

Abstract
Malaria, a major global health challenge worldwide, is accompanied by a severe anemia secondary to hemolysis and increased erythrophagocytosis. Iron is an essential functional component of erythrocyte hemoglobin and its availability is controlled by the liver-derived hormone hepcidin. We examined the regulation of hepcidin during malarial infection in mice using the rodent parasite Plasmodium berghei K173. Mice infected with Plasmodium berghei K173 develop a severe anemia and die after 18 to 22 days without cerebral malaria. During the early phase of blood-stage infection (days 1 to 5), a strong inflammatory signature was associated with an increased production of hepcidin. Between days 7 and 18, while infection progressed, red blood cell count, hemoglobin and hematocrit dramatically decreased. In the late phase of malarial infection, hepcidin production was reduced concomitantly to an increase in the messenger RNA expression of the hepcidin suppressor erythroferrone in the bone marrow and the spleen. Compared with wild-type mice, Erfe−/− mice failed to adequately suppress hepcidin expression after infection with Plasmodium berghei K173. Importantly, the sustained production of hepcidin allowed by erythroferrone ablation was associated with decreased parasitemia, providing further evidence that transient iron restriction could be beneficial in the treatment of malaria.
Introduction
Malaria remains a major health burden in intertropical countries. According to the annual World Malaria Report by the World Health Organization, an estimated 214 million people were clinically affected by malaria in 2015 and approximately 438 000 of these patients died due to severe complications.1 Infection initiates when Plasmodium sporozoites are injected together with anti-coagulant saliva during a blood meal of an infected Anopheles mosquito. Sporozoites migrate to the liver in search of a favorable niche in the hepatocyte where they replicate extensively. Thousands of merozoites are then produced and released into the circulation to invade red blood cells (RBCs),2 in which parasites further replicate during the symptomatic blood stage of the asexual developmental cycle.3
Nearly all forms of life, including plants and pathogens, utilize iron for fundamental processes such as DNA synthesis, oxygen transport and generation of ATP. Plasmodium species are no exception, as the replication of the parasite in the liver and in erythrocytes is highly dependent on iron.4 Indeed, iron chelators can inhibit Plasmodium growth in vitro,5 in murine models of malaria infection,6,7 and in malaria-infected monkeys.8 In humans, iron deficiency appears to protect against severe malaria,9–11 while iron supplementation increases the risks of infection and disease.12–15 Iron deficiency also provides protection against infection with Plasmodium berghei in mice.16,17
The host systemic iron availability is controlled by the iron regulatory hormone hepcidin,18–20 which could therefore influence the susceptibility to malaria. Iron is absorbed from the diet by intestinal enterocytes and recycled from senescent or damaged RBCs by macrophages.21 The export of iron across the basolateral membrane of enterocytes and from iron-recycling macrophages is ensured by the sole known iron exporter, ferroportin. Hepcidin binds to ferroportin and causes its internalization and degradation.19,20 The loss of ferroportin from the cell surface prevents iron efflux from intestinal enterocytes and from macrophages, leading to iron retention in these cells and subsequent hypoferremia.
Multiple studies have shown that hepcidin is upregulated during malarial infection in humans22–24 and in murine models.25,26 The underlying mechanisms may involve parasite-induced inflammatory pathways, but they are still unclear. Under the influence of high hepcidin concentration, as iron is redistributed to macrophages, the flow of iron into plasma is decreased, which routes iron away from the parasite and thereby prevents its multiplication. As a consequence, combined with RBC destruction by the parasite, this may worsen the host anemia because of restricted iron availability for erythropoiesis.
Although the majority of studies on hepcidin and malaria have demonstrated an increased production of hepcidin during malarial infection, three recent studies have shown that in certain circumstances, hepcidin suppression may also occur. One study reported that among all children presenting with malaria, those with severe anemia had the lowest hepcidin levels.27 Another study demonstrated that children with uncomplicated malaria had higher hepcidin levels than those who could be classified as either presenting with severe anemia or cerebral malaria.28 Finally, a group of children with severe malarial anemia exhibited very low serum hepcidin levels.29 Taken together, these studies clearly indicate that during severe malarial anemia, the signaling pathway that suppresses hepcidin can override the activation pathway associated with parasite-induced inflammation.
The mechanisms of hepcidin suppression in severe malaria syndromes are not well defined. In the two human studies that monitored erythropoietin (EPO) levels and serum hepcidin in malaria infection, EPO and hepcidin were negatively correlated.27,29 One animal study has also demonstrated a significant negative correlation between serum EPO and hepcidin in mice infected with Plasmodium berghei.24 We recently showed that high levels of EPO cause hepcidin suppression indirectly by inducing the production of the erythroid regulator erythroferrone (ERFE) by erythroid progenitors in the bone marrow and the spleen.30 ERFE is secreted in the circulation and acts on the liver to suppress hepcidin expression and increase dietary iron absorption and iron release from macrophages.30 To assess the role of ERFE in hepcidin suppression observed in severe malaria, we first checked whether Erfe expression was upregulated in a murine model of malaria induced by Plasmodium berghei K173 (PbK), a strain that causes severe malarial anemia. Mice injected with PbK-parasitized RBCs develop high parasitemia and severe anemia and die without cerebral symptoms 18–22 days post-infection.31 We then infected Erfe-deficient mice and examined the impact of Erfe deletion on hepcidin expression, parasitemia and RBC indices. By highlighting the contribution of ERFE in hepcidin regulation in malaria, this study may suggest new therapeutic strategies to combat malaria.
Methods
Mice and infection
Since iron parameters and hepcidin levels differ significantly between male and female mice,32,33 only male mice were used in this study. 9 week-old C57BL/6 wild-type (WT) mice (5 to 10 per time-point) were infected intraperitoneally with 106 PbK-infected RBCs, and given free access to tap water and standard laboratory mouse chow diet (250 mg iron/kg; SAFE, Augy, France) during the 18 days post-infection. All animals were kept in pathogen-free animal facilities in Toulouse, and analyzed at days 0, 1, 2, 3, 4, 5, 7, 9, 11, 13, 16 and 18 post-infection. PbK was serially passaged in vivo in C57BL/6 mice. Infected blood was harvested at day 5–7 post-infection and stored as frozen stabilates in Alsever’s solution containing 10% glycerol.
Erfe−/− mice on a C57BL/6 background were maintained at UCLA on a standard chow (200 ppm iron; Labdiet, MO, USA). Control WT C57BL/6J males were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). 8 week-old WT and Erfe−/− mice were infected intraperitoneally with 106 PbK-infected RBCs (6 mice per genotype). 50 ml of blood was sampled on days 0, 7, 10 and 13 to determine complete blood count and hepcidin levels, and mice were sacrificed on day 16. To monitor the effect of repetitive blood collection, control WT and Erfe−/− mice were injected intraperitoneally with normal saline and manipulated similarly to the infected mice (4 mice per genotype).
Ethic statement
Experimental protocols were approved by the Midi-Pyrénées Animal Ethics Committee, France (n° MP/07/59/10/11). Experiments including Erfe−/− mice were conducted in accordance with guidelines by the National Research Council and were approved by the University of California, Los Angeles, USA (protocol #2011-114-13C).
Measurement of hematologic parameters and parasitemia
Complete blood counts were obtained with a CELL-DYN Emerald Hematology System (Abbott Diagnostics, France) or with a HemaVet blood analyzer (Drew Scientific) for the experiment performed at UCLA. Serum iron concentration was determined using the IRON Direct Method kit (BIOLABO, Maizy, France). Parasitemia (i.e., the percentage of infected RBCs) was assessed at each time point by microscopic counts of thin blood smears stained with Giemsa solution (Diff-Quik, Medion Diagnostics, Switzerland).
Quantitation of messenger RNA (mRNA) levels
Total RNA from mouse liver, spleen, kidney or bone marrow was extracted using TRIzol (Invitrogen). Complementary DNA (cDNA) was synthesized using M-MLV RT (Promega). Quantitative polymerase chain reaction (qPCR) reactions were prepared with LightCycler® 480 DNA SYBR Green I Master reaction mix (Roche Diagnostics) and run in duplicate on a LightCycler® 480 System (Roche Diagnostics). For experiments in which WT and Erfe−/− mice were compared, cDNA was synthesized using iScript (Bio-rad). Quantitative PCR reactions were prepared with SsoAdvanced Universal Sybr Green supermix (Bio-Rad) and run in duplicate on a CFX Connect Instrument (Bio-Rad). Primer sequences are indicated in the Online Supplementary Table S1. Tnf, Ifn-γ, Hamp, Erfe, Gypa, Gdf15 and Twsg1 mRNA transcript abundance was normalized to the reference gene Rpl4. Epo mRNA expression was normalized to the reference gene Hprt. Results are expressed as −ΔCt ± standard error of the mean (i.e., the cycle threshold differences between reference and target genes within each group of mice).
Serum hepcidin enzyme-linked immunosorbent assays
Serum hepcidin and serum IL-6 levels were quantified using the Intrinsic LifeSciences (La Jolla, CA, USA) Hepcidin-Murine Compete™ competitive ELISA and the R&D Systems Mouse IL-6 Quantikine ELISA Kit, respectively, according to the manufacturer’s instructions.
Statistical analysis
The statistical significance of differences between groups was evaluated by one-way ANOVA followed by Dunnett’s multiple comparisons test using GraphPad Prism version 6.00 (GraphPad Software, La Jolla, CA, USA). The Student’s t-test was used to compare WT and Erfe−/− mice. A P value <0.05 in a two-tailed test was considered as statistically significant.
Results
Plasmodium berghei K173 infection causes inflammation and leads to severe anemia
We first assessed parasitemia and the hematological parameters 1 to 18 days after infection of mice with PbK. The parasites became detectable in the bloodstream 3 days after injection and multiplied exponentially to nearly 70% of parasitized RBCs 18 days after infection (Figure 1A). White blood cells count increased concomitantly with the parasitemia from day 7 to day 18 (Figure 1B). We next examined the expression of inflammatory cytokines during PbK infection. Tnf mRNA expression was induced 8-fold at day 1 and more than 30-fold at day 18 in the liver of PbK-infected mice compared to control mice (Figure 1C). Similarly, interferon-γ (Ifn-γ) expression was slightly increased at day 2 and maximally increased by 30- to 60-fold between day 5 and day 18 after infection with PbK compared to controls (Figure 1D). On the contrary, RBC, hemoglobin concentration, and hematocrit dramatically decreased between day 7 and day 18 after infection (Figure 2A–C), whereas mean corpuscular volume (MCV) increased between days 13 and 18 (Figure 2D). Collectively, these results indicate that infection with PbK leads to a severe malarial anemia with subsequent death after 18 days.
Figure 1.
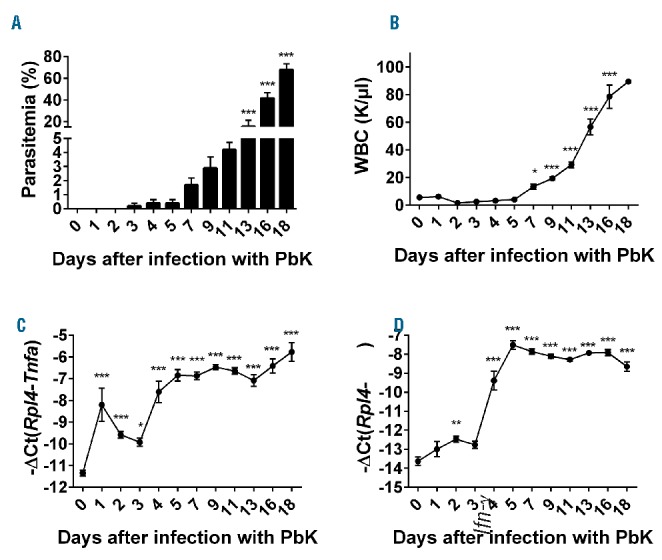
Plasmodium berghei K173 infection is accompanied by a strong inflammatory response. Parasitemia (A) and WBC count (B) progressively increased from day 7 to day 18. Hepatic Tnf mRNA expression (C) increased from day 1 to day 18 in the liver of mice infected with PbK. Similarly, Interferon-γ (Ifn-γ) expression (D) significantly increased by day 2 and reached a plateau at day 5 after infection with PbK. Data shown are mean ± sem and were compared for each time point to values for control mice at t=0 (n=5 to 10 mice per group except at day 18 where only 3 mice had survived) by one-way ANOVA. ***P<0.001, **P<0.01, *P<0.05. WBC: white blood cell; PbK: Plasmodium berghei K173.
Figure 2.
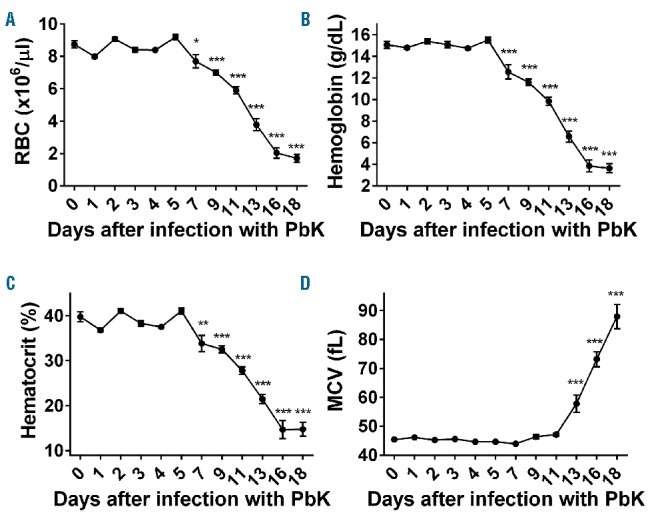
Mice infected with Plasmodium berghei K173 develop a severe anemia. RBC count (A), hemoglobin concentration (B) and hematocrit (C) decreased dramatically from day 7 to day 18. Conversely, MCV was increased 13 to 18 days after infection with PbK. Data shown are mean ± sem and were compared for each time point to values for control mice at t=0 (n=5 to 10 mice per group except at day 18 where only 3 mice had survived) by one-way ANOVA. ***P<0.001, **P<0.01, *P<0.05. RBC: red blood cell; PbK: Plasmodium berghei K173; MCV: mean corpuscular volume.
Hepcidin expression in Plasmodium berghei K173-infected mice is first induced by parasite-induced inflammation and, in a second phase, suppressed while anemia worsens
Increased production of the liver-produced hormone hepcidin has proven beneficial to prevent superinfections.25 However, hepcidin expression is stimulated by inflammation during infections, but also repressed by increased erythropoietic activity during anemia. We therefore measured circulating levels of hepcidin and liver hepcidin mRNA expression in PbK-infected mice. Consistent with the inflammatory status, serum hepcidin concentration and liver hepcidin mRNA expression were increased by approximately 2-fold 3 to 5 days after infection with PbK compared to control mice (Figure 3A,B). Since hepcidin expression is regulated by iron and inflammation, we next assessed the iron parameters and the production of IL-6 and activin B, two activators of hepcidin expression during inflammation. Serum iron concentration was only marginally increased 3 to 5 days after infection while liver Bmp6 mRNA expression decreased 4 to 5 days after infection (Figure 4A,B), suggesting that changes in circulating iron and liver iron content do not contribute to hepcidin upregulation during malarial infection. Changes in serum iron concentration are concomitant with the increase in parasitemia 3 days after infection. Erythrocytic forms of Plasmodium degrade approximately 60% to 80% of the total RBC hemoglobin content, which generates free heme and reactive oxygen species.34 We therefore surmise that the slight increase in serum iron observed at days 3 to 5 may be attributable to the release of free iron after hemoglobin degradation, or to positive interference of contaminating heme with the colorimetric procedure during the measurement.35 Compared to mice injected with lipopolysaccharide (LPS), serum IL-6 concentration and liver activin B (Inhbb) mRNA expression (Figure 4C,D) were only modestly increased after infection with PbK, and hepatocytes STAT3 phosphorylation was not detectable (Online Supplementary Figure S1). These results suggest that these molecules are not major contributors to the stimulation of hepcidin production during malaria. While anemia progressed, serum hepcidin concentration decreased below baseline levels 13 to 18 days after infection (Figure 3A). Hepcidin mRNA expression was also repressed by approximately 8- to 32-fold between 13 and 18 days after infection with PbK compared to control mice (Figure 3B).
Figure 3.
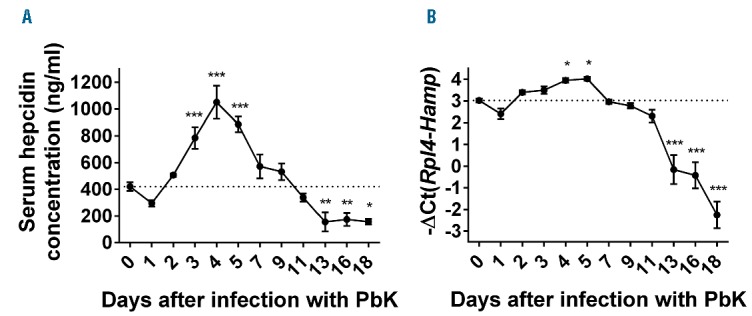
Biphasic regulation of hepcidin production during Plasmodium berghei K173 infection. Consistent with the inflammatory stimulation, serum hepcidin concentration (A) and hepcidin (Hamp) mRNA expression in the liver (B) were increased between day 3 and day 5 but progressively returned to normal between day 5 and 11 and were significantly reduced between day 13 and day 18. Data shown are mean ± sem and were compared for each time point to values for control mice at t=0 (n=5 to 10 mice per group except at day 18 where only 3 mice had survived) by one-way ANOVA. ***P<0.001, **P<0.01, *P<0.05. PbK: Plasmodium berghei K173.
Figure 4.
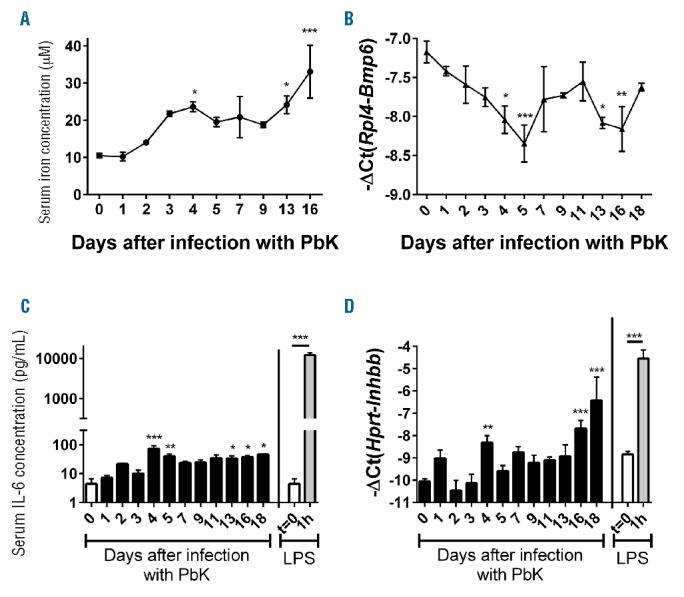
Serum iron and IL-6 concentration and liver Bmp6 and Inhbb mRNA expression during Plasmodium berghei K173 infection. Serum iron concentration (A) was slightly increased at days 4, 13 and 16 whereas Bmp6 mRNA expression (B) was decreased 5 days after infection with PbK. Serum IL-6 concentration (C) and liver activin B (Inhbb) mRNA expression (D) were marginally increased 4–5 days after infection with PbK compared to mice challenged with LPS.40 Data shown are mean ± sem and were compared for each time point to values for control mice at t=0 (n=5 to 10 mice per group except at day 18 where only 3 mice had survived) by one-way ANOVA. ***P<0.001, **P<0.01, *P<0.05. PbK: Plasmodium berghei K173; LPS: lipopolysaccharide; lL-6: interleukin 6.
Hepcidin suppression coincides with increased erythropoietic activity and erythroferrone production
In parallel, Epo mRNA expression in the kidney was upregulated between days 7 and 18 in mice infected with PbK compared to control mice (Figure 5A). Similarly, spleen index (spleen weight relative to body weight) increased from day 4 to day 18 after infection with PbK (Figure 5B) and glycophorin A (Gypa) mRNA expression, a marker of erythropoiesis, was elevated by approximately 5- to 64-fold in the spleen between days 7 and 18 (Figure 5C). Collectively, these results show a progressive increase in erythropoietic activity that is proportional to the degree of anemia in PbK-infected mice and accompanied by extramedullary erythropoiesis. Interestingly, Gypa mRNA expression was reduced 2 to 5 days after infection in the bone marrow and 3 days after infection in the spleen, presumably because of an inhibition of erythropoiesis by inflammatory cytokines36 during Plasmodium berghei infection. To decipher the mechanism involved in hepcidin suppression, we studied the expression of the known negative regulators of hepcidin: Erfe,30 Gdf15,37 and Twsg1.38 We found that Erfe mRNA expression mirrored circulating levels of EPO, and was induced in the bone marrow (~128-fold) and in the spleen (~2000-fold) of PbK-infected mice compared to control mice at day 18 (Figure 5D). The increase in Erfe mRNA expression in the spleen is mostly attributable to the expansion of the splenic erythroid compartment, as shown by Gypa mRNA expression (Figure 5C). On the contrary, Gdf15 and Twsg1 mRNA expression did not correlate with hepcidin suppression during malarial infection. Indeed, Gdf15 mRNA expression was only slightly increased between day 1 and day 9 in the bone marrow and the spleen of infected mice, and returned to normal before hepcidin suppression (Online Supplementary Figure S2). Similarly, Twsg1 mRNA expression showed only minor fluctuations in the spleen, and was significantly decreased in the bone marrow 11 and 13 days after infection compared to control mice (Online Supplementary Figure S2).
Figure 5.
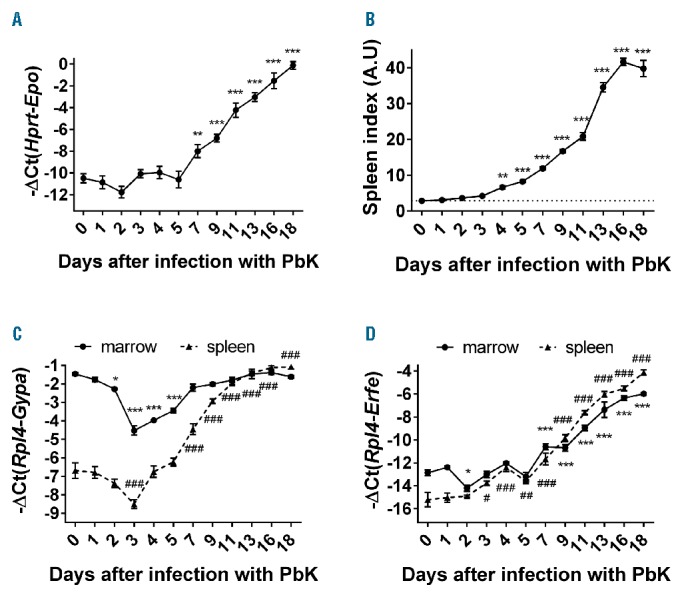
Erfe mRNA expression is highly increased during infection with Plasmodium berghei K173. Epo mRNA expression was highly induced in the kidney of infected mice (A). Increased EPO production was accompanied by a massive increase in spleen index (B), Gypa (C) and Erfe mRNA expression (D) in the bone marrow and the spleen. Gypa mRNA expression (C) decreased between day 0 and day 3 in the bone marrow and the spleen and increased in the spleen between day 15 and 18. Data shown are mean ± sem and were compared for each time point to values for control mice at t=0 (n=5 to 10 mice per group except at day 18 where only 3 mice had survived) by one-way ANOVA. (A, B) ***P<0.001, **P<0.01 and (C, D) ***P<0.001, *P<.05 for the marrow and ###P<0.001, ##P<0.01, #P<0.05 for the spleen. Spleen index = spleen weight in mg/body weight in g. PbK: Plasmodium berghei K173; A.U: arbitrary units.
Erythroferrone contributes to hepcidin suppression during malarial anemia, and its absence in Erfe−/− mice has a significant impact on parasitemia
To determine whether ERFE contributes to hepcidin suppression during malarial infection, we compared WT and Erfe−/− mice after infection with PbK. Six mice per genotype were infected with PbK, and blood was repetitively sampled on days 0, 7, 10 and 13 before the mice were analyzed on day 16. The repetitive blood collection did not influence serum hepcidin concentration (Figure 6A). We found that serum hepcidin concentration was similarly elevated in WT and Erfe-deficient mice 7 and 10 days after infection compared to baseline at day 0 (Figure 6B). However, whereas serum hepcidin concentration was reduced below baseline in WT mice 13 and 16 days after infection, hepcidin suppression was totally blunted in Erfe-deficient mice. In the same line, hepatic hepcidin mRNA expression was 6-fold higher in Erfe−/− mice compared to WT mice 16 days after infection with PbK (Figure 6C). As a consequence of higher circulating levels of hepcidin, Erfe−/− mice had higher spleen iron content compared to WT mice 16 days after infection (Figure 6D), which is consistent with hepcidin-induced degradation of the iron exporter ferroportin at the cell surface of macrophages. These results clearly demonstrate that ERFE contributes to hepcidin suppression and the mobilization of iron from the stores during malarial infection. Interestingly, determination of parasitemia revealed lower parasite count in Erfe-deficient mice compared to WT mice 13 days after infection (Figure 7A), suggesting that iron retention in the reticuloendothelial system temporarily impeded the multiplication of the parasite. Despite higher hemoglobin concentration and RBC in Erfe−/− compared to WT mice at day 10, the drop in hemoglobin (Figure 7B) and RBC (Online Supplementary Figure S3) was more prominent in Erfe−/− mice than in WT mice between day 10 and day 13 after infection with PbK (Figure 7B). Indeed, Erfe−/− mice had significantly lower hemoglobin at day 13 compared to WT mice. Similarly, MCV and mean corpuscular hemoglobin (MCH) were also lower in Erfe-deficient mice compared to WT mice at day 13 (Figure 7C,D), indicating a mild iron restriction in Erfe−/− mice. This shows that, by preventing the complete suppression of hepcidin, the absence of ERFE leads to transient iron restriction during Plasmodium berghei infection and has a significant impact on parasitemia.
Figure 6.
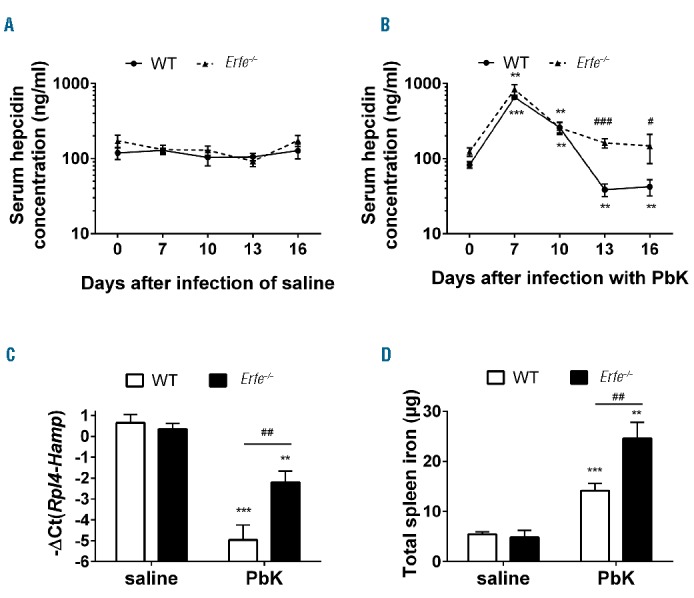
Ablation of Erfe leads to impaired regulation of hepcidin and iron retention in the stores during infection with Plasmodium berghei K173. Repetitive blood collection did not influence serum hepcidin concentration in saline controls (A). Serum hepcidin levels (B) were significantly decreased below baseline at days 13 and 16 after infection in WT mice (solid line, dotted symbols) but not in Erfe-deficient mice (dashed line, triangle symbols). At day 16 after infection with PbK, Hamp mRNA expression (C) was suppressed in WT but not in Erfe−/− mice. As a result, Erfe−/− mice had higher spleen iron content than WT mice (D). (A, B) The same mice were monitored between day 0 and 16 (n=6 mice per genotype for PbK-treated mice and 4 mice per genotype for saline controls). Data shown are mean ± sem and were compared for each mice at each time point to values at t=0 (***P<0.001, **P<0.01) and between WT and Erfe−/− mice (###P<0.001, ##P<0.01, #P<0.05) by two-tailed Student’s t-test (n=6 mice per group). (C-D) Data shown are mean ± sem and were compared for each genotype between mice injected with saline and PbK (***P<0.001, **P<0.01) and between WT and Erfe−/− mice (##P<0.01, #P<0.05) at day 16 by two-tailed Student’s t-test. WT: wild-type; PbK: Plasmodium berghei K173.
Figure 7.
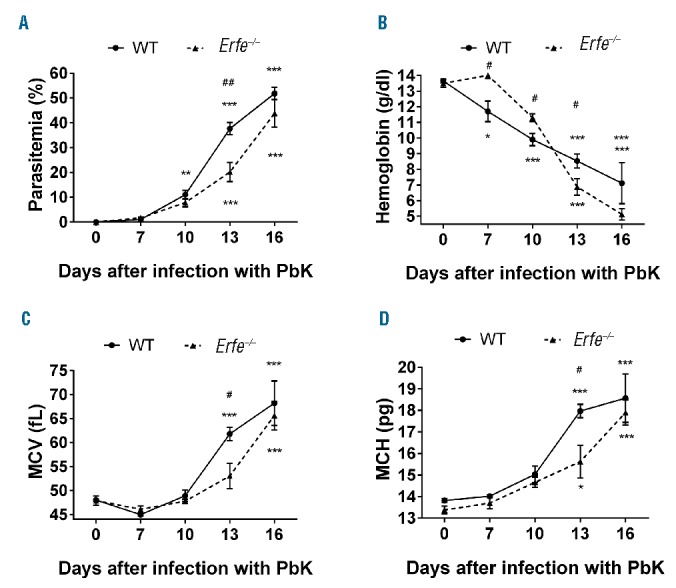
Parasitemia, hemoglobin, MCV and MCH in WT and Erfe−/− mice during infection with Plasmodium berghei K173. WT and Erfe−/− mice exhibited similar parasitemia at days 7 and 10 post-infection, but Erfe−/− had lower parasite count at day 13 (A). Hemoglobin (B) dramatically decreased during infection, whereas MCV (C) and MCH (D) increased between day 10 and 16. Compared to WT mice, the drop in hemoglobin between day 10 and 13 was more prominent in Erfe−/− mice, despite lower parasitemia. MCV and MCH were lower in Erfe−/− mice than in WT mice at day 13, suggesting iron restriction. The same mice were monitored between day 0 and 16 (n=6 mice per genotype for PbK-treated mice and 4 mice per genotype for saline controls). Data shown are mean ± sem and were compared for each mice at each time point to values at t=0 (***<0.001, **P<0.01, *P<0.05) by one-way ANOVA and between WT and Erfe−/− mice (##P<0.01, #P<0.05) by two-tailed Student’s t-test. WT: wild-type; PbK: Plasmodium berghei K173; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin.
Discussion
Malaria is currently one of the most geographically widespread and deadly diseases resulting in around half a million deaths every year.1 Iron, and its regulatory hormone hepcidin, play a crucial role in determining the multiplicity of malaria infections within the host. In accordance with previous studies on other murine models of malaria infection,25,26 we observed that the injection of PbK-infected RBCs into C57BL/6 mice led to a rapid upregulation of hepcidin production. The circulating levels of hepcidin more than doubled 3 to 5 days after infection, but the mechanisms involved in this upregulation remain unclear. Although IL-6 has been shown to be correlated with hepcidin in some studies,27–29 in another study serum IL-6 and urinary hepcidin were not significantly associated.23 Furthermore, in one ex vivo study, human peripheral blood mononuclear cells co-incubated with Plasmodium-infected RBCs exhibited a significant upregulation of hepcidin mRNA without a concomitant IL-6 mRNA increase.39 Finally, a study investigating the mechanisms by which blood-stage malaria infection can prevent the establishment of a liver-stage infection, which is thought to be modulated by hepcidin, found that liver-stage inhibition was preserved in mice treated with anti-IL-6 antibodies.25 Thus, the role of IL-6 in hepcidin induction in malaria remains controversial. Notably, in comparison to mice injected with LPS,40 we did not observe any physiologically relevant increase in serum IL-6 concentration 4 to 5 days after infection with PbK, and phosphorylated STAT3 was not detected in hepatocytes (Figure 4 and Online Supplementary Figure S1). Activin B is a ligand of the TGF-β superfamily that is upregulated by inflammatory stimuli such as LPS, and was shown to increase hepcidin expression through SMAD1/5/8 signaling in hepatoma cell lines.40 Similarly to IL-6, we detected only a mild induction of activin B mRNA expression compared to mice challenged with LPS (Figure 4). It is therefore unlikely that IL-6 and activin B stimulate hepcidin expression during Plasmodium berghei infection. Dissecting the pathways leading to early hepcidin upregulation in response to parasite exposure represents a major goal for future studies.
Although the mechanism stimulating hepcidin in malaria is unclear, increased hepcidin production appears to have several advantages for the host. First, as seen in this study, sustained hepcidin production leads to iron relocalization in macrophages, which is reflected by the increased splenic iron content in Erfe-deficient mice compared to WT mice. Although hepcidin upregulation also blocks iron absorption from the diet, which together with iron retention in macrophages probably contributes to the malarial anemia, it routes iron away from pathogens that could potentially exploit circulating iron, limiting Plasmodium growth and proliferation. Interestingly, the increased hepcidin caused by blood-stage infections was shown to significantly reduce the development of liver-stage Plasmodium infection by limiting the access of proliferating parasites to iron in hepatocytes.25 Clearance of an ongoing blood-stage infection with chloroquine returned hepcidin mRNA to normal levels and allowed the development of a secondary liver infection. Thus, the redistribution of iron caused by hepcidin is a key factor that can determine the outcome of liver Plasmodium infection, and may protect children living in endemic areas of malaria from superinfections.41
Iron sequestered in macrophages is not available for an efficient bone marrow response to increased erythrophagocytosis of both parasitized and non-parasitized RBCs.42 As a consequence, starting at day 7, mice infected with PbK present with a progressive decrease in the number of RBCs, hemoglobin, and hematocrit. A few days later, massive erythrocyte lysis due to elevated parasitemia and the negative effects of inflammatory cytokines, including TNF-a and IFN-γ, on reticulocyte production, leads to severe anemia in this experimental model. Stimulation of erythropoietic activity translates into a dramatic increase of EPO expression and extramedullary erythropoiesis. These manifestations are reminiscent of severe anemia observed in some children living in endemic areas of malaria,27,29 and is accompanied by a strong repression of hepcidin expression, both at the mRNA and the protein level. There is, therefore, a turning point post-infection where the signaling pathway that suppresses hepcidin overrides the activation pathway associated with blood-stage parasitemia.
Consistent with previous studies,27,29 we observed that the suppression of hepcidin starting at day 7 was negatively correlated with the increase in EPO. More interestingly, we show herein that this suppression is also negatively correlated with the expression of the recently described erythroid factor ERFE in the bone marrow and in the spleen. Given that ERFE suppresses hepcidin during increased erythropoietic activity, we used Erfe-deficient mice to demonstrate that ERFE was indeed involved in the suppression of hepcidin in mice infected with PbK. We showed that, in contrast to WT mice, Erfe-deficient mice failed to repress their hepcidin below the level of non-infected mice. As a consequence, they retain more iron in the cells of the reticuloendothelial system than WT mice, which translates into higher splenic iron content and lower RBC indices. Most interestingly, parasitemia is significantly lower at day 13 post-infection in Erfe-deficient mice compared with WT mice. These data indicate the potential benefit of maintaining even a moderate hepcidin production during the course of severe malaria.
We also observed a decrease in hepcidin production in Erfe-deficient mice between days 7 and 10 after infection, which may be attributable to the resolution of the inflammatory pathway stimulating hepcidin, or to potential additional regulators. Similar results were observed in mice treated with heat-killed Brucella abortus particles.43 However, hepcidin levels only returned toward baseline and we did not observe any suppression of hepcidin in Erfe−/− mice, indicating that ERFE represses hepcidin during malarial anemia. Further investigation will be necessary to identify the mechanism leading to hepcidin upregulation during malarial infection.
Surprisingly, Erfe−/− mice had higher RBC and hemoglobin levels compared to WT mice 7 to 10 days after infection, despite comparable parasitemia and hepcidin levels. We speculate that ERFE deficiency is somewhat protective against the ability of the parasite to invade or to replicate in RBCs during the early stage of infection.
Given the hepcidin suppressive effect of ERFE,30,43,44 and the clear advantage of Erfe-deficient mice during malaria infection, ERFE inhibitors would be expected to maintain hepcidin levels high enough to impede Plasmodium growth and proliferation and promise to be useful adjuvants in the treatment of malaria. However, the inhibition of ERFE during malarial infection may also worsen the anemia.30,43 Thus, after clearance of the parasite from the bloodstream, ERFE or related agonists could, in a second step, accelerate the recovery from malarial anemia by increasing intestinal iron absorption and mobilization of iron from the stores.
Importantly, our study focused on the blood-stage infection as the liver-stage was completely bypassed by the direct injection of infected RBCs into mice. However, the malaria parasite also requires iron during the exoerythrocytic phase of replication in the liver. Indeed, hepcidin peptide injection or hepcidin overexpression by transgene or viral vector can reduce parasite survival in the liver.25 It remains to be determined whether ERFE contributes to the asymptomatic liver-stage and the release of merozoites into the bloodstream.
Supplementary Material
Acknowledgments
The authors thank Elizabeta Nemeth (Los Angeles, CA, USA) for helpful discussions, Florence Capilla (Experimental Histopathology Platform, Toulouse Purpan, France), Antoine Berry (Toulouse, France) for the P. berghei K173 strain, and the members of the Inserm US006 facility (Toulouse, France) and Victoria Gabayan (Los Angeles, CA, USA) for their technical assistance and help in mouse breeding.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/1/60
Funding
This research was supported by the French National Research Agency (ANR-13-BSV3-0015-01), the Fondation pour la Recherche Médicale (FRM DEQ2000326528) to MPR, the NIH grant R01 DK 065029 to TG, the ASH scholar award and ANR-16-ACHN-0002-01 to LK, the PIA Parafrap Consortium (ANR-11-LABX0024) to NB and the Fondation pour l’Aide à la Recherche sur la Sclérose en Plaque (ARSEP) to MW and NB.
References
- 1.WHO. World Malaria Report 2015; 2015. [Google Scholar]
- 2.Haldar K, Murphy SC, Milner DA, Taylor TE. Malaria: mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu Rev Pathol. 2007;2: 217–249. [DOI] [PubMed] [Google Scholar]
- 3.Deroost K, Pham TT, Opdenakker G, Van den Steen PE. The immunological balance between host and parasite in malaria. FEMS Microbiol Rev. 2016;40(2):208–257. [DOI] [PubMed] [Google Scholar]
- 4.Spottiswoode N, Duffy PE, Drakesmith H. Iron, anemia and hepcidin in malaria. Front Pharmacol. 2014;5:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raventos-Suarez C, Pollack S, Nagel RL. Plasmodium falciparum: inhibition of in vitro growth by desferrioxamine. Am J Trop Med Hyg. 1982;31(5):919–922. [DOI] [PubMed] [Google Scholar]
- 6.Fritsch G, Treumer J, Spira DT, Jung A. Plasmodium vinckei: suppression of mouse infections with desferrioxamine B. Exp Parasitol. 1985;60(2):171–174. [DOI] [PubMed] [Google Scholar]
- 7.Ferrer P, Tripathi AK, Clark MA, Hand CC, Rienhoff HY, Jr, Sullivan DJ., Jr Antimalarial iron chelator, FBS0701, shows asexual and gametocyte Plasmodium falciparum activity and single oral dose cure in a murine malaria model. PLoS One. 2012; 7(5):e37171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pollack S, Rossan RN, Davidson DE, Escajadillo A. Desferrioxamine suppresses Plasmodium falciparum in Aotus monkeys. Proc Soc Exp Biol Med. 1987;184(2):162–164. [DOI] [PubMed] [Google Scholar]
- 9.Gwamaka M, Kurtis JD, Sorensen BE, et al. Iron deficiency protects against severe Plasmodium falciparum malaria and death in young children. Clin Infect Dis. 2012; 54(8):1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kabyemela ER, Fried M, Kurtis JD, Mutabingwa TK, Duffy PE. Decreased susceptibility to Plasmodium falciparum infection in pregnant women with iron deficiency. J Infect Dis. 2008;198(2):163–166. [DOI] [PubMed] [Google Scholar]
- 11.Nyakeriga AM, Troye-Blomberg M, Dorfman JR, et al. Iron deficiency and malaria among children living on the coast of Kenya. J Infect Dis. 2004;190(3):439–447. [DOI] [PubMed] [Google Scholar]
- 12.Sazawal S, Black RE, Ramsan M, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet. 2006; 367(9505):133–143. [DOI] [PubMed] [Google Scholar]
- 13.Smith AW, Hendrickse RG, Harrison C, Hayes RJ, Greenwood BM. The effects on malaria of treatment of iron-deficiency anaemia with oral iron in Gambian children. Ann Trop Paediatr. 1989;9(1):17–23. [DOI] [PubMed] [Google Scholar]
- 14.Oppenheimer SJ, Gibson FD, Macfarlane SB, et al. Iron supplementation increases prevalence and effects of malaria: report on clinical studies in Papua New Guinea. Trans R Soc Trop Med Hyg. 1986; 80(4):603–612. [DOI] [PubMed] [Google Scholar]
- 15.Clark MA, Goheen MM, Fulford A, et al. Host iron status and iron supplementation mediate susceptibility to erythrocytic stage Plasmodium falciparum. Nat Commun. 2014;5(4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koka S, Foller M, Lamprecht G, et al. Iron deficiency influences the course of malaria in Plasmodium berghei infected mice. Biochem Biophys Res Commun. 2007; 357(3):608–614. [DOI] [PubMed] [Google Scholar]
- 17.Matsuzaki-Moriya C, Tu L, Ishida H, et al. A critical role for phagocytosis in resistance to malaria in iron-deficient mice. Eur J Immunol. 2011;41(5):1365–1375. [DOI] [PubMed] [Google Scholar]
- 18.Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117(17):4425–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. [DOI] [PubMed] [Google Scholar]
- 20.Qiao B, Sugianto P, Fung E, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012;15(6):918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganz T, Nemeth E. Iron metabolism: interactions with normal and disordered erythropoiesis. Cold Spring Harb Perspect Med. 2012;2(5):a011668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howard CT, McKakpo US, Quakyi IA, et al. Relationship of hepcidin with parasitemia and anemia among patients with uncomplicated Plasmodium falciparum malaria in Ghana. Am J Trop Med Hyg. 2007;77(4): 623–626. [PubMed] [Google Scholar]
- 23.de Mast Q, Nadjim B, Reyburn H, Kemna EH, Amos B, Laarakkers CM, et al. Assesment of urinary concentrations of hepcidin provides novel insight into disturbances in iron homeostasis during malarial infection. J. Infect. Dis. 2009;199(2):253–262. [DOI] [PubMed] [Google Scholar]
- 24.de Mast Q, Syafruddin D, Keijmel S, et al. Increased serum hepcidin and alterations in blood iron parameters associated with asymptomatic P. falciparum and P. vivax malaria. Haematologica. 2010;95(7):1068–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Portugal S, Carret C, Recker M, et al. Host-mediated regulation of superinfection in malaria. Nat Med. 2011;17(6):732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang HZ, He YX, Yang CJ, Zhou W, Zou CG. Hepcidin is regulated during blood-stage malaria and plays a protective role in malaria infection. J Immunol. 2011; 187(12):6410–6416. [DOI] [PubMed] [Google Scholar]
- 27.Casals-Pascual C, Huang H, Lakhal-Littleton S, et al. Hepcidin demonstrates a biphasic association with anemia in acute Plasmodium falciparum malaria. Haematologica. 2012;97(11):1695–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burte F, Brown BJ, Orimadegun AE, et al. Circulatory hepcidin is associated with the anti-inflammatory response but not with iron or anemic status in childhood malaria. Blood. 2013;121(15):3016–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jonker FA, Calis JC, Phiri K, et al. Low hepcidin levels in severely anemic malawian children with high incidence of infectious diseases and bone marrow iron deficiency. PLoS One. 2013;8(12):e78964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014; 46(7):678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitchell AJ, Hansen AM, Hee L, et al. Early cytokine production is associated with protection from murine cerebral malaria. Infect Immun. 2005;73(9):5645–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Courselaud B, Troadec MB, Fruchon S, et al. Strain and gender modulate hepatic hepcidin 1 and 2 mRNA expression in mice. Blood Cells Mol Dis. 2004;32(2):283–289. [DOI] [PubMed] [Google Scholar]
- 33.Latour C, Kautz L, Besson-Fournier C, et al. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology. 2014;59(2): 683–694. [DOI] [PubMed] [Google Scholar]
- 34.Francis SE, Sullivan DJ, Jr, Goldberg DE. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol. 1997;51:97–123. [DOI] [PubMed] [Google Scholar]
- 35.Young DS. Effect of drugs on Clinical laboratory tests, 4th Ed. 1995;3-361 to 363-364. [Google Scholar]
- 36.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005; 352(10):1011–1023. [DOI] [PubMed] [Google Scholar]
- 37.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13(9):1096–1101. [DOI] [PubMed] [Google Scholar]
- 38.Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009; 114(1):181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armitage AE, Pinches R, Eddowes LA, Newbold CI, Drakesmith H. Plasmodium falciparum infected erythrocytes induce hepcidin (HAMP) mRNA synthesis by peripheral blood mononuclear cells. Br J Haematol. 2009;147(5):769–771. [DOI] [PubMed] [Google Scholar]
- 40.Besson-Fournier C, Latour C, Kautz L, et al. Induction of activin B by inflammatory stimuliupregulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood. 2012; 120(2): 431–439 [DOI] [PubMed] [Google Scholar]
- 41.Portugal S, Drakesmith H, Mota MM. Superinfection in malaria: Plasmodium shows its iron will. EMBO Rep. 2011; 12(12):1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamikanra AA, Brown D, Potocnik A, Casals-Pascual C, Langhorne J, Roberts DJ. Malarial anemia: of mice and men. Blood. 2007;110(1):18–28. [DOI] [PubMed] [Google Scholar]
- 43.Kautz L, Jung G, Nemeth E, Ganz T. Erythroferrone contributes to recovery from anemia of inflammation. Blood. 2014;124(16):2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kautz L, Jung G, Du X, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood. 2015;126(17):2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.