

Abstract
The pleiotropic actions of the renin-angiotensin system (RAS) depend on the availability of angiotensinogen (AGT) which generates angiotensin I (ANG I) when cleaved by renin. Thus, quantification of the intact AGT (iAGT) concentrations is important to evaluate the actual renin substrate available. The iAGT conformation exists as oxidized AGT (oxi-AGT) and reduced AGT (red-AGT) in a disulfide bond, and oxi-AGT has a higher affinity for renin, which may exacerbate RAS-associated diseases. Accordingly, we determined iAGT, oxi-AGT, and red-AGT levels in plasma from rats and mice. Blood samples were obtained by cardiac puncture and then immediately mixed with an inhibitor solution containing a renin inhibitor. Total AGT (tAGT) levels were measured by tAGT ELISA which detects both cleaved and iAGT. iAGT levels were determined by iAGT ELISA which was found to only detect red-AGT. Thus, it was necessary to treat samples with dithiothreitol, a reducing agent, to quantify total iAGT concentration. tAGT levels in rat and mouse plasma were 1,839 ± 139 and 1,082 ± 77 ng/ml, respectively. iAGT levels were 53% of tAGT in rat plasma but only 22% in mouse plasma, probably reflecting the greater plasma renin activity in mice. The ratios of oxi-AGT and red-AGT were ∼4:1 (rat) and 16:1 (mouse). Plasma iAGT consists of oxi-AGT and red-AGT, suggesting that oxidative stress can influence ANG I generation by the AGT conformation switch. Furthermore, the lower availability of plasma iAGT in mice suggests that it may serve as a limiting factor in ANG I formation in this species.
Keywords: intact AGT, ELISA, renin-angiotensin system, plasma RAS
angiotensinogen (AGT) is the only known substrate for renin that is the rate-limiting enzyme responsible for angiotensin (Ang) production (2, 17). The enzymatic processing of AGT produces ANG I, a decapaptide at N-terminus of AGT from intact AGT (iAGT). Overexpression of AGT in the liver, brain, and kidneys augments systemic and local ANG II levels and causes the development of hypertension (15, 20, 21). In human genetic studies, a linkage has been established between the AGT gene and hypertension (3–5, 8, 12, 18). Thus, AGT is a key factor in the development and progression of hypertension and in other renin-angiotensin system (RAS)-associated diseases (7, 13).
It was recently demonstrated that there are two types of plasma iAGT conformation which are oxidized AGT (oxi-AGT) and reduced AGT (red-AGT) in a disulfide bond (22). Results from this study indicate that oxi-AGT has a higher affinity for renin than red-AGT (22), suggesting that changes in redox status can induce conformational alterations of AGT determining capability of ANG generation. Indeed, many pathophysiological conditions in which an activated RAS plays crucial roles, such as hypertension and diabetes mellitus, are associated with increased oxidative stress and consequent changes in redox status (9, 11). On the other hand, a recent study demonstrated that artificial expression of Cys18- and Cys137-mutated AGT, which does not form a disulfide bond, in liver-specific AGT knockout mice exhibited similar plasma ANG II and blood pressure levels with artificial expression of wild-type AGT (19). These findings suggest that pathophysiological roles of the conformational alterations of AGT require further detailed studies that determine absolute concentrations of the varying AGT conformations. Therefore, establishment of measurements quantifying iAGT, oxi-AGT, and red-AGT levels is important for accurate assessments of intact renin substrate in both its forms.
Rahgozar and colleagues (14) established an ELISA system to determine oxi-AGT and red-AGT levels. In the ELISA, biotins bind to free-thiol groups in AGT by disulfide bonds, which enables detection of only red-AGT via formation of avidin-biotin complex (14). While this assay system can be a useful tool, this method detects red-AGT in both iAGT and des-ANG I AGT. It is important to determine oxi-AGT and red-AGT levels of iAGT to evaluate levels of renin substrate and its affinity to renin. We recently developed direct quantitative methods to measure total AGT (tAGT) in plasma of humans (18) as well as of rodents (24). In addition, there are commercial iAGT ELISAs (IBL) in which a sandwich ELISA technique using both anti-ANG I and anti-AGT antibodies is employed. However, in the course of our studies, we discovered that the iAGT ELISA only measured the reduced form of AGT. Thus, we developed a procedure that involves treating samples with a reducing agent to obtain an accurate measurement for total iAGT. In the current study, we report the iAGT, oxi-AGT, and red-AGT levels in rat and mouse plasma utilizing this modification of the commercially available ELISA kits.
METHODS
Animals and sample collection.
Male C57BL/6J mice (18-wk-old) and Sprague-Dawley rats (8-wk-old) were used. The animal experimental protocol was approved by the Animal Care and Use Committee of Tulane University. The rats were housed in a constant temperature room with 12:12-h dark-light cycle with free access to food and water until the experiments were conducted.
Blood samples were collected from conscious animals by cardiac puncture using 1-ml syringe (BD, Franklin Lakes, NJ) with 23-gauge needle (Fisher Scientific, Asheville, NC). The syringes were filled with 150 μl of cold proteinase inhibitor cocktail containing 10 mM EDTA, 2.5 mM phenanthridinone, 20 μM PMSF, 80 μM enalaprilat (an ACE inhibitor), and 80 μM pepstatin A (an inhibitor against aspartic proteases including renin) before the blood collection. Blood (150 μl) was collected and immediately mixed with the cold proteinase inhibitor cocktail to prevent cleavage of AGT by plasma renin during sample processing. Plasma samples were obtained by centrifugation of the blood samples and each volume of collected plasma was measured by using a micropipetter. A dilution factor of each plasma sample by mixing with the inhibitor cocktail was calculated based on the plasma volume and hematocrit ratio that was determined separately. The dilution factor in each sample was used to calculate tAGT, iAGT, oxi-AGT, and red-AGT levels after ELISA measurements.
Western blot and dot blot analyses.
Immunoreactivity of anti-AGT (IBL America, Minneapolis, MN) and anti-ANG I (Santa Cruz Biotechnology, Dallas, TX) antibodies against denatured plasma AGT was tested using Western blot. The anti-ANG I antibody recognizes the ANG I region at N-terminus of AGT. A mouse plasma sample prepared by the aforementioned method was used for Western blot analysis. One microliter of the plasma sample was incubated at 70°C with lithium dodecyl sulfate (LDS) and 50 mM dithiothreitol (DTT), a reducing reagent, to denature plasma AGT and dissociate disulfide bonds in AGT. Another sample was prepared without DTT. In the Western blot, reducing reagents were not added to running buffer and transfer buffer to maintain disulfide bonds in the nonreduced sample. The samples were applied to a precast NuPAGE 4–12% gel (Invitrogen, Carlsbad, CA), and the Western blot was performed as previously described (16). AGT proteins on nitrocellulose membranes (Bio-Rad, Hercules, CA) were detected by the anti-AGT and anti-ANG I antibodies.
Dot blot assay was employed to investigate immunoreactivity of anti-AGT and anti-ANG I antibodies against semidenatured plasma AGT. Mouse plasma sample (0.5 μl) was treated with/without 50 mM DTT. This DTT treatment dissociates disulfide bonds in AGT, but it does not linearize AGT protein. The samples were directly spotted onto nitrocellulose membranes. Thereafter, the same immunoblotting was performed as described above. All immunoblots were visualized by the Odyssey System (Li-Cor, Lincoln, NE).
tAGT ELISA.
tAGT levels including des-ANG I-AGT and iAGT in plasma were measured by using rat and mouse tAGT ELISAs (IBL America; Fig. 1) as previously described (6).
Fig. 1.

Figure depicting the system of total angiotensinogen (tAGT) ELISA and intact AGT (iAGT) ELISA. Red line in AGT molecule indicates ANG I region. Dotted line shows a disulfide bond in AGT molecule. In tAGT ELISA, two different anti-AGT antibodies are used. In iAGT ELISA, a combination of an anti-ANG I antibody and an anti-AGT antibody was used. iAGT ELISA without dithiothreitol (DTT) treatment detects only endogenous reduced intact AGT (red-iAGT).
Plasma iAGT, oxi-AGT, and red-AGT assay.
Plasma iAGT, oxi-AGT, and red-AGT levels were determined by modifying commercially available iAGT ELISAs (IBL America). In these ELISAs, an anti-ANG I antibody is coated in 96-well plates. This anti-ANG I antibody catches AGT protein in specimen by recognizing ANG I region of AGT. Accordingly, only iAGT is retained in the plate and des-ANG I-AGT is washed out during washing steps (Fig. 1). Caught iAGT is detected by anti-AGT antibody; namely, it is a sandwich ELISA using anti-ANG I and anti-AGT antibodies. The epitope of the anti-rat/mouse AGT antibody is in COOH terminus of rodent AGT (AA: 405–420 in mouse AGT, NH2-IGDTNPRVGEVLNSIL-COOH) as previously reported (6). Rat and mouse plasma samples were treated with/without 50 mM DTT. Thereafter, the plasma samples were diluted (1:20) with ELISA buffer contained in the ELISA kit. These diluted plasma samples treated with/without DTT were applied to ELISAs. Since the final concentration of DTT in the plasma samples was 2.5 mM, standard protein provided by the kits also received the same concentration of DTT. The ELISAs were performed following the manufacturer's instructions. Briefly, 100 μl of standard or sample were added to the proper well and incubated for 1 h at 37°C. During this incubation step, iAGT binds to an anti-ANG I antibody coated on the well. Then, the wells were washed four times with a washing buffer to remove unbound proteins including des-ANG I-AGT. A horseradish peroxidase (HRP)-conjugated anti-AGT antibody was added to the wells and incubated 30 min at 37°C. After free-antibody was washed out, HRP activity was shown by reacting with 3,3′,5,5′-tetramethylbenzidine. The enzymatic reaction was stopped by a sulfuric acid-based stop solution provided by the kits. The absorbance was detected at an optical density at 450 nm using FLUOstar micoplate-reader (BMG Labtech, Cary, NC).
Statistical analysis.
Data are expressed as means ± SE. The data were analyzed using Student's t-test. A value of P < 0.05 was considered statistically significant.
RESULTS
Immunoreactivity of plasma oxi-AGT and red-AGT to an anti-ANG I antibody.
Rat, mouse, and human AGT possess a cysteine residue at position 18. This cysteine forms an intramolecular disulfide bond with another cysteine in AGT. It has been proposed that the binding is promoted by elevated oxidative stress via oxidation of cysteine residues (22). Since the Cys18 is located at a proximal region of ANG I (position 1–10), we hypothesized that formation of the disulfide bond interferes with access of the anti-ANG I antibody to the ANG I region of AGT. To address this hypothesis, Western blot and dot blot analyses were performed.
In the Western blot analysis, mouse plasma AGT linearized by LDS and reduced by DTT treatments was detected by an anti-AGT antibody (Fig. 2A). In the same volume of mouse plasma (1 μl), AGT linearized by LDS but not reduced was also detected on the membrane (Fig. 2A). Intensities of the detected bands in Fig. 2A were identical, suggesting equal loading. These bands exhibited slightly different migrations in the gel (Fig. 2A; ∼48 and 50 kDa, respectively) because of conformational alterations due to dissociation of disulfide bonds. These plasma samples were used to test immunoreactivity to an anti-ANG I antibody (Fig. 2B). The reducing treatment of the plasma sample with DTT markedly increased immunoreactivity of AGT to the anti-ANG I antibody (Fig. 1B), suggesting that the disulfide bond in AGT limits accessibility of the anti-ANG I antibody to the ANG I moiety in the oxi-AGT.
Fig. 2.
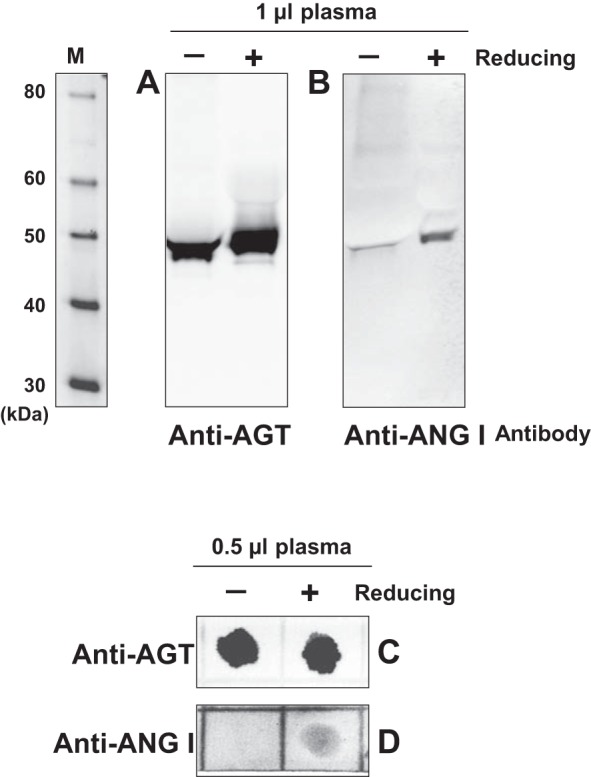
Immunoreactivity of plasma oxidized AGT (oxi-AGT) and red-AGT to an anti-ANG I antibody. The immunoreactivity of oxi-AGT and red-AGT to an anti-AGT antibody and an anti-ANG I antibody was tested. Mouse plasma was used in the experiments. One microliter of the plasma sample was incubated at 70°C with lithium dodecyl sulfate (LDS) and 50 mM DTT, a reducing reagent, to denature plasma AGT and dissociate disulfide bonds in AGT. Another sample was prepared without DTT. After gel electrophoresis and protein transfer from the gel to a membrane in the absence of DTT, AGT proteins were detected by an anti-AGT antibody (A) and anti-ANG I antibody (B). M, molecular weight markers. Furthermore, dot blot assays were also performed using these antibodies (C and D).
In the Western blot described above, immunoreactivity to the anti-ANG I antibody was tested using (semi-) linearized AGT by LDS. Therefore, immunoreactivity of nonlinearized AGT to the antibody was evaluated in a dot blot. Mouse plasma samples (0.5 μl) were treated with/without DTT in the absence of LDS to maintain the three-dimensional structure of AGT as much as possible except for changes in disulfide bond formation. The DTT treatment did not change immunoreactivity of AGT to an anti-AGT antibody (Fig. 2C). However, the immunoreactive spot was not detected by the anti-ANG I antibody when the plasma did not receive the reducing treatment (Fig. 2D). On the other hand, the immunoreactivity to the anti-ANG I antibody was clearly observed in DTT-treated plasma samples (Fig. 2D).
tAGT and iAGT levels in rodent plasma.
Rat and mouse plasma tAGT and iAGT levels were determined by tAGT ELISAs and iAGT ELISAs, respectively. Considered together with the findings presented above (Fig. 2), plasma samples were pretreated with DTT in the iAGT assay to obtain the maximum immunoreactivity to anti-ANG I antibody used in the ELISA. Total AGT levels in rat plasma were 1,839.2 ± 138.8 ng/ml (n = 12; Fig. 3A). iAGT levels in rat plasma were 983.3 ± 56.3 ng/ml (n = 12; Fig. 3A), which is 53.5% of the tAGT levels. In mouse plasma (n = 12), tAGT and iAGT levels were 1,081.9 ± 76.8 and 243.2 ± 16.6 ng/ml, respectively (Fig. 3B). Thus, 22.5% of the tAGT was iAGT in mouse plasma. The DTT treatment maintained the linearity of standard curve in the iAGT ELISA (Fig. 3C).
Fig. 3.
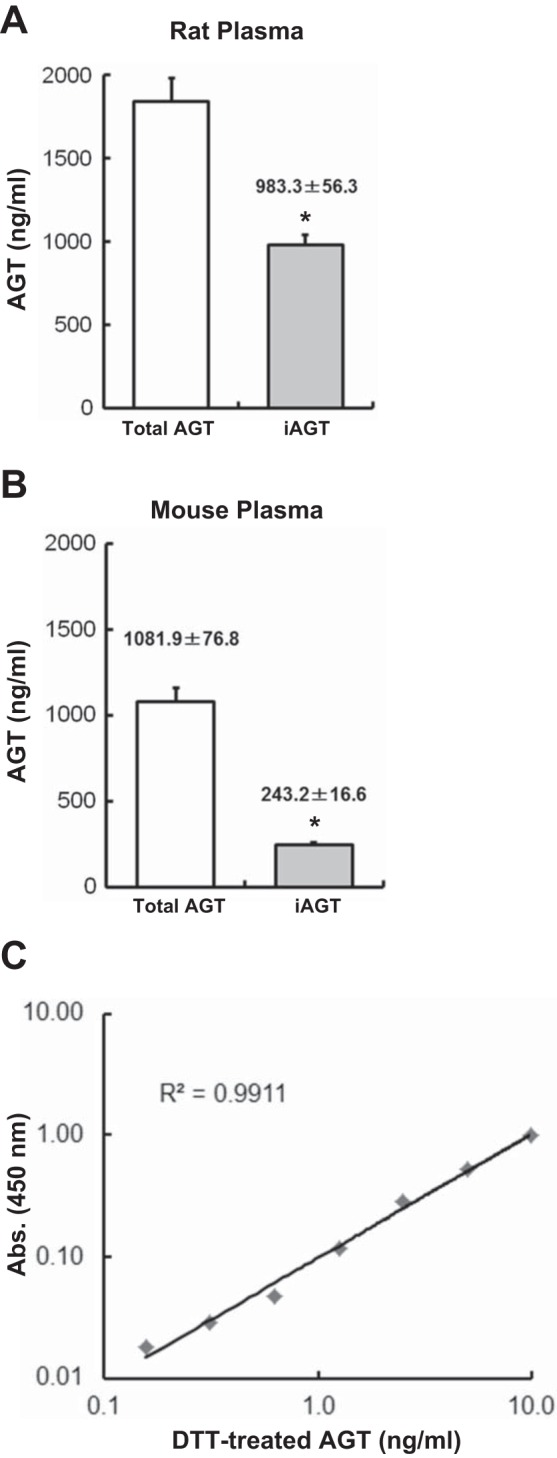
tAGT and iAGT levels in rodent plasma. Plasma tAGT and iAGT levels in rats (A, n = 12) and mice (B, n = 12) were measured by using tAGT and iAGT ELISAs. To determine iAGT levels, AGT in the plasma samples was treated by DTT. C: typical standard curve in iAGT ELISA. Data are expressed as means ± SE. *P < 0.05 indicates significant difference compared with each tAGT level.
oxi-AGT and red-AGT levels in rodent plasma.
Additional measurements of iAGT in rat and mouse plasma without a reducing treatment of plasma samples were performed to detect endogenous plasma red-AGT which does not require the reducing treatment to be detected by an anti-ANG I antibody in the ELISA. Naturally occurring red-AGT levels in rat plasma were 204.8 ± 11.5 ng/ml (n = 12; Fig. 4A). By subtraction of the red-AGT level from the rat iAGT level (Fig. 3) in individual animals, oxi-AGT levels were calculated. The rat plasma oxi-AGT levels were 778.5 ± 49.5 ng/ml (n = 12; Fig. 4A). Thus, the ratio of oxi-AGT and red-AGT was ∼4:1 in rat plasma iAGT. Mouse plasma oxi-AGT and red-AGT were 227.7 ± 16.4 and 15.5 ± 1.5 ng/ml, respectively (n = 12; Fig. 4B). The ratio of oxi-AGT and red-AGT was ∼16:1 for mouse plasma iAGT.
Fig. 4.
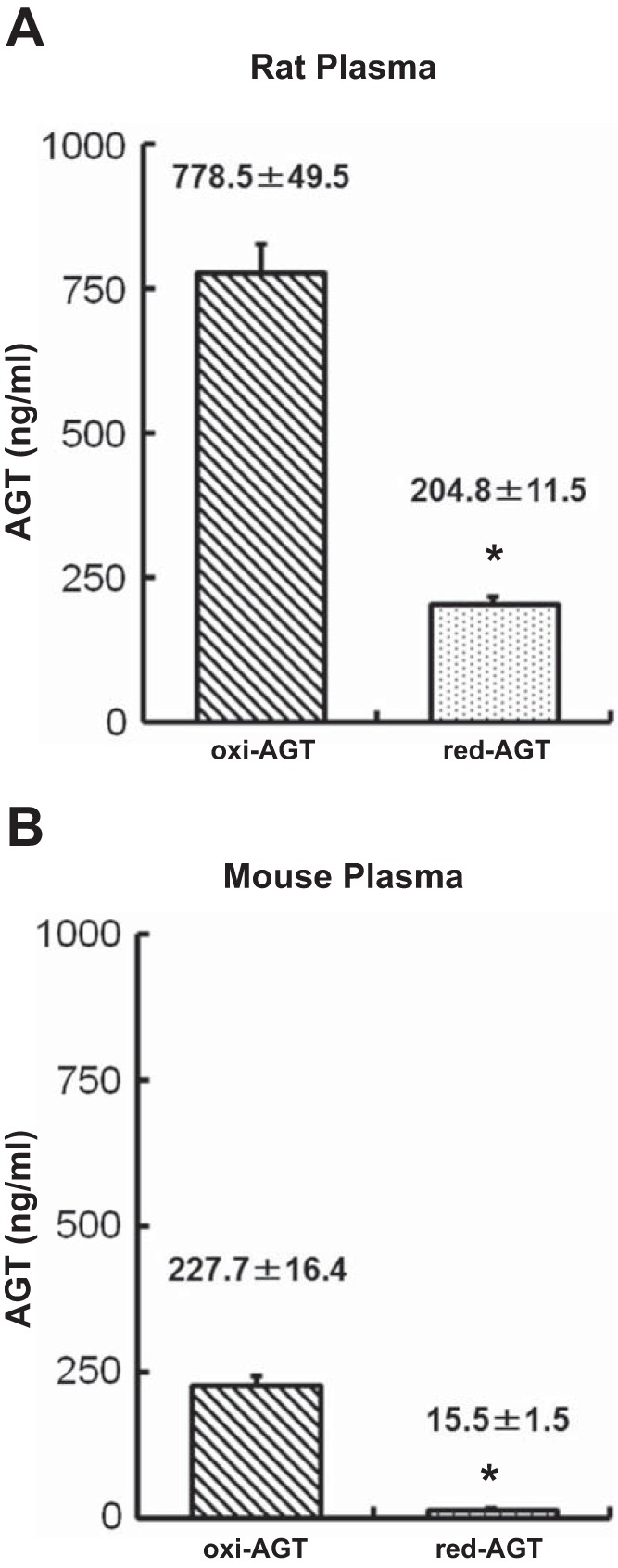
oxi-AGT and red-AGT levels in rodent plasma. Plasma oxi-AGT and red-AGT levels in rats (A, n = 12) and mice (B, n = 12) were quantified by using iAGT ELISA. Naturally occurring red-AGT levels were determined using non-DTT-treated plasma samples. oxi-AGT levels were calculated by subtracting the red-AGT level from the iAGT level in individual animals. Data are expressed as means ± SE. *P < 0.05 indicates significant difference compared with each oxi-AGT level.
DISCUSSION
Accurate measurement of the total amount of iAGT is necessary for estimating the actual amount of renin substrate that is available. Moreover, a recent study proposed pathophysiological functions of des-ANG I AGT (10). Thus, the establishment of measurements for iAGT and des-ANG I AGT levels can delineate changes in the levels of these two forms of iAGT, and help establish their respective roles in RAS-associated diseases. Investigators previously determined iAGT levels by ANG I conversion assays in which generated ANG I levels in the presence of excess renin in vitro were assessed. Although these techniques have provided important evidence in RAS-associated pathophysiological events, there are technical difficulties with accurate ANG I measurements. This study determined iAGT levels in rodent plasma by a modified method using commercially available ELISAs. tAGT levels in rat plasma were greater than in mouse plasma (1,839.2 vs. 1,081.9 ng/ml). In rat plasma, total AGT contained 53% of iAGT. Interestingly, iAGT was only 22.5% of tAGT in mouse plasma. The lower relative level of iAGT in mouse plasma reflects the greater plasma renin activity in mice than rats (1).
Since the results of Western blot analyses demonstrated that the anti-ANG I antibody exhibits the immunoreactivity only to red-AGT, plasma oxi-AGT and red-AGT levels were quantified using reduced and nonreduced plasma samples and iAGT ELISA. In rat and mouse plasma, the amount of iAGT detected by the ELISA without the reducing treatment using DTT was very low (204.8 ng/ml in rat and 15.5 ng/ml in mouse). These numbers suggested very low levels of endogenous red-AGT in the plasma. In particular, the ratio of oxi-AGT and red-AGT in mouse plasma iAGT was 16:1, suggesting that much of iAGT is oxidized in mouse plasma under normal conditions. The results of Western blot and dot blot analyses also showed very low levels of AGT band (Fig. 2B) and no AGT spot (Fig. 2D) detected by an anti-ANG I antibody when plasma did not receive the reducing treatment using DTT, supporting the low levels of naturally occurring red-AGT in rodent plasma observed in ELISAs. Since oxi-AGT exhibits higher affinity to renin than red-AGT (22), the high ratio of oxi-AGT in mouse plasma may lead to greater levels of AGT cleavage by renin, explaining the lower levels of iAGT in mouse than rat. In contrast to the results in this study, previous studies have reported high ratio of red-AGT in human blood; ∼40% (22) and >90% (14) of red-AGT in tAGT. The inconsistency of the findings may be because in the previous study measured red-AGT and oxi-AGT were factored by tAGT, which included cleaved des-ANG I-AGT, and the possible differences between human and rodent circulating AGT.
In conclusion, this study describes a specific method to quantify total iAGT and to estimate the ratio of oxi-AGT and red-AGT in rodent plasma by modifying the commercially available ELISAs. We document that total circulating AGT concentrations in mice are lower than in rats and that the fraction of iAGT is greater in rat plasma than mouse plasma. The lower availability of plasma iAGT in mice than rats suggests that it may be a limiting factor in ANG I formation in this species. Accumulation of experimental evidence will be required to precisely evaluate oxi-AGT and red-AGT levels under different physiological and disease conditions.
GRANTS
This study was supported by grants from the National Institute of General Medical Sciences IDeA Program (COBRE, P30GM103337) and the National Institute of Diabetes and Digestive and Kidney Diseases (Grant R01DK107694-01).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
R.S., H.K., A.K., K.M., and L.G.N. conception and design of research; R.S., H.K., A.K., and K.M. performed experiments; R.S., H.K., A.K., K.M., and L.G.N. analyzed data; R.S., H.K., A.K., K.M., and L.G.N. interpreted results of experiments; R.S., H.K., A.K., K.M., and L.G.N. prepared figures; R.S., H.K., A.K., K.M., and L.G.N. drafted manuscript; R.S., H.K., A.K., K.M., and L.G.N. edited and revised manuscript; R.S., H.K., A.K., K.M., and L.G.N. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Yoshiaki Hagiwara and Kazuya Miyashita (Immuno-Biological Laboratories, Fujioka, Gunma, Japan) for providing technical advices.
REFERENCES
- 1.Catanzaro DF, Mullins JJ, Morris BJ. The biosynthetic pathway of renin in mouse submandibular gland. J Biol Chem 258: 7364–7368, 1983. [PubMed] [Google Scholar]
- 2.Dickson ME, Sigmund CD. Genetic basis of hypertension: revisiting angiotensinogen. Hypertension 48: 14–20, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Dickson ME, Zimmerman MB, Rahmouni K, Sigmund CD. The −20 and −217 promoter variants dominate differential angiotensinogen haplotype regulation in angiotensinogen-expressing cells. Hypertension 49: 631–639, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Ishigami T, Umemura S, Tamura K, Hibi K, Nyui N, Kihara M, Yabana M, Watanabe Y, Sumida Y, Nagahara T, Ochiai H, Ishii M. Essential hypertension and 5′ upstream core promoter region of human angiotensinogen gene. Hypertension 30: 1325–1330, 1997. [DOI] [PubMed] [Google Scholar]
- 5.Jeunemaitre X, Inoue I, Williams C, Charru A, Tichet J, Powers M, Sharma AM, Gimenez-Roqueplo AP, Hata A, Corvol P, Lalouel JM. Haplotypes of angiotensinogen in essential hypertension. Am J Hum Genet 60: 1448–1460, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobori H, Katsurada A, Miyata K, Ohashi N, Satou R, Saito T, Hagiwara Y, Miyashita K, Navar LG. Determination of plasma and urinary angiotensinogen levels in rodents by newly developed ELISA. Am J Physiol Renal Physiol 294: F1257–F1263, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Kumar A, Li Y, Patil S, Jain S. A haplotype of the angiotensinogen gene is associated with hypertension in African Americans. Clin Exp Pharmacol Physiol 32: 495–502, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40: 511–515, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu H, Wu C, Howatt DA, Balakrishnan A, Moorleghen JJ, Chen X, Zhao M, Graham MJ, Mullick AE, Crooke RM, Feldman DL, Cassis LA, Vander Kooi CW, Daugherty A. Angiotensinogen exerts effects independent of angiotensin II. Arterioscler Thromb Vasc Biol 36: 256–265, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyata K, Ohashi N, Suzaki Y, Katsurada A, Kobori H. Sequential activation of the reactive oxygen species/angiotensinogen/renin-angiotensin system axis in renal injury of type 2 diabetic rats. Clin Exp Pharmacol Physiol 35: 922–927, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakajima T, Wooding S, Sakagami T, Emi M, Tokunaga K, Tamiya G, Ishigami T, Umemura S, Munkhbat B, Jin F, Guan-Jun J, Hayasaka I, Ishida T, Saitou N, Pavelka K, Lalouel JM, Jorde LB, Inoue I. Natural selection and population history in the human angiotensinogen gene (AGT): 736 complete AGT sequences in chromosomes from around the world. Am J Hum Genet 74: 898–916, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension 39: 316–322, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rahgozar S, Amirian T, Qi M, Shahshahan Z, Entezar EGM, Ghasemi Tehrani H, Miroliaei M, Krilis SA, Giannakopoulos B. Improved assay for quantifying a redox form of angiotensinogen as a biomarker for pre-eclampsia: a case-control study. PLos One 10: e0135905, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sachetelli S, Liu Q, Zhang SL, Liu F, Hsieh TJ, Brezniceanu ML, Guo DF, Filep JG, Ingelfinger JR, Sigmund CD, Hamet P, Chan JS. RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney Int 69: 1016–1023, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Satou R, Miyata K, Gonzalez-Villalobos RA, Ingelfinger JR, Navar LG, Kobori H. Interferon-gamma biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J 26: 1821–1830, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi N, Lopez ML, Cowhig JE Jr, Taylor MA, Hatada T, Riggs E, Lee G, Gomez RA, Kim HS, Smithies O. Ren1c homozygous null mice are hypotensive and polyuric, but heterozygotes are indistinguishable from wild-type. J Am Soc Nephrol 16: 125–132, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Watkins WS, Hunt SC, Williams GH, Tolpinrud W, Jeunemaitre X, Lalouel JM, Jorde LB. Genotype-phenotype analysis of angiotensinogen polymorphisms and essential hypertension: the importance of haplotypes. J Hypertens 28: 65–75, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu C, Xu Y, Lu H, Howatt DA, Balakrishnan A, Moorleghen JJ, Vander Kooi CW, Cassis LA, Wang JA, Daugherty A. Cys18-Cys137 disulfide bond in mouse angiotensinogen does not affect Ang II-dependent functions in vivo. Hypertension 65: 800–805, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu P, Wang Y, Sterner-Kock A, Bader M, Schultheiss HP, Walther T. Excessive hypertension and end-organ damage in a transgenic mouse line carrying the rat angiotensinogen gene. J Cardiovasc Pharmacol 53: 38–43, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Ying J, Stuart D, Hillas E, Gociman BR, Ramkumar N, Lalouel JM, Kohan DE. Overexpression of mouse angiotensinogen in renal proximal tubule causes salt-sensitive hypertension in mice. Am J Hypertens 25: 684–689, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou A, Carrell RW, Murphy MP, Wei Z, Yan Y, Stanley PL, Stein PE, Broughton Pipkin F, Read RJ. A redox switch in angiotensinogen modulates angiotensin release. Nature 468: 108–111, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]