experiences of the Myology Institute (B. Eymard, T. Stojkovic, P. Laforet, P. Richard et al.) where 11 patients (6 male and 5 female) were identified with FHL1 mutations. Among the male patients, two were EDMD and one XMPMA cases with missense or truncating mutations affecting only FHL1A and FHL1B isoforms, one atypical case presenting with HCM and severe diaphragmatic defects (F-19949 in [8]) and one childhood RBM cases with a typical missense in LIM2 domain (p.C132F) affecting all FHL1 isoforms. The remaining case presented with atypical premature aging features associated with a complex FHL1 mutation currently under investigation. The female cases were relatives of male patients presenting either as asymptomatic or with mild myopathic and/or cardiac features. The following discussion confirmed that EDMD, XMPMA and HCM seem to share the same type of FHL1 mutation, i.e. essentially (non LIM2 domain) missense or truncating mutations affecting to different range the three isoforms.
Christian Geier reported cardiac involvement in reducing body negative FHL1 patients. In a large German family with HCM, mild to moderate left ventricular hypertrophy was observed in six male patients with EDMD disease caused by a FHL1 p.C209R mutation located in LIM3 domain [23]. ECG showed repolarization abnormalities in all affected patients. Two patients developed atrial fibrillation in their forties and one patient had asymptomatic non-sustained ventricular tachycardia recorded on Holter ECG. At least one patient had died suddenly. Another patient had died in his thirties from acute renal and cardiac failure. Dr. Geier also highlighted the cardiac features of HCM patients recently reported by the team of Lucie Carrier [8]. A de novo single nucleotide deletion (c.134delA, p.K45Sfs) in exon 3 was reported in a young Italian adult with septal hypertrophy and outflow tract obstruction. Isolated HCM was also reported in another young Italian adult carrying a missense mutation (p.C276S) located in exon 8 that was also present in his clinically unaffected mother. A family of French descent with syndromic HCM caused by a FHL1 nonsense mutation (p.C153X) in exon 5 was peculiar in that a cardiac HCM phenotype was also found in heterozygous females. In contrast, the skeletal muscle phenotype remained restricted to male patients. Onset of cardiac disease in FHL1 patients was mostly recognized in adolescents or young adults. Myocardial hypertrophy was variable in extent and localization, but most patients presented with mild to moderate asymmetric hypertrophy. Repolarization abnormalities with inverted T-waves were the most common ECG abnormalities. Conduction defects may occur, but do not seem to be a consistent marker of FHL1 disease. Late gadolinium enhancement indicating myocardial fibrosis was seen in some FHL1 patients with hypertrophic cardiomyopathy.
Reviewing the published cases of FHL1 associated cardiomyopathy raises the suspicion that heart failure and sudden cardiac death might be more frequent in this condition compared to their incidence in HCM caused by mutations in sarcomeric proteins.
5. Muscle pathology and other evaluation tools
Norma B. Romero reported a detailed histochemical, immunohistochemical, ultrastructural, and immunoelectron microscopy comparative analysis of a large combined series of 18 FHL1 mutated patients. Patients were divided in two groups according to their clinical phenotype: a first one composed of 14 patients presenting RBM (group 1), and a second group composed of three patients with EDMD and one patient with HCM and muscular hypertrophy (group 2). Among the patients from group 1 a recurrent striking asymmetrical muscle weakness and atrophy pattern was described in five subjects (38%). As mentioned above in other series, Dr. Romero considers that this is a clinical clue that could help in the identification of FHL1 mutated patients. Muscle biopsies in group 1 showed reducing bodies (RB) associated with cytoplasmic bodies as a constant feature. RB had a prominent FHL1 immunoreactivity. Desmin, αB-crystallin and myotilin immunoreactivity was observed surrounding RB. Under electron microscopy (EM), RB were composed of electron dense tubulofilamentous material arising from the I-band, M-line, and near the Z-line, that progressively spreads between the myofibrils and around myonuclei (Fig. 1b and c). Using immunoelectron microscopy they demonstrated that in patients from group 1, FHL1 protein is found uniquely inside RB. Samples from group 2 showed mild dystrophic abnormalities, without RBs. Only minor non-specific myofibrillar abnormalities were observed under EM. Molecular analysis revealed missense mutations in the FHL1 LIM2 domain in group 1 patients and ins/del or missense mutations within the FHL1 LIM4 domain in patients from group 2. This study expand the morphological features of RBM, clearly demonstrating the localization of FHL1 in RB, and further illustrating major morphological differences between different FHL1-related myopathies [24].
Caroline Sewry discussed aspects of the pathology of FHL1 cases and presented data on some recently molecularly identified patients. The menadione NBT method without substrate is recognized as a useful technique for identifying reducing bodies which stain intensely. In addition, whole fibers or large areas of fibers may stain and probably reflect areas of material that has accumulated several proteins, including FHL1. Areas of disrupted myofibrillar material in various other disorders can also show staining with menadione–NBT but it was agreed in discussion that this is generally less intense than in FHL1-related cases. Dr. Sewry illustrated this with an example of a case with a mutation in the gene encoding myotilin in which pale menadione–NBT staining was seen in the extensive areas of myofibrillar material. In one molecularly unresolved case, targets were positive for menadione NBT. Other cases with target fibers however did not show this staining of targets. It was pointed out that a case published by Sharma and colleagues with acid maltase deficiency showed dense bodies that were positive for menadione–NBT [25]. With EM the appearance of the RB can be variable and they may contain glycogen and are thus PAS positive. Reducing bodies may be discrete granular bodies of variable size resembling a nucleus in shape or be round dense structures. Granular material may occupy large areas of a fiber or surround nuclei and is distinct from areas of non-specific myofibrillar disruption resembling core-like areas. Cytoplasmic bodies are known to be common in FHL1 cases with RB and they have the typical dense center with a halo. Although this halo is pale, the radiating filaments of desmin are not as apparent as in typical cytoplasmic bodies. Some cases may show membrane bound autophagic vacuoles as seen in Danon’s disease and X-linked myopathy with excess autophagy but the material between the plasma membrane and the basal lamina that is typical of these disorders has not been seen in FHL1 cases.
Caroline Sewry also presented some additional cases with FHL1 mutations. Three cases had a severe phenotype, typical clinical features of FHL1 and had a mutation the LIM2 domain of FHL1. All 3 cases showed RB with menadione–NBT, confirmed with EM. A fourth case, also with a LIM2 domain mutation with elbow contractures and a rigid spine, showed no RB with menadione–NBT, nor accumulation of FHL1 protein in one block, but examination (by Rahul Phadke) of a second block of tissue embedded in wax confirmed the focal pathology that can occur in FHL1 cases and focal inflammation, and the presence of RBs with electron microscopy. Dr. Sewry also pointed out that it is not known if the case of “reducing body myopathy” published by Dubowitz and Brooke in 1972 has a mutation in the FHL1 gene but attempts to extract DNA from the few remaining sections of the muscle biopsy are being attempted.
Duygu Selcen reported on a study of five patients with FHL1 myopathy [26]. Because some pathologic features of the FHL1-muscular dystrophies and the myofibrillar myopathies overlap, Selcen and coworkers searched for mutations in FHL1 in a cohort of 50 patients with myofibrillar myopathy and detected two novel and one previously identified missense mutations in five. Two mutations were in the second LIM domain. All but one patient presented with progressive muscle weakness; one had hypertrophied muscles, rigid spine, and joint contractures, and one also had a peripheral neuropathy. Only patients with mutations in the second LIM domain displayed menadione–NBT positive RB. On high resolution EM, RB were composed of 13-nm tubulofilaments that appeared to initially emanate from Z-disks. At a more advanced stage, there was myofibrillar disintegration, appearance of myriad RB in the sarcoplasm and in many nuclei, accumulation of cytoplasmic degradation products, and aggregation of endoplasmic reticulum and sarcotubular profiles. They conclude that the pathology of some patients with FHL1 myopathy is typical of myofibrillar myopathy. Mutations in the second LIM domain are associated with RB composed of distinct tubulofilaments. Finally, the mutation not in the 2nd LIM domains resulted in a milder phenotype with late onset, typical myofibrillar myopathy pathology, but no RB.
6. Emerging pathophysiological mechanisms
Andrea A. Domenighetti exploited a mouse model to evaluate the loss of FHL1 function on skeletal muscle development and homeostasis [27]. FHL1-null mice were engineered to disrupt FHL1 gene through a LacZ/Neo knock-in strategy, resulting in global loss of FHL1 expression in all tissues. Their data provide compelling evidence that loss of FHL1 function is sufficient to induce development of a progressive myopathy associated with myofibrillar and intermyofibrillar disorganization in mice. Specifically, they demonstrate that FHL1 deficiency in vivo is associated with (1) defects in the structure of the sarcomeric reticulum and mitochondrial content, with appearance of inclusions, but no reducing bodies, and susceptibility to autophagy in both oxidative and glycolytic muscles; (2) development of myofibrillar disarray and decreased aerobic activity in muscles that expressed higher oxidative capacity; (3) decreased survival rates, associated with age-dependent impairment of muscle contractile function and lower exercise capacity. In addition they show expression of a variant of the FHL1A transcript (variant 2). This variant is required for proper muscle fiber differentiation and maturation in vitro. Along with the unique endogenous fiber-type specific expression pattern of FHL1A, which is elevated in oxidative fibers, this study suggests an important correlation between FHL1A and skeletal muscle disease pathogenesis in muscles bearing increased oxidative capacity.
Meagan McGrath presented on the exploration of pathophysiological mechanisms of FHL1 myopathies, in particular the role of RBM-mutant FHL1 protein overexpression and aggregate formation. A defining histopathological feature of these FHL1 myopathies is the presence of RB aggregates, which contain mutant FHL1 protein in the skeletal muscle of affected patients. However, emerging evidence suggests the categorizing of FHL1 myopathies into two distinct sub-groups, those associated with reducing body aggregates (early onset RBM and later onset RBM with scapuloperoneal presentation) versus those in which aggregates are not typically observed (XMPMA, EDMD and HCM). Therefore, a key question which is related to the disease pathology of FHL1 myopathies, and remains unanswered is whether all FHL1 mutant proteins can form aggregates. To address this question Meagan discussed the development of a C2C12 myotube model for expression of FHL1 mutants, which can be used to examine their pathogenic effects.
Lucie Carrier evaluated the hypothesis of the involvement of the ubiquitin–proteasome system (UPS) in the pathogenesis of FHL1-related hypertrophic cardiomyopathy, through the analysis of the FHL1 mutations they reported in Friedrich et al. [8]: i.e. two different mutations (p.K45Sfs and p.C276S) in two unrelated families with isolated HCM, and one mutation (p.C153X) in a family that presented either with only HCM or with both HCM and skeletal muscle weakness in some cases. The three mutations were unstable after gene transfer in C2C12 myoblasts or in neonatal mouse cardiac myocytes. As most of the proteins are degraded by the UPS, transfected cells were treated with the proteasome inhibitor MG132. The levels of HCM mutant proteins (including missense) were stabilized, and truncated proteins formed aggregates in cardiac myocytes. This suggested the involvement of the UPS in the regulation of the expression of HCM-FHL1 mutations. To evaluate whether the HCM mutations induced a phenotype, gene transfer was performed using an adeno-associated virus encoding the WT and different FHL1 mutant in fibrin-based engineered heart tissue (EHT). The two HCM-specific mutants (p.K45Sfs, p.C276S) induced alterations of both the force and the kinetics of contraction of the EHTs. Importantly, FHL1 is not the only example of HCM mutations for which the UPS is involved. The group of Lucie Carrier had also shown that the expression of HCM-ANKRD1 or HCM-MYBPC3 is regulated by the UPS [28,29]. Taken together, these data suggest that in HCM, (i) the UPS is very active in degrading mutant proteins leading to haploinsufficiency or complete loss in males in the case of X-linked disease, and (ii) the presence of external stress with an overwhelmed UPS precipitates the system into impairment. Similar mechanisms could be involved in the pathogenesis of HCM-FHL1 mutations. Further analyses are required to validate this hypothesis for different HCM genes. Moreover, Dr. Carrier believes that the analyses of the expression of “muscle-specific” FHL1 mutations in cardiac myocytes will help to confirm whether their hypothesis concerns only the “HCM-specific” mutations or not.
Anthony Gramolini presented the method his group has developed to analyze protein interactomes systematically in mammalian cells and discussed their attempts to apply these tools to investigate the critical FHL1 protein interactions [30]. Experiments were presented using affinity-tagged full length FHL1 in a 2-step affinity purification based on the small molecular weight streptavidin binding- and calmodulin-binding affinity tags, to obtain highly purified FHL1 proteins interacting protein partners. Protein complex purification samples were analyzed by gel-free, liquid chromatography mass spectrometry (LC–MS) using a Thermo Orbitrap Mass Spectrometer. They identified a total of 310 different proteins from a total of 9 different purifications including 3 experimental replicates, and by applying stringent filtering criteria they eliminated all proteins found in any of the controls and only allowed those that were detected in two or more bait purifications. This resulted in identifying 34 high-confidence potential binding partners of FHL1. Potential interactors were then verified by immunoprecipitation assays from mouse heart ventricles and interactions were visualized in adult cardiomyocytes using 3D fluorescence microscopy. They showed that FHL1 exists as part of a complex that binds with PDZ, LIM Domain1 (PDLIM1), Gelsolin (GSN) and actinin-1 (ACTN1). The potential of additional studies to investigate FHL1 interactions using skeletal and cardiac muscle cells through in vitro and in vivo viral delivery methods was then discussed.
Joachim Schessl reported on the proteomic approach to identify the protein composition in the aggregates in RBM. An initial study of the proteomic characterization of pathological protein aggregates in skeletal muscle biopsies from normal human muscle and patients with MFM-causing gene mutations was shown. The novel technical strategy was based on the microdissection of aggregates and normal fibers from the patients by laser dissection microscopy, filter-aided sample preparation, iTRAQ-labeling, and analysis on the peptide level using offline nano-LC and MALDI-TOF-TOF MS/MS for protein identification and quantification [31]. Using this method FHL1 protein was identified as the component showing the highest level in the aggregates in two patients with a p.C224W mutation in the 2nd LIM domain, confirming the validity of this approach.
7. Management of the cardiac involvement of FHL1 disease
Christian Geier reported on the cardiac management of FHL1 myopathy. In general, all FHL1 mutation carriers should have regular cardiac evaluation once a year including 12-lead ECG, Holter-ECG and echocardiography even when there is no hint for cardiac involvement. Even shorter evaluation intervals might be appropriate for patients with already overt cardiac phenotype.
According to current knowledge, management of FHL1-related HCM should be done according to the accepted general standards of care for HCM [32]. This includes pharmacotherapy to control HCM related symptoms. Septal reduction therapy is indicated only in patients with significant outflow tract obstruction who remain symptomatic despite maximal pharmacotherapy. Particular attention should be paid to detect arrhythmias. Atrial fibrillation or flutter seems to be common complications in FHL1 patients putting them at risk to suffer cardio-embolic complications. Particular care is also warranted to early detect systolic heart failure that may complicate FHL1 associated HCM. A staged pharmacotherapy according to the standards for dilated cardiomyopathy is indicated if systolic heart failure occurs. Other causes of heart failure requiring specific therapy, i.e. heart failure of hypertensive, ischemic, or valvular origin should be excluded.
Whether the established risk factors to detect patients at high risk for sudden cardiac death are sufficient and adequate remains unknown. Since myocardial fibrosis has been reported in FHL1 patients with moderate left ventricular hypertrophy and a positive family history of sudden cardiac death, cardiac MRI with Gadolinium contrast to detect late enhancement may be useful as an additional risk marker. Hopefully, a registry of FHL1 related disease might clarify whether FHL1 patients are at higher risk to suffer sudden cardiac death independent of the general risk markers.
8. Conclusions
This workshop was an excellent occasion to summarize the current knowledge available on FHL1-related myopathy. There was a general agreement that the majority of the available data justifies a subdivision of these disorders in two main categories, (i) those with LIM2 missense mutations typically (but not exclusively) affecting the zinc coordinating histidine and cysteine, equally altering the three FHL1 isoforms on one hand, and (ii) those modifying differently the FHL1 isoforms, being either missense mutations located in exon 7 or 8 encoding the C-terminal domain of FHL1 isoforms, or nonsense ins/del, or splice site mutations leading to frame shift mutations and premature stop codon and thus various truncated mutant FHL1 isoforms. From a clinical point of view, this includes the LIM2 missense mutation category RBM (with a later manifesting milder variant presenting as SPM) and the second category with truncating mutations, EDMD, XMPMA and HCM plus mild muscle features. From a pathology point of view, RB are present in the first category, and seem to be absent in the second one, with the exception of the family reported by Joachim Schessl, although, future report may confirm or refute this. Finally, from a pathomechanism point of view, more work is needed to examine the effect of mutation, e.g. haploinsufficiency versus dominant negative effect of mutant proteins and the effects of the aggregate formation on muscle cells. A fundamental question posed by this meeting is how and to which extent each FHL1 mutated isoform contributes to disease phenotypes.
There was general agreement for the establishment of an international FHL1 consortium, and the first initiative that was validated during this meeting was to share the biological material available between the consortium teams, as well as the existing protocols especially those related to the use of FHL1 antibodies. Meagan McGrath provided to the consortium an updated list of the currently available FHL1 antibodies (FHL1A, FHL1B and FHL1C-specific) and their validity for different types of experiments (western-blot and immunofluorescence).
Acknowledgments
This workshop was made possible thanks to the financial support of the European Neuromuscular Centre (ENMC) and ENMC main Sponsors: Association Française contre les Myopathies (France), Deutsche Gesellschaft für Muskelkranke (Germany), Muscular Dystrophy Campain (UK), Muskelvindfonden (Denmark), Prinses Beatrix Spierfonds (the Netherlands), Schweizerische Stiftung für die Erforschung der Muskelkrankheiten (Switzerland), Telethon Foundation (Italy), Spierziekten Nederland (The Netherlands), and Associated members: Finnish Neuromuscular Association (Finland).
Footnotes
- Roberta Battini (Pisa, Italy)
- Anne T. Bertrand (Paris, France)
- Gisèle Bonne (Paris, France)
- Carsten Bönnemann (Bethesda, USA)
- Lucie Carrier (Hamburg, Germany)
- Friederike Cuello (Hamburg, Germany)
- Andrea A. Domenighetti (San Diego, California, USA)
- Christian Geier (Berlin, Germany)
- Anthony Gramolini (Toronto, Canada)
- Yukiko Hayashi (Tokyo, Japan)
- Michio Hirano (New York, USA)
- Meagan McGrath (Melbourne, Victoria, Australia)
- Christina Mitchell (Melbourne, Victoria, Australia)
- Jachinta Rooney (Bethesda, USA)
- Norma B Romero (Paris, France)
- Joachim Schessl (Munich, Germany)
- Duygu Selcen (Rochester, USA)
- Caroline Sewry (London, United Kingdom)
- Sherine Shalaby (Cairo, Egypt)
- Christian Windpassinger (Graz, Austria)
References
- 1.Cowling BS, Cottle DL, Wilding BR, et al. Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: a comprehensive review of the clinical, histological and pathological features. Neuromuscul Disord. 2011 doi: 10.1016/j.nmd.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 2.McGrath MJ, Cottle DL, Nguyen MA, et al. Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly. J Biol Chem. 2006;281:7666–83. doi: 10.1074/jbc.M512552200. [DOI] [PubMed] [Google Scholar]
- 3.Cowling BS, McGrath MJ, Nguyen MA, et al. Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J Cell Biol. 2008;183:1033–48. doi: 10.1083/jcb.200804077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118:904–12. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Windpassinger C, Schoser B, Straub V, et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet. 2008;82:88–99. doi: 10.1016/j.ajhg.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quinzii CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet. 2008;82:208–13. doi: 10.1016/j.ajhg.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gueneau L, Bertrand AT, Jais JP, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2009;85:338–53. doi: 10.1016/j.ajhg.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedrich FW, Wilding BR, Reischmann S, et al. Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum Mol Genet. 2012;21:3237–54. doi: 10.1093/hmg/dds157. [DOI] [PubMed] [Google Scholar]
- 9.Sarkozy A, Bushby K, Beroud C, Lochmuller H. 157th ENMC international workshop: patient registries for rare, inherited muscular disorders 25–27 January 2008 Naarden, The Netherlands. Neuromuscul Disord. 2008;18:997–1001. doi: 10.1016/j.nmd.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Schoser B, Goebel HH, Janisch I, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology. 2009;73:543–51. doi: 10.1212/WNL.0b013e3181b2a4b3. [DOI] [PubMed] [Google Scholar]
- 11.Schessl J, Taratuto AL, Sewry C, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain. 2009;132:452–64. doi: 10.1093/brain/awn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feldkirchner S, Walter MC, Muller S, et al. Proteomic characterization of aggregate components in an intrafamilial variable FHL1-associated myopathy. Neuromuscul Disord. 2013;23:418–26. doi: 10.1016/j.nmd.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Schreckenbach T, Henn W, Kress W, et al. Novel FHL1 mutation in a family with reducing body myopathy. Muscle Nerve. 2013;47:127–34. doi: 10.1002/mus.23500. [DOI] [PubMed] [Google Scholar]
- 14.Goebel HH, Halbig LE, Goldfarb L, et al. Reducing body myopathy with cytoplasmic bodies and rigid spine syndrome: a mixed congenital myopathy. Neuropediatrics. 2001;32:196–205. doi: 10.1055/s-2001-17374. [DOI] [PubMed] [Google Scholar]
- 15.Schessl J, Columbus A, Hu Y, et al. Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: identification of a second LIM domain mutation in FHL1. Neuropediatrics. 2010;41:43–6. doi: 10.1055/s-0030-1254101. [DOI] [PubMed] [Google Scholar]
- 16.Astrea G, Schessl J, Clement E, et al. Muscle MRI in FHL1-linked reducing body myopathy. Neuromuscul Disord. 2009;19:689–91. doi: 10.1016/j.nmd.2009.06.372. [DOI] [PubMed] [Google Scholar]
- 17.Shalaby S, Hayashi YK, Goto K, et al. Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1) Neuromuscul Disord. 2008;18:959–61. doi: 10.1016/j.nmd.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Shalaby S, Hayashi YK, Nonaka I, Noguchi S, Nishino I. Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology. 2009;72:375–6. doi: 10.1212/01.wnl.0000341311.84347.a0. [DOI] [PubMed] [Google Scholar]
- 19.Komagamine T, Kawai M, Kokubun N, et al. Selective muscle involvement in a family affected by a second LIM domain mutation of fhl1: an imaging study using computed tomography. J Neurol Sci. 2012;318:163–7. doi: 10.1016/j.jns.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 20.Liewluck T, Hayashi YK, Ohsawa M, et al. Unfolded protein response and aggresome formation in hereditary reducing-body myopathy. Muscle Nerve. 2007;35:322–6. doi: 10.1002/mus.20691. [DOI] [PubMed] [Google Scholar]
- 21.Binder JS, Weidemann F, Schoser B, et al. Spongious hypertrophic cardiomyopathy in patients with mutations in the four-and-a-half LIM domain 1 gene. Circ Cardiovasc Genet. 2012;5:490–502. doi: 10.1161/CIRCGENETICS.111.962332. [DOI] [PubMed] [Google Scholar]
- 22.Poparic I, Schreibmayer W, Schoser B, et al. Four and a half LIM protein 1C (FHL1C): a binding partner for voltage-gated potassium channel K(v1.5) PLoS One. 2011;6:e26524. doi: 10.1371/journal.pone.0026524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knoblauch H, Geier C, Adams S, et al. Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann Neurol. 2010;67:136–40. doi: 10.1002/ana.21839. [DOI] [PubMed] [Google Scholar]
- 24.Malfatti E, Olive M, Taratuto AL, et al. Skeletal muscle biopsy analysis in reducing body myopathy and other FHL1-related disorders. J Neuropathol Exp Neurol. 2013;72:833–45. doi: 10.1097/NEN.0b013e3182a23506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma MC, Goebel HH. Protein aggregate myopathies. Neurol India. 2005;53:273–9. doi: 10.4103/0028-3886.16921. [DOI] [PubMed] [Google Scholar]
- 26.Selcen D, Bromberg MB, Chin SS, Engel AG. Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology. 2011;77:1951–9. doi: 10.1212/WNL.0b013e31823a0ebe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domenighetti AA, Chu PH, Wu T, et al. Loss of FHL1 induces an age-dependent skeletal muscle myopathy associated with myofibrillar and intermyofibrillar disorganization in mice. Hum Mol Genet. 2014;23:209–25. doi: 10.1093/hmg/ddt412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crocini C, Arimura T, Reischmann S, et al. Impact of ANKRD1 mutations associated with hypertrophic cardiomyopathy on contraction parameters of engineered heart tissue. Basic Res Cardiol. 2013;108:349. doi: 10.1007/s00395-013-0349-x. [DOI] [PubMed] [Google Scholar]
- 29.Sarikas A, Carrier L, Schenke C, et al. Impairment of the ubiquitin– proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 30.Sharma P, Shathasivam T, Ignatchenko V, Kislinger T, Gramolini AO. Identification of an FHL1 protein complex containing ACTN1, ACTN4, and PDLIM1 using affinity purifications and MS-based protein–protein interaction analysis. Mol Biosyst. 2011;7:1185–96. doi: 10.1039/c0mb00235f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldkirchner S, Schessl J, Muller S, Schoser B, Hanisch FG. Patient-specific protein aggregates in myofibrillar myopathies: laser microdissection and differential proteomics for identification of plaque components. Proteomics. 2012;12:3598–609. doi: 10.1002/pmic.201100559. [DOI] [PubMed] [Google Scholar]
- 32.American College of Cardiology Foundation/American Heart Association Task Force on Practice G, American Association for Thoracic S, American Society of E et al. ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American college of cardiology foundation/American heart association task force on practice guidelines. J Thorac Cardiovasc Surg. 2011;142:1303–38. doi: 10.1016/j.jtcvs.2011.10.019. [DOI] [PubMed] [Google Scholar]