

Abstract
Podocytes are the key target for injury in proteinuric glomerular diseases that result in podocyte loss, progressive focal segmental glomerular sclerosis (FSGS), and renal failure. Current evidence suggests that the initiation of podocyte injury and associated proteinuria can be separated from factors that drive and maintain these pathogenic processes leading to FSGS. In nephrotic urine aberrant glomerular filtration of plasminogen (Plg) is activated to the biologically active serine protease plasmin by urokinase-type plasminogen activator (uPA). In vivo inhibition of uPA mitigates Plg activation and development of FSGS in several proteinuric models of renal disease including 5/6 nephrectomy. Here, we show that Plg is markedly increased in the urine in two murine models of proteinuric kidney disease associated with podocyte injury: Tg26 HIV-associated nephropathy and the Cd2ap−/− model of FSGS. We show that human podocytes express uPA and three Plg receptors: uPAR, tPA, and Plg-RKT. We demonstrate that Plg treatment of podocytes specifically upregulates NADPH oxidase isoforms NOX2/NOX4 and increases production of mitochondrial-dependent superoxide anion (O2−) that promotes endothelin-1 synthesis. Plg via O2− also promotes expression of the B scavenger receptor CD36 and subsequent increased intracellular cholesterol uptake resulting in podocyte apoptosis. Taken together, our findings suggest that following disruption of the glomerular filtration barrier at the onset of proteinuric disease, podocytes are exposed to Plg resulting in further injury mediated by oxidative stress. We suggest that chronic exposure to Plg could serve as a “second hit” in glomerular disease and that Plg is potentially an attractive target for therapeutic intervention.
Keywords: plasminogen, podocytes, oxidative stress
normal urine is essentially protein free. Proteinuria of increased severity is associated with progression of chronic kidney disease (CKD) independent of the etiology and baseline glomerular filtration rate (45, 58). Specialized endothelial cells, the glomerular basement membrane (GBM), and podocytes work in partnership to preserve the permselectivity of the kidney filter (24, 67, 74, 79). Podocytes, the component of the glomerulus most susceptible to injury (8, 20, 22, 24, 36, 74), are highly differentiated cells that adhere to the GBM. Podocytes are critical for maintaining the integrity of the kidney filter (13, 22, 24, 36, 54, 60, 67). Independent of the cause of injury, podocyte response follows similar pathways including disorganization of the actin cytoskeleton, foot process effacement, loss of the slit diaphragm (SD), detachment and death (24, 36, 67, 74, 75, 93). Podocytes' ability to undergo cell division and compensate for injury and loss is limited (20, 24, 36, 67, 74, 92, 93). When podocyte loss exceeds 30%, persistent nonselective proteinuria, progressive glomerulosclerosis, and progressive CKD ensue (20, 36, 91–93).
Clinically, proteinuria accompanied by continued podocyte loss and CKD progression is a hallmark of many glomerular diseases of different etiology including diabetic nephropathy and focal segmental glomerular sclerosis (FSGS) (8, 13, 24, 36, 54, 60, 67). To date, it remains unclear whether persistent injury is driven by persistence of the initial pathogenic process or by a superimposed pathogenic process, a so-called “second hit model of podocyte injury.” The latter raises the question of whether continuous trans-glomerular passage of large circulating macromolecules might contribute to podocyte damage as a “second hit” (20, 24, 36, 46, 47, 54, 68, 93). In this context, studies have shown that albuminuria itself may contribute to tubule-interstitial disease and to glomerulosclerosis (11, 53).
Clinically, most of the mechanisms linking proteinuria to CKD progression remain to be fully defined (9, 13, 22, 54, 68). Data from clinical and animal models have documented significant quantities of biologically active urinary plasminogen (Plg) and the protease plasmin in nephrotic range proteinuria of diverse etiologies (6, 81). Furthermore, in patients in remission from nephrotic syndrome the urinary Plg/plasmin content is minimal (1), thereby supporting the concept that under physiologic conditions podocytes are not exposed to large concentrations of Plg; a fact further supported by its molecular size (81 kDa), which is larger than albumin (3).
It is well-established that after binding onto cell surface receptors (42, 51, 52, 59) Plg is protected from inhibitors, undergoes proteolytic activation, and converts to the serine protease plasmin. In many cell types, plasmin initiates responses linked to both its proteolytic as well as its nonproteolytic actions including cleavage of matrix proteins (62, 87), inactivation of vascular endothelial growth factor (69), induction of reactive oxygen species (ROS) (85), as well as activation of transforming growth factor (TGF)-β (23, 34, 43).
Here, we demonstrate that Plg is increased in the urine in two murine models of proteinuric kidney disease associated with podocyte injury: the Tg26 HIV-associated nephropathy mouse (37) and the Cd2ap−/− model of focal segmental glomerulosclerosis (76). We show that immortalized human podocytes constitutively express and synthesize urokinase-type plasminogen activator (uPA) as well as the Plg receptors uPAR, tPA, and the novel trans-membrane receptor Plg R-KT.
Our studies show, to our knowledge for the first time, that podocyte binding of Plg upregulates NADPH oxidase isoforms NOX2 and NOX4 and elicits mitochondrial production of superoxide anion (O2−) accompanied by 1) upregulation of the multifunctional scavenger receptor CD36 (77, 96) resulting in increased podocyte uptake of oxidized LDL (oxLDL) with subsequent apoptosis and 2) induction of podocyte synthesis of endothelin-1, a molecule that acts in a paracrine/autocrine fashion and has been implicated in podocyte-endothelial cell cross talk and glomerular disease progression (14, 35, 41, 61). Epsilon aminocaproic acid (EACA), an inhibitor of cellular Plg binding and activation (51, 52), prevented all the injurious actions elicited by exposure of podocytes to Plg. In addition, we demonstrated that Apocynin, AMPkinase, and Mito Tempo, molecules that mitigate production of ROS (18, 19, 40), provided similar podocyte protection.
These data strongly support the conclusion that Plg-plasmin is a molecule that via previously unrecognized mechanisms that include oxidative stress can play a role as a promoter/contributor of a “second hit” type of podocyte injury in proteinuric nephropathies. Our studies may contribute to development of therapeutic interventions in proteinuric kidney disease.
METHODS
Mouse studies.
Urine was collected from two proteinuric kidney disease models that have been well-characterized: 5-wk-old Cd2ap−/− mice (25, 72, 76, 90, 94) and 6-wk-old Tg26 mice (2, 4, 21, 32, 37, 38, 65). Urinary Plg was measured using 2 μl urine with Elisa Kit (ICL E-90PMG) read at absorbance at 450 nm. Mouse creatinine was measured with an enzymatic assay kit (Crystal Chem: no. 80350).
Human podocyte culture.
Human podocytes initially generated by MA Saleem Children's Renal Unit and Academic Renal Unit, University of Bristol, UK (71) were generously provided by S. Merscher-Gomez and A. Fornoni, University of Miami, who also provided initial technical supervision of podocyte culture as reported (50, 64, 71). Briefly, human podocytes were cultured and differentiated in RPMI 1640 culture medium containing 10% FBS, 1% penicillin/streptomycin with or without 1% ITS, 50 U/ml human interferon as described (50, 71). The immortalized normal human podocytes were propagated at 33°C and then thermoshifted for differentiation for 14 days at 37°C. Terminally differentiated podocytes were starved in serum-free RPMI 1640 medium for 24 h before the experiments were performed (24, 50, 64, 71).
Determination of O2− production.
O2− production was determined by using lucigenin-enhanced chemiluminescence as described in our previous publications (96). Human podocytes were treated with Plg (1 μmol/l), the concentration of Plg found in human serum (51, 52), for 6 h as determined in experiments of time responses to Plg up to 24 h (not shown).
To determine the source of ROS, the cells were preincubated with the following compounds for 30 min before incubation with Plg: 1) NADPH oxidase inhibitors diphenyleneiodonium (DPI, 1 μmol/l, Sigma) and APOCY (100 μmol/l, Sigma) (12, 39, 40, 96), 2) AICAR, an agonist to AMP-activated protein kinase (100 μmol/l, Sigma) (19, 52, 86), 3) EACA (0.2 mol/l, Sigma) (42, 52), and 4) amiloride (75 μM, Sigma).
Immunoblot analyses.
The cells were harvested with lysis buffer containing a cocktail of protein inhibitor (Complete Mini, Roche Diagnostics GmbH, cat. no. 11836153001). Protein was quantified by Bio-Rad assay, and 30 mg of total protein were first subjected to SDS-PAGE and then transferred to nitrocellulose membranes. The membranes were incubated overnight in a cold room with primary rabbit anti-CD36, anti-tPA, anti-WT1, and anti-uPAR (Santa Cruz Biotechnology), anti-phospho-AMPK, anti-AMPK, rabbit anti-NOX 4 (Santa Cruz Biotechnology), and anti-NOX2 (AbCam) followed by incubation with a peroxidase-conjugated secondary antibody for 1 h. All the Ab used equivalence-of-protein loading and transfer was confirmed by reblotting the samples with anti-β-actin antibody. Immune reactive bands were detected by chemiluminescence and quantified by densitometry. Relative quantities of each protein were normalized by β-actin (Santa Cruz Biotechnology) and expressed as fold increase vs. control (96).
Determination of total cellular cholesterol content.
Human podocytes in six-well dishes were incubated with or without Plg (1 μmol/l) for 24 h followed by incubation with oxLDL (50 μg/ml) for another 24 h (96). Total cellular cholesterol was extracted by adding 1 ml hexane/isopropanol (3:2, vol:vol) to the wells. The samples were evaporated by using a SpeedVac to remove the solvent and then redissolved in 50 ml ethanol. Cholesterol was determined with an enzyChrom AF cholesterol assay kit according to the manufacturer's instructions (Bioassay Systems, Hayward, CA). The assay results were normalized according to the sample protein content.
Apoptosis assay.
A total of 2 × 106 cells was seeded in 75-cm2 flasks and treated for 24 h with one of the following conditions: vehicle, Plg (1 μmol/l), oxLDL (50 μg/ml), or incubation with plasminogen followed by oxLDL (50 μg/ml). The cells were trypsinized and washed with PBS twice. Apoptosis was measured using the annexin V-fluorescein isothiocyanate apoptosis detection kit (Roche Diagnostics, Indianapolis, IN) followed by flow cytometry.
Statistics.
The results were expressed as means ± SD. Statistical analyses were performed by ANOVA with Bonferonni's correction for multiple comparisons, followed by Scheffé's test. Significance was assumed at P < 0.05.
RESULTS
Urinary plasminogen is increased in proteinuric mouse models.
We measured urinary Plg normalized to urinary creatinine in two independent models of proteinuric kidney disease. Cd2ap−/− mice provide a well-characterized model of focal segmental glomerulosclerosis with proteinuria developing between 2 and 3 wk of age. Urinary Plg from 5-wk-old Cd2ap−/− mice (mean urine protein/cr ratio = 125.4 mg/mg) was markedly elevated compared with wild-type littermate controls (30.96 vs. 0.09 ng/mg; P: 0.007; Fig. 1). We also detected elevated urine Plg in 6-wk-old mice from the Tg26 model (mean urine protein/creatinine ratio 83.1 mg/mg) of HIV nephropathy (2.56 vs. 0.49 ng/mg; P: 0.008).
Fig. 1.
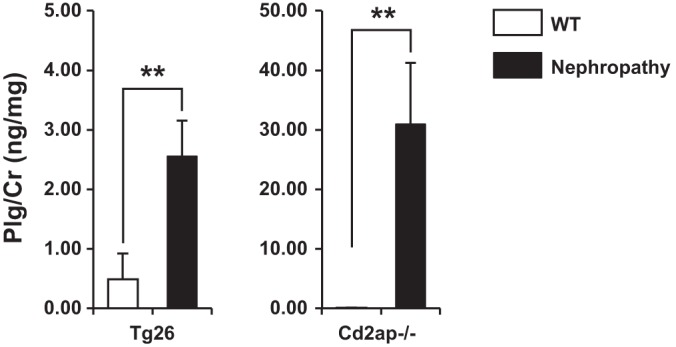
Urine plasminogen (Plg) levels are increased in mouse models of proteinuric kidney disease. Urine Plg/Cr is significantly increased in 6-wk-old Tg26 and 5-wk-old Cd2ap−/− mice. WT, wild-type. **P < 0.005.
Plasminogen receptor expression and superoxide upregulation in podocytes.
In cultured human podocytes, we established the expression of uPA as well as three well-characterized Plg receptors that are known to be blocked by the lysine analog EACA: uPAR, tPA, and the novel receptor R-KT (Fig. 2A) (42, 51, 52, 59). Previous studies in vivo and in vitro demonstrated that several isoforms of NADPH oxidase including NOX2 and NOX4 are present in podocytes and that ROS originating from NADPH oxidase play an important role in ROS-mediated podocyte injury. NOX4 is the most abundant isoform in podocytes (10, 19, 31, 80). RTPCR (Fig. 2, B and C) and Western blotting (Fig. 2, D and E) showed that Plg significantly upregulated podocyte NOX4 and NOX2 expression. This was prevented by prior incubation with EACA, thereby demonstrating that Plg binding and activation were required (51, 85).
Fig. 2.

Plasminogen upregulates NADPH oxidase isoforms NOX2 and NOX4: Plg upregulated mRNA and protein expression of NOX2 and NOX4 isoforms of NADPH oxidase that was significantly prevented by inhibition of Plg binding/activation by epsilon aminocaproic acid (EACA). M = marker. Data were expressed as means ± SE. *P < 0.05 vs. control. #P < 0.05 vs. plasminogen.
Exposure of podocytes to Plg increased NADPH oxidase-mediated O2− production (Fig. 3A) that was prevented by the inhibitors of NADPH oxidase diphenyleneiodonium (DPI) and by apocynin (APOCY) (39, 40).
Fig. 3.
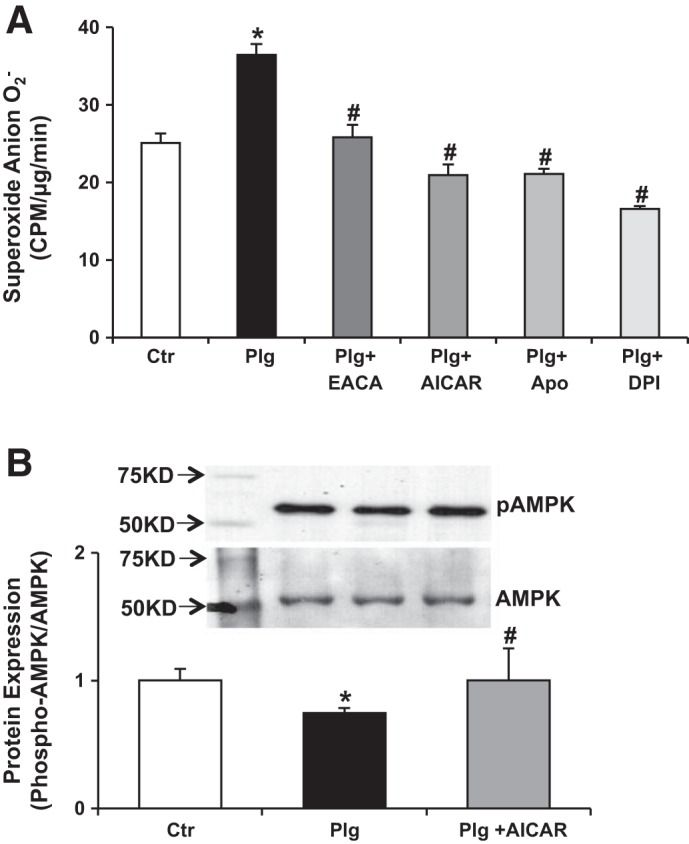
Plasminogen induces production of superoxide anion in human podocytes. A: Plg binds to human podocytes, is activated, and induces production of superoxide anion. Plg (1 μmol/l, the Plg concentration in serum) significantly increased podocyte O2− production; increased O2− production was prevented by the NADPH oxidase and flavin-containing oxidases diphenyleneiodonium (DPI) 1 μmol/l, the NADPH oxidase inhibitor APOCY (100 μmol/l), the agonist of AMP-activated protein kinase AICAR (100 μmol/l), as well as the inhibitor of Plg/plasmin activation EACA (0.2 mol/l). Data was expressed as means ± SE. *P < 0.05 vs. control. #P < 0.05 vs. plasminogen; n = 6. B: pAMPK is active AMPK. Exposure of podocytes to Plg reduced pAMPK/AMPK ratio, and increased NADPH oxidase-mediated O2− production. Activation of AMPK by AICAR, 5-aminoimidazole-4-carboxamide-1-riboside (52–54, 63) significantly prevented these effects by increasing the pAMPK/AMPK ratio and concomitantly reducing O2− production. AMPK is a ubiquitous serine/threonine kinase that acts as an energy sensor and is expressed in the plasma membrane of podocytes; pAMPK suppresses NADPH oxidase (52–54, 63). Data are expressed as means ± SE. *P < 0.05 vs. control. #P < 0.05 vs. plasminogen; n = 5.
Plg concomitantly reduced the pAMPK/AMPK ratio (Fig. 3B). AICAR, 5-aminoimidazole-4-carboxamide-1-riboside, is known to phosphorylate and activate AMPK (18, 86). Pretreatment of podocytes with AICAR before exposure to Plg significantly prevented the reduction in pAMPK/AMPK ratio and concurrently inhibited O2− production (Fig. 3A). This suggests an important role of this stress-sensing kinase in the biology of podocyte injury associated with oxidative stress (18, 86).
Expression of the scavenger receptor CD36 is upregulated by Plg.
The B scavenger receptor CD36, which is present in many cells including podocytes (49), recognizes, binds, and internalizes oxLDL (77, 96). Plg significantly upregulated podocyte expression of CD36 (Fig. 4A). This paralleled the increased production of O2− induced by Plg (Fig. 3A). The upregulation of CD36 was prevented by EACA as well as by the NADPH inhibitor APOCY and AICAR, the agonist of AMPK (18, 19, 86). Importantly, the significant inhibition of Plg upregulation of CD36 by EACA, APOCY, and AICAR indicates the interdependence between Plg activation, increased oxidative stress, and CD36 upregulation in podocytes.
Fig. 4.
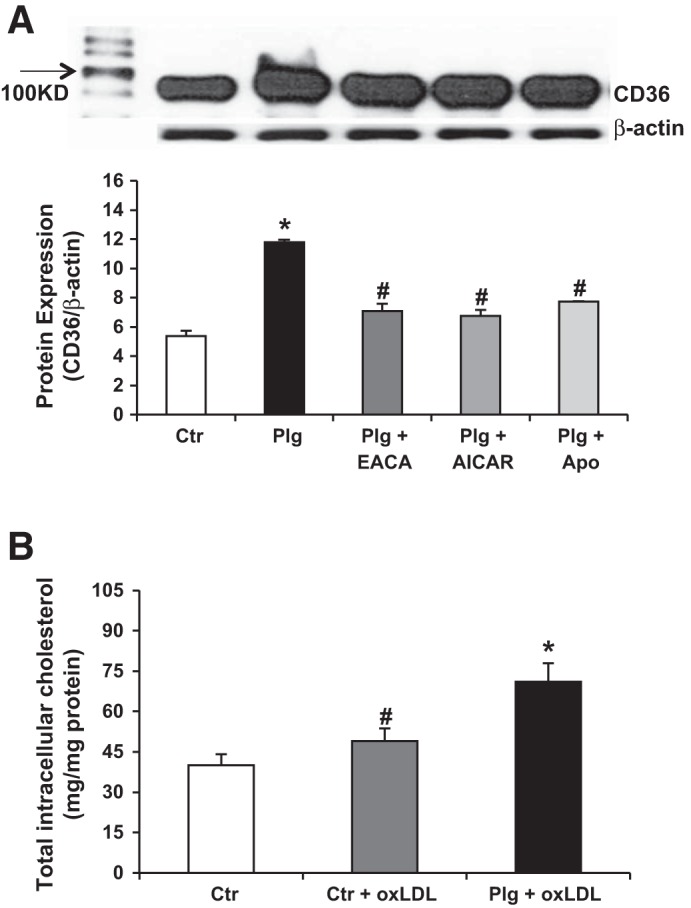
Expression of the scavenger receptor CD36 is upregulated by plasminogen. A: effects of Plg on scavenger receptor CD36 expression in human podocytes. Plg significantly increased CD36 expression, which was prevented by the NADPH oxidase inhibitor APOCY (10 μmol/l), AICAR (10 μmol/l) an agonist of AMP-activated protein kinase, as well as the inhibitor of Plg (plasmin) activation EACA (0.2 mol/l). Data are expressed as means ± SE. *P < 0.05 vs. control. #P < 0.05 vs. plasminogen; n = 5. B: effect of Plg and oxidized LDL (oxLDL) on total intracellular cholesterol content in human podocytes. Incubation of human PODC with oxLDL (50 mg/ml) resulted in a small but significant increase in total intracellular cholesterol content. Addition of Plg in the presence of oxLDL further increased intracellular cholesterol content. Data are expressed as means ± SE. *P < 0.05 vs. control and control + oxLDL. #P < 0.05 vs. control group; n = 6 in each group.
Our group and others have shown that upregulation of CD36 in human macrophages increases uptake of oxLDL accompanied by increased synthesis of ROS (77, 96). It is presently unknown whether upregulation of CD36 plays a similar role in podocytes. Similar to macrophages, podocytes treated with oxLDL for 24 h showed a significant increase in cholesterol accumulation (Fig. 4B) (96), which was further increased in podocytes treated with Plg plus oxLDL thereby suggesting that the increased oxLDL uptake was linked to Plg upregulation of CD36 (77, 96). The off-target effects of the diuretic amiloride as an inhibitor of uPA are well-documented (30, 48, 88). We found that amiloride inhibited Plg-induced upregulation of CD36 (Fig. 5A) and ROS superoxide generation (Fig. 5B).
Fig. 5.
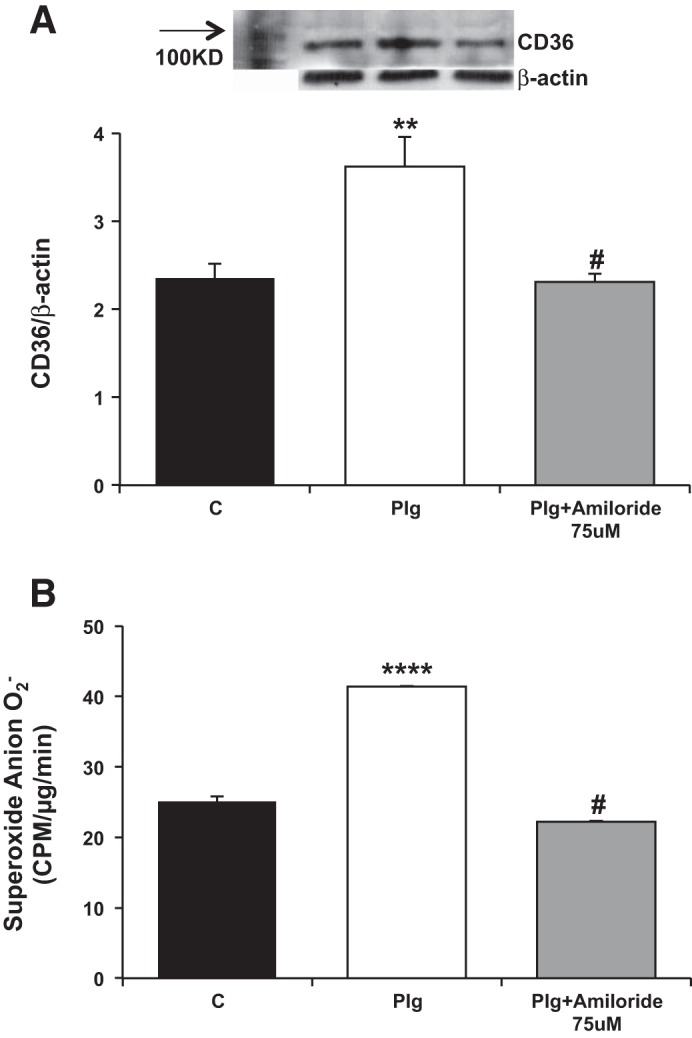
Amiloride regulation of Plg-induced CD36 and reactive oxygen species (ROS) generation in podocytes. Amiloride abrogates Plg induction of CD36 (A) and ROS (B) in podocytes. **P < 0.005, #P < 0.05 vs. plasminogen, ****P < 0.0001.
Plg enhances oxLDL-mediated podocyte apoptosis.
Based on our novel findings that Plg upregulates CD36 (Fig. 4A) and increases oxLDL uptake (Fig. 4B), we investigated by flow cytometry the effects of oxLDL and Plg individually as well as together with oxLDL on podocyte apoptosis. As shown in Fig. 6, both Plg as well as oxLDL alone slightly increased podocyte apoptosis. Importantly, the combination of Plg and oxLDL had a clear additive effect that further significantly augmented podocyte apoptosis.
Fig. 6.

Exposure of human podocytes to Plg plus oxLDL has additive effects in promoting podocyte apoptosis. A: both Plg and oxLDL induced slight but significant podocyte apoptosis after 24 h. The combination of Plg and oxLDL had a clear additive effect on podocyte apoptosis driven by increased oxLDL uptake associated with Plg upregulation of scavenger receptor CD36 (Fig. 4, A and B). B: podocyte apoptosis measured by flow cytometry. Data were expressed as means ± SE. *P < 0.05 vs. control.
Plg promotes endothelin-1 synthesis.
Studies in vivo and in vitro showed that endothelin-1 (ET1), acting in paracrine/autocrine fashion, induces oxidative stress and injury of glomerular endothelial cells and podocytes (14, 35, 41). It is currently unknown whether Plg promotes ET1 synthesis by podocytes and whether it is linked to oxidative stress by acting as a second messenger. As shown in Fig. 7B, we demonstrated that Plg promotes synthesis-release of ET1 that was prevented by EACA, APOCY, and by the selective inhibitor of mitochondria ROS Mito Tempo (Fig. 7, A and B) (14). This demonstrated for the first time the important role of Plg-induced ROS, particularly mitochondrial ROS in promoting podocyte ET1 synthesis.
Fig. 7.
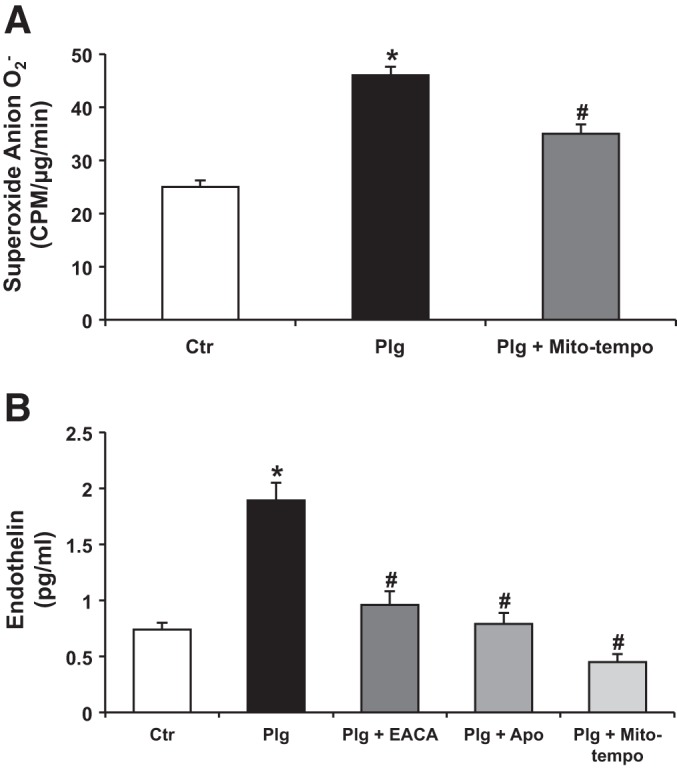
Plasminogen-promoted synthesis of endothelin 1 was specifically prevented by Mito-Tempo an inhibitor of mitochondrial ROS. A: Plg induced O2− production in podocytes that was partially prevented (40%) by Mito Tempo, a selective inhibitor of mitochondrial oxidative stress. B: endothelin 1 synthesis/release was measured by ELISA in the supernatants of the various conditions. Plg increased podocyte synthesis of endothelin 1 that was inhibited by APOCY and by Mito-Tempo. Important to note is that while Mito-Tempo only reduced ROS by 40% (A), it completely prevented endothelin 1 synthesis in response to Plg, suggesting that O2− from mitochondria plays a critical role in modulating podocyte endothelin 1 synthesis. Data were expressed as means ± SE. *P < 0.05 vs. control. #P < 0.05 vs. plasminogen.
DISCUSSION
Independent of the etiology, chronic severe nonselective proteinuria is a harbinger of progression of CKD (24, 27, 67, 74, 79). It has long been suspected that the relentless trans-glomerular passage of selected plasma macromolecules may contribute to podocyte injury as a “second hit.” Studies have shown that albuminuria itself may contribute to glomerular injury directly and indirectly via podocytes uptake of fatty acids (FFA) bound to albumin (11) as well as by sequestration of retinoic acid, a molecule shown to be critical in protecting podocytes from injury (44, 57). Data from both patients and experimental animals with nephrotic syndrome have consistently shown their urine to contain large quantities of biologically active Plg and the protease plasmin (1, 5, 6, 81–84, 95).
It has been demonstrated that plasmin in nephrotic urine proteolytically activates ENaC, an effect that disappears during remission of the nephrotic syndrome (1, 5, 6, 81–84, 95). In vitro and in vivo studies demonstrated the ability of plasmin to contribute to cellular injury and to inflammation via its proteolytic as well as its nonproteolytic actions (85). Interestingly, previous studies in animals and in patients with proteinuric nephropathies treated with EACA, the inhibitor of Plg activation to plasmin (51, 52, 85), reported a marked reduction of proteinuria and glomerular injury (28).
We utilized two independent well-characterized models of progressive proteinuric kidney disease, the Cd2ap−/− mouse model of FSGS and the Tg26 model of HIVAN. In both, urinary Plg was significantly elevated (Fig. 1). We also used cultured human podocytes (71) to further assist us in determining whether the Plg/plasmin system could potentially participate as a “second hit” type of podocyte injury linked to proteinuria.
In human podocytes, we confirmed the expression of urokinase plasminogen activator uPA that is synthesized by podocytes and of Plg receptors uPAR, tPA, and Plg R-KT (51, 52) (Fig. 2A). uPAR has high-affinity for uPA, but has no intracellular domain, which limits its signaling capacity. Plg R-KT colocalizes with uPAR and is a unique trans-membrane protein that activates Plg because it exposes a COOH-terminal lysine on the cell's surface (51, 52). In addition, Plg R-KT specifically interacts with tPA, which secures Plg on the cell surface and promotes its activation to plasmin that once bound to the cell surface is protected from its physiological inhibitors (51, 52, 85).
Here, we demonstrated that Plg upregulated mRNA and protein expression of the podocyte NOX2 and NOX4 (Fig. 2) (10, 31), accompanied by significant production of the ROS superoxide anion O2− as determined by lucigenin chemiluminescence. EACA prevented NOX2 and NOX4 upregulation as well as O2− production, thereby demonstrating that these effects are associated with Plg binding and activation (Fig. 2, B–E). Next, we determined that Apocynin, an inhibitor of NADPH oxidases, which are considered to be the major source of ROS-mediated podocyte injury (10, 31), prevented the increase in O2− production induced by Plg (Fig. 3A).
AMPK is a serine/threonine kinase that is present in podocytes and acts as an energy sensor (7, 47). Once AMPK is phosphorylated (pAMPK), it becomes activated and enables cells to reduce NADPH oxidase-dependent ROS formation (18, 19, 70, 86). We determined that exposure of podocytes to Plg reduced pAMPK (Fig. 3B) and that AICAR, an activator of AMPK (18, 19, 70), normalized pAMPK and concomitantly inhibited Plg-driven O2− production (Fig. 3, A and B). Of note, AICAR has been shown to have beneficial effects in diabetic nephropathy via similar mechanisms to those reported here (18, 19, 86). Hence, AMPK may have a universal role in mitigating podocyte pathobiology linked to oxidative stress (10, 18, 19, 70, 86).
Podocyte ROS originate in the cytosol and particularly in mitochondria, a major source of intracellular ROS production under physiologic and pathologic conditions (10, 14, 18, 31). Many studies have shown that sustained mitochondria ROS accumulation results in mitochondrial dysfunction (10, 14, 18). Here, we showed that Mito Tempo, a selective inhibitor of mitochondrial ROS, reduced by 40% podocyte O2− induced by Plg (Fig. 6A). Based on our novel findings with AICAR and Mito Tempo, we propose that both agents may have synergistic therapeutic benefits in proteinuric podocytopathies.
CD36 is an innate multifunctional membrane scavenger receptor that plays an important mechanistic role in atherosclerosis and in inflammation (77, 96). The B scavenger receptor CD36 is constitutive in many cells including podocytes, and recognizes, binds, and internalizes oxLDL and FFA (49, 56, 77, 96). We showed in human macrophages that ROS upregulate CD36 (96) and foster increased uptake of OxLDL and foam cell formation. Mayrhofer et al. (49) showed in preparations of glomeruli isolated from normal rats that CD36 is constitutively expressed in podocytes; in the same studies, they demonstrated that in rats with PAN nephrosis podocyte CD36 was upregulated and promoted uptake of FFA and oxLDL resulting in podocyte injury and development of FSGS. Furthermore, a recent study by Souza et al. (78) demonstrated that the apolipoprotein A-I-mimetic peptide 5A, a CD36 antagonist, reduced glomerular and tubular injury as well as CKD progression independent of blood pressure in 5/6 nephrectomized mice subject to ANG II infusion (78). Similar renoprotection was observed in 5/6 nephrectomized CD36KO mice (78). In the aggregate, these in vivo studies support the notion that CD36 plays an important role in the pathogenesis of progressive CKD.
Here, we determined that Plg significantly upregulated CD36 expression (Fig. 4A). This was prevented by inhibition with EACA of Plg binding as well as by inhibition of O2− synthesis by either APOCY or AICAR (Fig. 4A). Importantly, in subsequent experiments we determined that podocytes take up OxLDL and that incubation with Plg had an additive effect that resulted in increased podocyte oxLDL uptake (Fig. 4B) accompanied by podocyte apoptosis (Fig. 6).
The off target effect of amiloride as an inhibitor of uPA was documented in early studies reported in the cancer literature (66, 89). In vivo experimental studies in stroke-prone spontaneously hypertensive rats, Dahl salt-sensitive, and in 5/6 nephrectomy rats showed that amiloride significantly reduces glomerular injury and proteinuria independent of blood pressure (26, 33, 73). In patients with type 2 diabetic nephropathy, amiloride lowered urine plasminogen excretion and activation, and albumin/creatinine ratio (55). In the current studies, we determined that amiloride inhibits Plg-induced O2− production and CD36 upregulation in human podocytes (Fig. 5). This might explain, at least in part, the mechanisms involved in the salutary effects of amiloride in clinical and experimental proteinuric renal disease. These studies also provide strong support to our hypotheses regarding the potential role of Plg-plasmin in proteinuric renal diseases.
Elegant studies of biopsies from patients with FSGS presented evidence of endothelial-mitochondrial oxidative stress associated with podocyte-endothelial cell cross talk mediated by ET1 synthesized by podocytes (14, 15, 35, 41). Here, we demonstrated that Plg-induced synthesis and release of ET1 were inhibited by pretreatment of podocytes with either EACA or the NADPH inhibitor APOCY (Fig. 7B) (39, 40). Importantly, we made the new observation that selective inhibition of mitochondrial ROS with Mito-Tempo was sufficient to completely prevent Plg-induced ET1 synthesis (Fig. 7B) (14).
Cross talk between mitochondria and NADPH oxidases is well-documented and likely represents a feed-forward vicious cycle of ROS production in which activation of NADPH oxidases increases production of mitochondrial ROS and vice versa (16, 17).
In podocytes, Nox4 is predominately localized in mitochondria (19, 31) and has been shown to produce hydrogen peroxide (H2O2). Our results with apocyanin that inhibits NOX2 and Mito Tempo would suggest (Fig. 8) that Plg activation of NOX2 generates O2−, which activates mitochondrial Nox4 that reciprocally activates NOX2 via H2O2 (16, 17).
Fig. 8.
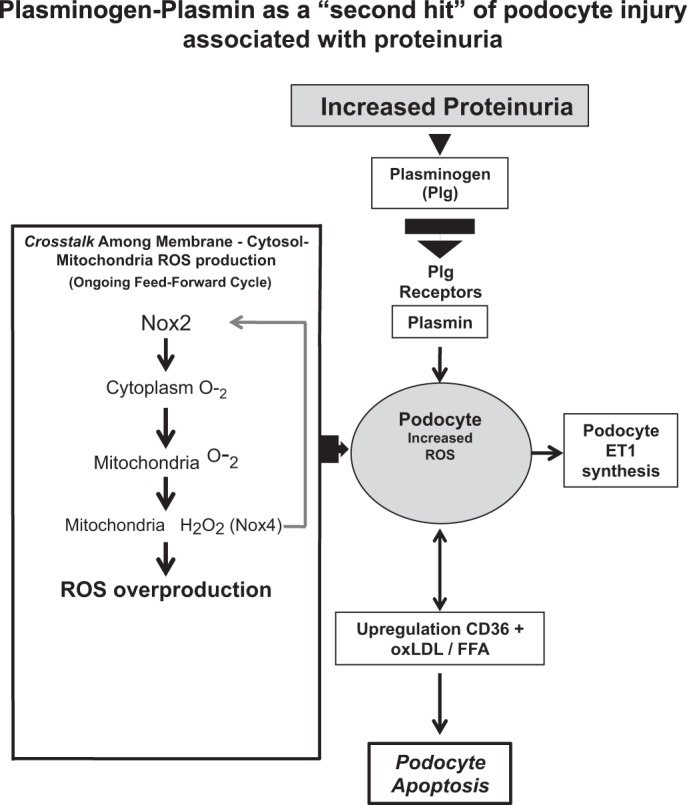
Working model of the participation of plasminogen-plasmin as a “second hit” type of podocyte injury associated with proteinuria.
Overall, our studies elucidated important new mechanisms that identified apoptosis, linked to the interaction of Plg-CD36-ox-LDL as a potential “second hit” in podocyte injury and death associated with proteinuria (Fig. 8) (20, 36, 47, 54, 68, 91, 93). Based on our studies, we conclude that 1) Plg activation on podocyte membranes can play a significant role in a “second hit” type of podocyte injury associated with chronic proteinuria independent of the initial etiology and 2) that oxidative stress generated by Plg via NADPH oxidases plays a central role in these processes.
At the present time, specific therapies for podocyte injury are not available. Our novel studies suggest that to slow CKD progression of proteinuric glomerular diseases would require multipronged strategies combining pharmacologically new molecules added to other agents such as biologicals that specifically target podocyte antigens, mitochondrial targeted antioxidants, activators of AMPKinase, ET1 receptor blockers/synthesis inhibitors (14, 15, 18, 19, 35, 70), certainly RAAS inhibitors/blockers, (8, 29, 63), and amiloride, cautiously monitoring serum potassium. Further studies will be required to further define the pathogenic role of circulating Plg and potential benefits of its removal or inhibition. In summary, we suggest that our studies provide a new biologic framework for the development of strategies for “comprehensive treatments” to arrest progression of proteinuric podocytopathies.
GRANTS
This work was supported by funds from the South Florida Veterans Affairs Foundation For Research and Education (SFVAFRE) and National Institutes of Health Grant R01DK103022 to K. N. Campbell.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
L.R. and K.N.C. conception and design of research; L.R., R.T., J.S.W., and J.C.H. analyzed data; L.R. interpreted results of experiments; L.R. drafted manuscript; L.R. and K.N.C. edited and revised manuscript; L.R. and K.N.C. approved final version of manuscript; R.T., J.S.W., and J.C.H. performed experiments; R.T., J.S.W., and J.C.H. prepared figures.
ACKNOWLEDGMENTS
Cd2ap−/− mice were received from Dr. Andrey Shaw (Washington University School of Medicine and Genentech). We thank Kim Jaimes for manuscript preparation. We also thank Drs. Alessia Fornoni and Sandra Merscher for providing the initial set of podocytes as well as initial technical advice to Dr. Raij's laboratory.
REFERENCES
- 1.Andersen RF, Buhl KB, Jensen BL, Svenningsen P, Friis UG, Jespersen B, Rittig S. Remission of nephrotic syndrome diminishes urinary plasmin content and abolishes activation of ENaC. Pediatr Nephrol 28: 1227–1234, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Barisoni L, Bruggeman LA, Mundel P, D'Agati VD, Klotman PE. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int 58: 173–181, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Barlow GH, Summaria L, Robbins KC. Molecular weight studies on human plasminogen and plasmin at the microgram level. J Biol Chem 244: 1138–1141, 1969. [PubMed] [Google Scholar]
- 4.Bruggeman LA, Dikman S, Meng C, Quaggin SE, Coffman TM, Klotman PE. Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J Clin Invest 100: 84–92, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buhl KB, Friis UG, Svenningsen P, Gulaveerasingam A, Ovesen P, Frederiksen-Moller B, Jespersen B, Bistrup C, Jensen BL. Urinary plasmin activates collecting duct ENaC current in preeclampsia. Hypertension 60: 1346–1351, 2012. [DOI] [PubMed] [Google Scholar]
- 6.Buhl KB, Oxlund CS, Friis UG, Svenningsen P, Bistrup C, Jacobsen IA, Jensen BL. Plasmin in urine from patients with type 2 diabetes and treatment-resistant hypertension activates ENaC in vitro. J Hypertens 32: 1672–1677, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Cammisotto PG, Bendayan M. Adiponectin stimulates phosphorylation of AMP-activated protein kinase alpha in renal glomeruli. J Mol Histol 39: 579–584, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Campbell KN, Raij L, Mundel P. Role of angiotensin II in the development of nephropathy and podocytopathy of diabetes. Curr Diabetes Rev 7: 3–7, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cellesi F, Li M, Rastaldi MP. Podocyte injury and repair mechanisms. Curr Opin Nephrol Hypertens 24: 239–244, 2015. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Meng XF, Zhang C. Role of NADPH oxidase-mediated reactive oxygen species in podocyte injury. Biomed Res Int 2013: 839761, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung JJ, Huber TB, Godel M, Jarad G, Hartleben B, Kwoh C, Keil A, Karpitskiy A, Hu J, Huh CJ, Cella M, Gross RW, Miner JH, Shaw AS. Albumin-associated free fatty acids induce macropinocytosis in podocytes. J Clin Invest 125: 2307–2316, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couser WG, Remuzzi G, Mendis S, Tonelli M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int 80: 1258–1270, 2011. [DOI] [PubMed] [Google Scholar]
- 13.D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med 365: 2398–2411, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Daehn I, Casalena G, Zhang T, Shi S, Fenninger F, Barasch N, Yu L, D'Agati V, Schlondorff D, Kriz W, Haraldsson B, Bottinger EP. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest 124: 1608–1621, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Zeeuw D, Coll B, Andress D, Brennan JJ, Tang H, Houser M, Correa-Rotter R, Kohan D, Lambers Heerspink HJ, Makino H, Perkovic V, Pritchett Y, Remuzzi G, Tobe SW, Toto R, Viberti G, Parving HH. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J Am Soc Nephrol 25: 1083–1093, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51: 1289–1301, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassegue B, Griendling KK, Harrison DG, Dikalova AE. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal 20: 281–294, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eid AA, Ford BM, Block K, Kasinath BS, Gorin Y, Ghosh-Choudhury G, Barnes JL, Abboud HE. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem 285: 37503–37512, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, Abboud HE. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes 58: 1201–1211, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukuda A, Wickman LT, Venkatareddy MP, Sato Y, Chowdhury MA, Wang SQ, Shedden KA, Dysko RC, Wiggins JE, Wiggins RC. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int 81: 40–55, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gharavi AG, Ahmad T, Wong RD, Hooshyar R, Vaughn J, Oller S, Frankel RZ, Bruggeman LA, D'Agati VD, Klotman PE, Lifton RP. Mapping a locus for susceptibility to HIV-1-associated nephropathy to mouse chromosome 3. Proc Natl Acad Sci USA 101: 2488–2493, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm–from a thin grey line to a complex signaling hub. Nature Rev Nephrol 9: 587–598, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Grainger DJ, Wakefield L, Bethell HW, Farndale RW, Metcalfe JC. Release and activation of platelet latent TGF-beta in blood clots during dissolution with plasmin. Nat Med 1: 932–937, 1995. [DOI] [PubMed] [Google Scholar]
- 24.Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol 74: 299–323, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grunkemeyer JA, Kwoh C, Huber TB, Shaw AS. CD2-associated protein (CD2AP) expression in podocytes rescues lethality of CD2AP deficiency. J Biol Chem 280: 29677–29681, 2005. [DOI] [PubMed] [Google Scholar]
- 26.He C, Zhang B, Xie S, Yang Y, Ma J, Shi W. Amiloride reduces proteinuria and inhibits podocyte uPAR in the 5/6 nephrectomy rats. Nan Fang Yi Ke Da Xue Xue Bao 34: 1654–1657, 2014. [PubMed] [Google Scholar]
- 27.Heerspink HJ, Kropelin TF, Hoekman J, de Zeeuw D, Reducing Albuminuria as Surrogate Endpoint C. Drug-induced reduction in albuminuria is associated with subsequent renoprotection: a meta-analysis. J Am Soc Nephrol 26: 2055–2064, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hruby Z, Wendycz D, Kopec W, Czerchawski L, Jozefowiak M, Rabczynski J. Effect of antiproteolytic drugs: epsilon-aminocaproic acid (EACA) and aprotinin on experimental anti-GBM nephritis. Nephrol Dial Transplant 11: 32–39, 1996. [PubMed] [Google Scholar]
- 29.Hsu TW, Liu JS, Hung SC, Kuo KL, Chang YK, Chen YC, Hsu CC, Tarng DC. Renoprotective effect of renin-angiotensin-aldosterone system blockade in patients with predialysis advanced chronic kidney disease, hypertension, and anemia. JAMA Intern Med 174: 347–354, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Jankun J, Skrzypczak-Jankun E. Molecular basis of specific inhibition of urokinase plasminogen activator by amiloride. Cancer Biochem Biophys 17: 109–123, 1999. [PubMed] [Google Scholar]
- 31.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25: 1237–1254, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kajiyama W, Kopp JB, Marinos NJ, Klotman PE, Dickie P. Glomerulosclerosis and viral gene expression in HIV-transgenic mice: role of nef. Kidney Int 58: 1148–1159, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, Shiraishi N, Adachi M, Zhang Z, Masilamani S, Tomita K. Aberrant ENaC activation in Dahl salt-sensitive rats. J Hypertens 27: 1679–1689, 2009. [DOI] [PubMed] [Google Scholar]
- 34.Khalil N, Corne S, Whitman C, Yacyshyn H. Plasmin regulates the activation of cell-associated latent TGF-beta 1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am J Respir Cell Mol Biol 15: 252–259, 1996. [DOI] [PubMed] [Google Scholar]
- 35.Kohan DE, Barton M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int 86: 896–904, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopp JB. Glomerular homeostasis requires a match between podocyte mass and metabolic load. J Am Soc Nephrol 23: 1273–1275, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, Eckhaus M, Bryant JL, Notkins AL, Klotman PE. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci USA 89: 1577–1581, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korgaonkar SN, Feng X, Ross MD, Lu TC, D'Agati V, Iyengar R, Klotman PE, He JC. HIV-1 upregulates VEGF in podocytes. J Am Soc Nephrol 19: 877–883, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30: 653–661, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenoir O, Milon M, Virsolvy A, Henique C, Schmitt A, Masse JM, Kotelevtsev Y, Yanagisawa M, Webb DJ, Richard S, Tharaux PL. Direct action of endothelin-1 on podocytes promotes diabetic glomerulosclerosis. J Am Soc Nephrol 25: 1050–1062, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lighvani S, Baik N, Diggs JE, Khaldoyanidi S, Parmer RJ, Miles LA. Regulation of macrophage migration by a novel plasminogen receptor Plg-R KT. Blood 118: 5622–5630, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol 106: 1659–1665, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mallipattu SK, He JC. A new mechanism for albuminuria-induced podocyte injury. J Am Soc Nephrol 24: 1709–1711, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mann JF, Yi QL, Gerstein HC. Albuminuria as a predictor of cardiovascular and renal outcomes in people with known atherosclerotic cardiovascular disease. Kidney Int Suppl 92: S59–S62, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Matsusaka T, Kobayashi K, Kon V, Pastan I, Fogo AB, Ichikawa I. Glomerular sclerosis is prevented during urinary tract obstruction due to podocyte protection. Am J Physiol Renal Physiol 300: F792–F800, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsusaka T, Sandgren E, Shintani A, Kon V, Pastan I, Fogo AB, Ichikawa I. Podocyte injury damages other podocytes. J Am Soc Nephrol 22: 1275–1285, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matthews H, Ranson M, Kelso MJ. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: an orally active anti-cancer drug waiting for its call-of-duty? Int J Cancer 129: 2051–2061, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Mayrhofer C, Krieger S, Huttary N, Chang MW, Grillari J, Allmaier G, Kerjaschki D. Alterations in fatty acid utilization and an impaired antioxidant defense mechanism are early events in podocyte injury: a proteomic analysis. Am J Pathol 174: 1191–1202, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merscher-Gomez S, Guzman J, Pedigo CE, Lehto M, Aguillon-Prada R, Mendez A, Lassenius MI, Forsblom C, Yoo T, Villarreal R, Maiguel D, Johnson K, Goldberg R, Nair V, Randolph A, Kretzler M, Nelson RG, Burke GW 3rd, Groop PH, Fornoni A, FinnDiane Study G. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes 62: 3817–3827, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miles LA, Lighvani S, Baik N, Andronicos NM, Chen EI, Parmer CM, Khaldoyanidi S, Diggs JE, Kiosses WB, Kamps MP, Yates JR 3rd, Parmer RJ. The plasminogen receptor, Plg-R(KT), and macrophage function. J Biomed Biotechnol 2012: 250464, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miles LA, Parmer RJ. Plasminogen receptors: the first quarter century. Semin Thromb Hemost 39: 329–337, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morigi M, Buelli S, Angioletti S, Zanchi C, Longaretti L, Zoja C, Galbusera M, Gastoldi S, Mundel P, Remuzzi G, Benigni A. In response to protein load podocytes reorganize cytoskeleton and modulate endothelin-1 gene: implication for permselective dysfunction of chronic nephropathies. Am J Pathol 166: 1309–1320, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mundel P, Reiser J. Proteinuria: an enzymatic disease of the podocyte? Kidney Int 77: 571–580, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oxlund CS, Buhl KB, Jacobsen IA, Hansen MR, Gram J, Henriksen JE, Schousboe K, Tarnow L, Jensen BL. Amiloride lowers blood pressure and attenuates urine plasminogen activation in patients with treatment-resistant hypertension. J Am Soc Hypertens 8: 872–881, 2014. [DOI] [PubMed] [Google Scholar]
- 56.Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest 119: 136–145, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peired A, Angelotti ML, Ronconi E, la Marca G, Mazzinghi B, Sisti A, Lombardi D, Giocaliere E, Della Bona M, Villanelli F, Parente E, Ballerini L, Sagrinati C, Wanner N, Huber TB, Liapis H, Lazzeri E, Lasagni L, Romagnani P. Proteinuria impairs podocyte regeneration by sequestering retinoic acid. J Am Soc Nephrol 24: 1756–1768, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peterson JC, Adler S, Burkart JM, Greene T, Hebert LA, Hunsicker LG, King AJ, Klahr S, Massry SG, Seifter JL. Blood pressure control, proteinuria, and the progression of renal disease. The modification of diet in renal disease study. Ann Intern Med 123: 754–762, 1995. [DOI] [PubMed] [Google Scholar]
- 59.Plow EF, Doeuvre L, Das R. So many plasminogen receptors: why? J Biomed Biotechnol 2012: 141806, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pollak MR, Quaggin SE, Hoenig MP, Dworkin LD. The glomerulus: the sphere of influence. Clin J Am Soc Nephrol 9: 1461–1469, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pollock DM. 2013 Dahl Lecture: American Heart Association council for high blood pressure research clarifying the physiology of endothelin. Hypertension 63: e110–e117, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Quigley JP, Gold LI, Schwimmer R, Sullivan LM. Limited cleavage of cellular fibronectin by plasminogen activator purified from transformed cells. Proc Natl Acad Sci USA 84: 2776–2780, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raij L. End-organ susceptibility as a determinant of renal disease in hypertension. Kidney Int 64: 1923–1932, 2003. [DOI] [PubMed] [Google Scholar]
- 64.Raij L, Runxia T, Acevedo B, Peev V, Merscher SM, Fornoni A, Zhou MS. Podocytes are a target of nicotine: link to proteinuria and progression of renal failure (Abstract). J Am Soc Nephrol 25, 2014. Poster FR-PO496, Page 483A. [Google Scholar]
- 65.Ratnam KK, Feng X, Chuang PY, Verma V, Lu TC, Wang J, Jin Y, Farias EF, Napoli JL, Chen N, Kaufman L, Takano T, D'Agati VD, Klotman PE, He JC. Role of the retinoic acid receptor-alpha in HIV-associated nephropathy. Kidney Int 79: 624–634, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ray P, Bhatti R, Gadarowski J, Bell N, Nasruddin S. Inhibitory effect of amiloride on the urokinase plasminogen activators in prostatic cancer. Tumour Biol 19: 60–64, 1998. [DOI] [PubMed] [Google Scholar]
- 67.Reiser J, Sever S. Podocyte biology and pathogenesis of kidney disease. Annu Rev Med 64: 357–366, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Remuzzi G, Benigni A, Remuzzi A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J Clin Invest 116: 288–296, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roth D, Piekarek M, Paulsson M, Christ H, Bloch W, Krieg T, Davidson JM, Eming SA. Plasmin modulates vascular endothelial growth factor-A-mediated angiogenesis during wound repair. Am J Pathol 168: 670–684, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 123: 2764–2772, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saleem MA, O'Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13: 630–638, 2002. [DOI] [PubMed] [Google Scholar]
- 72.Schiffer M, Mundel P, Shaw AS, Bottinger EP. A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem 279: 37004–37012, 2004. [DOI] [PubMed] [Google Scholar]
- 73.Sepehrdad R, Chander PN, Oruene A, Rosenfeld L, Levine S, Stier CT Jr. Amiloride reduces stroke and renalinjury in stroke-prone hypertensive rats. Am J Hypertens 16: 312–318, 2003. [DOI] [PubMed] [Google Scholar]
- 74.Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int 69: 2131–2147, 2006. [DOI] [PubMed] [Google Scholar]
- 75.Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int 72: 26–36, 2007. [DOI] [PubMed] [Google Scholar]
- 76.Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286: 312–315, 1999. [DOI] [PubMed] [Google Scholar]
- 77.Silverstein RL, Li W, Park YM, Rahaman SO. Mechanisms of cell signaling by the scavenger receptor CD36: implications in atherosclerosis and thrombosis. Trans Am Clin Climatol Assoc 121: 206–220, 2010. [PMC free article] [PubMed] [Google Scholar]
- 78.Souza AC, Bocharov AV, Baranova IN, Vishnyakova TG, Huang YG, Wilkins KJ, Hu X, Street JM, Alvarez-Prats A, Mullick AE, Patterson AP, Remaley AT, Eggerman TL, Yuen PS, Star RA. Antagonism of scavenger receptor CD36 by 5A peptide prevents chronic kidney disease progression in mice independent of blood pressure regulation. Kidney Int 89: 809–822, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stoycheff N, Pandya K, Okparavero A, Schiff A, Levey AS, Greene T, Stevens LA. Early change in proteinuria as a surrogate outcome in kidney disease progression: a systematic review of previous analyses and creation of a patient-level pooled dataset. Nephrol Dial Transplant 26: 848–857, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55: 225–233, 2006. [PubMed] [Google Scholar]
- 81.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C, Skott O. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol 20: 299–310, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Svenningsen P, Friis UG, Bistrup C, Buhl KB, Jensen BL, Skott O. Physiological regulation of epithelial sodium channel by proteolysis. Curr Opin Nephrol Hypertens 20: 529–533, 2011. [DOI] [PubMed] [Google Scholar]
- 83.Svenningsen P, Friis UG, Versland JB, Buhl KB, Moller Frederiksen B, Andersen H, Zachar RM, Bistrup C, Skott O, Jorgensen JS, Andersen RF, Jensen BL. Mechanisms of renal NaCl retention in proteinuric disease. Acta Physiol (Oxf) 207: 536–545, 2013. [DOI] [PubMed] [Google Scholar]
- 84.Svenningsen P, Skott O, Jensen BL. Proteinuric diseases with sodium retention: is plasmin the link? Clin Exp Pharmacol Physiol 39: 117–124, 2012. [DOI] [PubMed] [Google Scholar]
- 85.Syrovets T, Lunov O, Simmet T. Plasmin as a proinflammatory cell activator. J Leukoc Biol 92: 509–519, 2012. [DOI] [PubMed] [Google Scholar]
- 86.Towler DA. Mitochondrial ROS deficiency and diabetic complications: AMP[K]-lifying the adaptation to hyperglycemia. J Clin Invest 123: 4573–4576, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tzarfaty-Majar V, Lopez-Alemany R, Feinstein Y, Gombau L, Goldshmidt O, Soriano E, Munoz-Canoves P, Klar A. Plasmin-mediated release of the guidance molecule F-spondin from the extracellular matrix. J Biol Chem 276: 28233–28241, 2001. [DOI] [PubMed] [Google Scholar]
- 88.Vassalli JD, Belin D. Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett 214: 187–191, 1987. [DOI] [PubMed] [Google Scholar]
- 89.Wang Y, Dang J, Liang X, Doe WF. Amiloride modulates urokinase gene expression at both transcription and posttranscription levels in human colon cancer cells. Clin Exp Metastasis 13: 196–202, 1995. [DOI] [PubMed] [Google Scholar]
- 90.Weins A, Wong JS, Basgen JM, Gupta R, Daehn I, Casagrande L, Lessman D, Schwartzman M, Meliambro K, Patrakka J, Shaw A, Tryggvason K, He JC, Nicholas SB, Mundel P, Campbell KN. Dendrin ablation prolongs life span by delaying kidney failure. Am J Pathol 185: 2143–2157, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005. [DOI] [PubMed] [Google Scholar]
- 92.Wickman L, Afshinnia F, Wang SQ, Yang Y, Wang F, Chowdhury M, Graham D, Hawkins J, Nishizono R, Tanzer M, Wiggins J, Escobar GA, Rovin B, Song P, Gipson D, Kershaw D, Wiggins RC. Urine podocyte mRNAs, proteinuria, and progression in human glomerular diseases. J Am Soc Nephrol 24: 2081–2095, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71: 1205–1214, 2007. [DOI] [PubMed] [Google Scholar]
- 94.Woroniecki RP, Schiffer M, Shaw AS, Kaskel FJ, Bottinger EP. Glomerular expression of transforming growth factor-beta (TGF-beta) isoforms in mice lacking CD2-associated protein. Pediatr Nephrol 21: 333–338, 2006. [DOI] [PubMed] [Google Scholar]
- 95.Zachar RM, Skjodt K, Marcussen N, Walter S, Toft A, Nielsen MR, Jensen BL, Svenningsen P. The epithelial sodium channel γ-subunit is processed proteolytically in human kidney. J Am Soc Nephrol 26: 95–106, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou MS, Chadipiralla K, Mendez AJ, Jaimes EA, Silverstein RL, Webster K, Raij L. Nicotine potentiates proatherogenic effects of oxLDL by stimulating and upregulating macrophage CD36 signaling. Am J Physiol Heart Circ Physiol 305: H563–H574, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]