

Abstract
In most patients with primary myelofibrosis, one of three mutually exclusive somatic mutations is detected. In approximately 60% of patients, the Janus kinase 2 gene is mutated, in 20%, the calreticulin gene is mutated, and in 5%, the myeloproliferative leukemia virus gene is mutated. Although patients with mutated calreticulin or myeloproliferative leukemia genes have a favorable outcome, and those with none of these mutations have an unfavorable outcome, prognostication based on mutation status is challenging due to the heterogeneous survival of patients with mutated Janus kinase 2. To develop a prognostic model based on mutation status, we screened primary myelofibrosis patients seen at the MD Anderson Cancer Center, Houston, USA, between 2000 and 2013 for the presence of Janus kinase 2, calreticulin, and myeloproliferative leukemia mutations. Of 344 primary myelofibrosis patients, Janus kinase 2V617F was detected in 226 (66%), calreticulin mutation in 43 (12%), and myeloproliferative leukemia mutation in 16 (5%); 59 patients (17%) were triple-negatives. A 50% cut-off dichotomized Janus kinase 2-mutated patients into those with high Janus kinase 2V617F allele burden and favorable survival and those with low Janus kinase 2V617F allele burden and unfavorable survival. Patients with a favorable mutation status (high Janus kinase 2V617F allele burden/myeloproliferative leukemia/calreticulin mutation) and aged 65 years or under had a median survival of 126 months. Patients with one risk factor (low Janus kinase 2V617F allele burden/triple-negative or age >65 years) had an intermediate survival duration, and patients aged over 65 years with an adverse mutation status (low Janus kinase 2V617F allele burden or triple-negative) had a median survival of only 35 months. Our simple and easily applied age- and mutation status-based scoring system accurately predicted the survival of patients with primary myelofibrosis.
Introduction
Primary myelofibrosis (PMF) is a myeloproliferative neoplasm characterized by bone marrow fibrosis and extramedullary hematopoiesis, resulting in variable degrees of splenomegaly, leukocytosis, anemia, thrombocytopenia, and impaired quality of life.1 The median survival of patients with PMF is five years from diagnosis,2,3 but the clinical course is variable. Some patients succumb to the disease within one year, whereas others survive for more than ten years.2–4
Several prognostic scoring systems have been developed for PMF that are based on clinical characteristics and blood counts.2,5 The international prognostic scoring system (IPSS) stratifies patients into 4 risk groups (low, intermediate-1, intermediate-2, and high) based on age (>65 years), the presence of constitutional symptoms, hemoglobin less than 10 g/dL, white blood cell (WBC) count of more than 25×109/L, and circulating blast cells of 1% or more at time of diagnosis.5 Based on the IPSS, a dynamic IPSS (DIPSS) was developed, which accounts for acquisition of risk factors over time.6 A refinement of the DIPPS that incorporates adverse karyotype, transfusion dependency, and thrombocytopenia has been suggested by the Mayo Clinic.7
In most patients with PMF, one of three mutually exclusive hematopoietic cell somatic mutations is commonly identified.8–10 In approximately 60% of PMF patients, an activating substitution mutation at position 617 of the pseudo kinase domain of Janus kinase 2 (JAK2V617F) is detected.11–13 In 20%–25% of patients, frameshift mutations caused either by deletions or insertions in the last exon of the calreticulin (CALR) gene are detected. CALR encodes a Ca++ binding protein that is primarily localized to the endoplasmic reticulum (ER). When CALR is mutated, the ER C-terminal ER retention signal (KDEL) is lost and, as a result, the protein is no longer localized to the ER.8,9 In 5% of patients, an activating mutation in the myeloproliferative leukemia virus (MPL) (thrombopoietin receptor) gene is found.14
Patients with a mutated JAK2 present a more aggressive disease than patients with mutated CALR. However, the overall survival (OS) of patients with high JAK2V617F allele burden is better than that of patients with a low JAK2V617F allele burden.15,16
In this study, we developed an easy-to-use scoring system that integrates age and mutation status, and accurately predicts the survival of patients with newly diagnosed PMF.
Methods
Included in our study were patients with PMF who were referred to MD Anderson Cancer Center, Houston, USA, between June 2000 and July 2013; the diagnosis was established in accordance with World Health Organization (WHO) criteria.17 Demographic and clinical information at the time of presentation was obtained from patients’ medical records by using a retrospective chart review protocol that was approved by the MD Anderson Institutional Review Board. The IPSS5 and DIPSS scores were assigned to each patient, as previously described.6
After obtaining patients’ informed consent, we analyzed residual blood and/or bone marrow cells which had been obtained from the patients for diagnostic purposes and stored, in accordance with a research protocol that was approved by the MD Anderson Institutional Review Board. Before freezing, all samples were fractionated with use of the Ficoll Hypaque 1077 (Sigma-Aldrich, St. Louis, MO, USA). Low-density cells were recovered from the Ficoll interface and collected by centrifugation. Genomic DNA was extracted by using Puregene DNA purification reagents (Gentra, Minneapolis, MN, USA).
To detect JAK2V617F mutation and measure the JAK2V617F allele burden, we extracted 50 ng of total genomic DNA and performed quantitative allele-specific suppressive polymerase chain reaction (PCR) with the use of the 7900HT FAST platform sequence detection system (Applied Biosystems, Foster City, CA, USA), as previously described.18
Detection of frame shift mutations in exon 9 of CALR was performed as previously described8 by using the following primer pairs: Forward: 5′ -FAM-GGCAAGGCCCTGAGGTGT; reverse: GGCCTCAGTCCAGCCCTG. This reaction captures the two frameshift type 1 (52 bp deletion) and type 2 (5-bp deletions) mutations. To detect mutations in exon 10 of MPL, we amplified genomic DNA by using the following primer set: MPL13474-F; GTGACCGCTCTGCATCTAGTG, MPL13726-R; GTGGGCGTGTTAGAG TGT. The resulting 250-bp PCR product was purified with use of a Qiagen PCR purification kit (Qiagen, Valencia, CA, USA) and was subjected to Sanger sequencing by using the above primers on a 3300 Genetic Analyzer (Applied Biosystems). The DNA sequencing fragments were analyzed with the use of Lasergene 11 (DNASTAR, Madison, WI, USA).
Statistical analysis
Patients’ characteristics were summarized by using frequencies (percentages) for categorical variables and median and range for continuous variables. To compare patients on the basis of categorical variables, we used the χ2 test. To compare medians, we used the Mann-Whitney test. To determine the optimal survival cut-off point that dichotomized patients according to their JAK2V617F allele burden, we used the X-Tile statistical software (http://www.tissuearray.org/rimmlab). The cut-off point used corresponds to the maximum χ2 value of the Mantel-Cox test for OS between groups above and below the cut-off point threshold.19 The probability of OS was estimated by the Kaplan-Meier method. The log-rank test was used to compare patients’ survival. Univariable and multivariable Cox proportional hazard regression models were fit to assess the association between mutation status and OS. The Wald test was used to assess the significance of covariates in Cox models. To compare competing models, we used the log-likelihood ratio. The replicability of the prognostic scoring system was tested by bootstrap resampling. One thousand samples, the same size as the original series, were built through random extraction with reposition. To predict the risk of transformation based on mutation status, we applied a logistic regression model and used the Exp (β) to estimate the odds ratio and the 95% confidence interval (CI) around it. Statistical analyses were performed with SPSS software (version 21, SPSS Inc., Chicago, IL, USA) and Graph Pad Prism (version 6.0, San Diego, CA, USA).
Results
JAK2V617F, CALR, and MPL mutation frequency
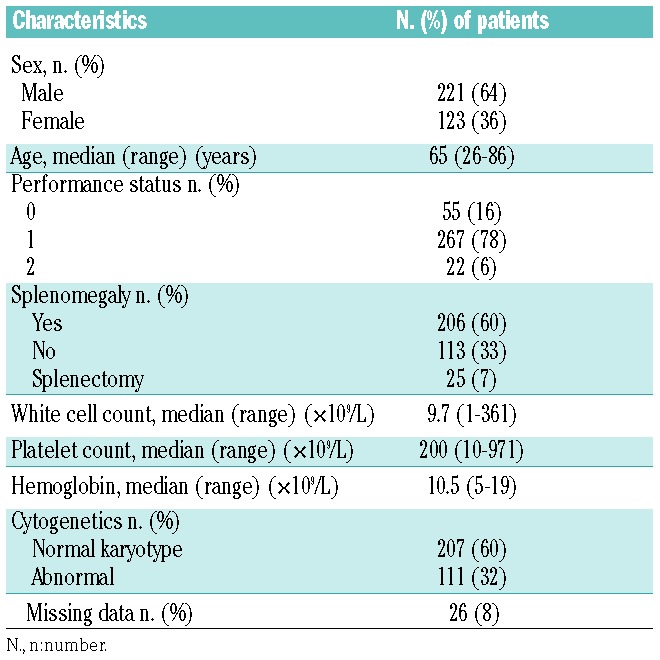
A total of 344 PMF patients, aged 26 to 86 years (median: 65 years; 64% males) were included. Patients’ characteristics are shown in Table 1. Of the 344 patients, 226 (66%) had a JAK2V617F mutation, 43 (12%) had a CALR mutation (40 patients had 50–52-bp deletions and 3 patients had 5–10 bp insertion), and 16 (5%) had an MPL mutation. Fifty-nine patients (17%) had none of these mutations and were designated ‘triple-negative’.
Table 1.
Baseline characteristics of the 344 patients with primary myelofibrosis.
JAK2V617 allele burden and survival
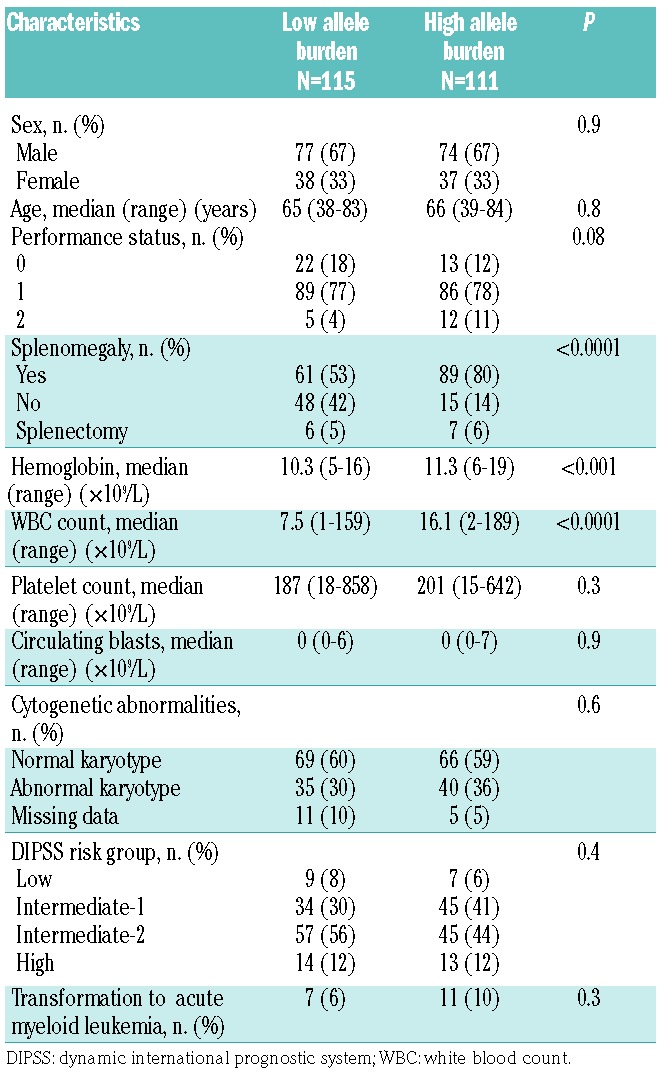
When used as a continuous variable, JAK2V617F allele burden (ranging from 0% to 98%) had only marginal power to predict OS [Hazard Ratio (HR): 0.997, 95% Confidence Interval (CI): 0.990–1.00]. However, a 50% cut off of the JAK2V617F allele burden dichotomized patients into two groups with different survival outcomes. Patients with a JAK2V617F allele burden of 50% or over had a median OS of 80 months (95%CI: 51–109 months), whereas patients with a JAK2V617F allele burden of less than 50% had a median OS of 50 months (95%CI: 40–60 months) (P=0.01) (Figure 1A). Remarkably, patients with a high (≥50%) JAK2V617F allele burden had a larger spleen, a higher hemoglobin level, and a higher WBC count than did patients with a low (<50%) JAK2V617F allele burden (Table 2).
Figure 1.
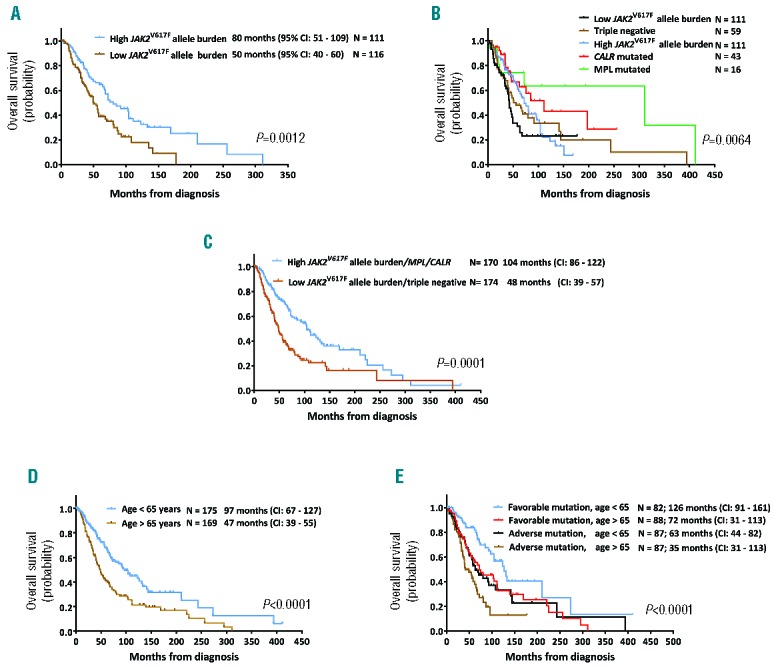
Survival of 344 patients with primary myelofibrosis (PMF). (A) Overall survival of PMF patients with high (≥50%) and low (<50%) JAK2V617F allele burden. (B) Overall survival (OS) of PMF patients according to mutation status. (C) Overall survival of PMF patients with favorable and adverse mutation status. Patients with either high JAK2V617F allele burden, mutated CALR, or mutated MPL had a better OS than patients with low JAK2V617F allele burden or triple-negative mutation status. (D) Overall survival according to patients’ age (over or under 65 years of age). (E) OS of patients with PMF based on risk stratification according to age and mutation status.
Table 2.
Clinical characteristics of 226 primary myelofibrosis patients with a high (≥50%) and low (<50%) JAK2V617F allele burden.
Mutation status and survival outcome
The longest OS was observed in patients with mutated MPL (median survival 221 months, 95%CI: 40–401 months), followed by patients with mutated CALR (median survival 131 months, 95%CI: 100–160 months), high JAK2V617F burden (median survival 80 months, 95%CI: 51–109 months), triple-negatives (median survival 56 months, 95%CI: 35–77 months), and low JAK2V617F burden (median survival 50 months, 95%CI: 38–62 months) (Figure 1B). The incorporation of high- and low-JAK2V617F mutation status divides PMF patients into two groups: patients with either high JAK2V617F allele burden, mutated CALR, or mutated MPL had a median survival of 104 months (95%CI: 86–122 months), whereas patients with low JAK2V617F allele burden or triple-negative mutation status had a median survival of 48 months (95%CI: 39–57 months) (Figure 1C).
Development of an age- and mutation status-based prognostic model for survival
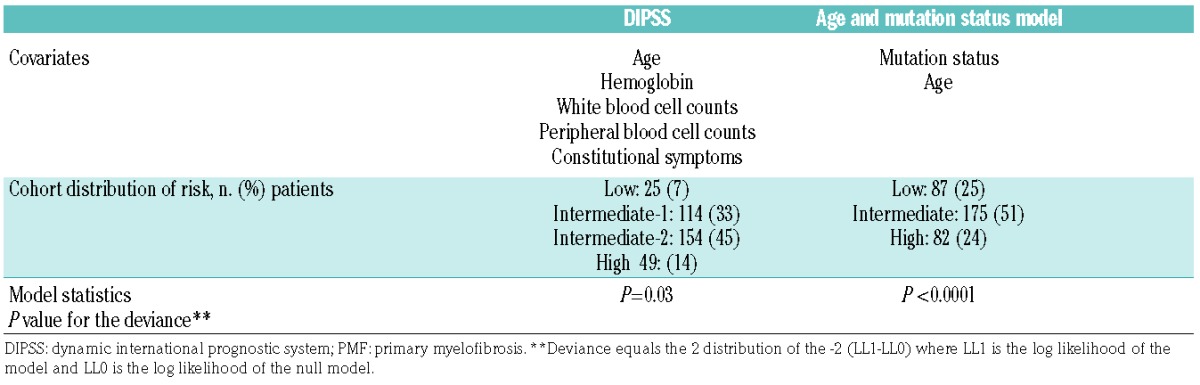
When mutation status and DIPSS variables were included as covariates in a multivariable analysis, only unfavorable mutation status, older age, and a high percentage of peripheral blood blasts predicted a shorter survival (Table 3). Age dichotomized patients into two groups with different survival outcomes. Patients aged 65 years or under had a median OS of 97 months (95%CI: 67–127 months; n=175), whereas patients older than 65 had a median OS of 47 months (95% CI: 39–55 months; n=169) (P<0.0001) (Figure 1D). However, a model that included 2 parameter estimates (age and mutation status) was superior in fitting the data, as had the largest log-likelihood ratio, and divided the cohort into 4 groups of almost equal size. Bootstrapping resampling procedure confirmed the stability of the model. Patients with a favorable mutation status (high JAK2V617F allele burden, CALR, or MPL mutations) and aged 65 years or under had a median OS of 126 months (95%CI: 91–161 months; n=82). Patients with one risk factor, either age over 65 years (n=88) or adverse mutation status (n=87) had an intermediate OS of 72 months, whereas patients with two risk factors, e.g. age over 65 years and an adverse mutation status (low JAK2V617F allele burden or triple-negative; n=87) had a median OS of 35 months (95%CI: 31–113 months) (Table 4 and Figure 1E). In comparison, the DIPSS uses 5 risk factors to classify patients into one of 4 groups. In our cohort, most patients (n=228, 78%) were classified as either intermediate-1 or -2 (Table 5) and had similar survival outcome.
Table 3.
Cox regression model of mortality including age and mutation status and DIPSS variables as covariates.
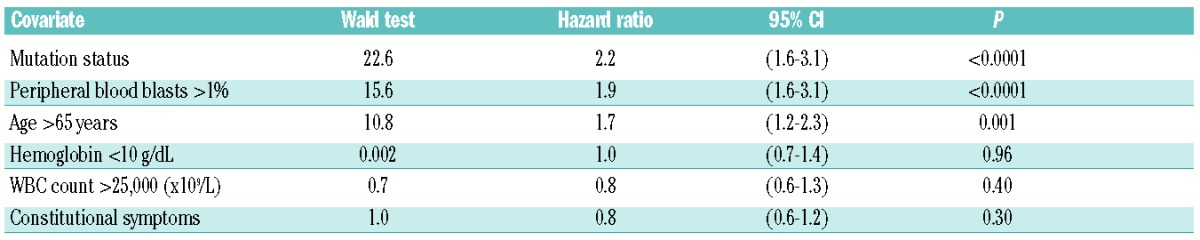
Table 4.
Cox regression model to assess the age and mutation status model of mortality in patients with PMF.
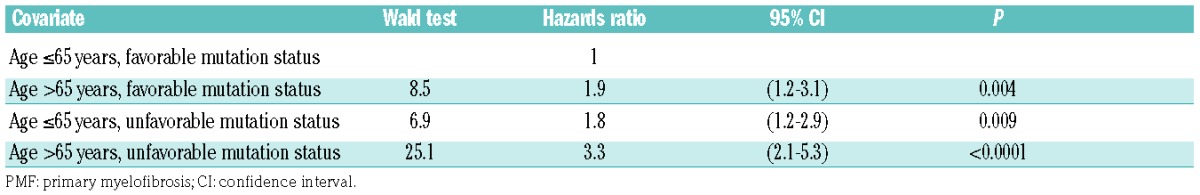
Table 5.
Comparison of the DIPSS and genetic-based Cox proportions models for prediction of survival in patients with PMF.
Mutation status and the risk of transformation
Thirty-two patients (9%) transformed to acute myeloid leukemia. Median time to transformation was 33 months (range 1–271 months). None of the mutations predicted transformation to acute myeloid leukemia.
Discussion
The clinical outcome of patients with PMF is partially dictated by mutually exclusive driver mutations in the genes JAK2, CALR, or MPL.20 Here we show that patients’ mutation status can be integrated into a prognostic model.
Although the survival of patients with a JAK2V617F mutation is heterogeneous, dividing these patients into subgroups of high and low JAK2V617F allele burden enabled the development of a prognostic model that integrates genetic information. Patients with low JAK2V617F allele burden or a triple-negative mutation status had a shorter OS than patients in the other groups. Because we found that patients’ age is of prognostic significance, similar to other investigators’ findings, we integrated the patients’ mutation status and age, and analyzed 4 equally-sized cohorts. Patients with no risk factors (aged 65 years or under with a favorable mutation status) had the longest survival (median OS 126 months); patients with a single risk factor (over 65 years of age or an adverse mutation status) had an intermediate survival duration (median OS 72 months); and patients with two risk factors (over 65 years of age and an adverse mutation such as JAK2V617F allele burden or triple-negative mutation status) had the worst prognosis (median OS 35 months). Although the percentage of circulating blasts emerged as a prognostic indicator, it did not contribute to the overall variance and was not included in the final model. Given that our hospital is a tertiary care cancer center, our PMF patient cohort has a high proportion of high-risk patients; this PMF patient population is particularly suitable for this analysis. Notably, only 7% of our patients were low-risk patients according to the DIPSS, compared with 44% in the IPSS. Hence, while our prognostic scale divides our patient cohort into 4 groups of equal size, it is possible that lower-risk patients were under-represented. Nevertheless, because prognostic scales for patients with PMF are routinely used to identify high-risk patients who are suitable for allogeneic transplantation, this scale might prove to be very useful.
The identification of mutually exclusive mutations in most patients with PMF8–10 suggests that at least three distinct pathways play a role in disease acquisition.
In our cohort, 13% of PMF patients had triple-negative mutation status. It is possible that these patients carry mutations in yet unidentified genes or that triple-negative status may represent a late event in clonal evolution that gives proliferation and/or survival advantage to a dominant neoplastic clone that is no longer dependent on the initial ‘driver’ mutagenic event.
The survival of patients with a high (≥50%) JAK2V617F allele burden was significantly better than that of patients with a low JAK2V617F allele burden. It has been reported that PMF patients with a homozygous JAK2 mutation have distinct clinical features such as splenomegaly.21 Here we show that the clinical features on presentation of patients with a high JAK2V617F allele burden were reminiscent of patients with polycythemia vera (PV); they had discernible splenomegaly, leukocytosis, and higher hemoglobin levels compared with the group with low JAK2V617F allele burden. Therefore, it is possible that this group consists, at least in part, of patients with post-PV myelofibrosis that evolved from an undiagnosed PV. Interestingly, in a study of 68 patients with post-PV myelofibrosis, all patients carried a high JAK2V617F allele burden, and 78% had an allele burden of more than 50%.6,22 CALR mutations have been divided into two types. In PMF the type 1/type 1-like mutations are the most common ones.23–25 In our patient cohort only 3 patients had type 2 mutations. Therefore, our study was not powered to determine the prognostic value of CALR mutation subtypes.
Since the initial publication of the IPSS prognostic score,5 several refinements have been proposed, most of which attempt to incorporate recurrent gene mutations that have been identified in patients with PMF.26 Some mutations, such as those in DNTM327 or TET2,28 have not been shown to correlate with survival outcome. Conversely, mutations in ASXL1, SRSF2, and EZH2 predicted short survival in a large cohort of patients, and only the ASXL1 mutation remained statistically significant when added to the IPSS prognostic score.29 A report by Tefferi et al.10 points to the CALR−/ASXL1+ profile as the most detrimental mutation profile in PMF.
The applicability of our prognostic scale depends on screening for mutations in CALR and MPL and quantification of the JAK2V617F allele burden. Recently, the WHO added CALR and MPL mutations to the PMF diagnostic criteria30 and, as a result, most diagnostic laboratories perform these tests. Moreover, most diagnostic laboratories assess the presence of JAK2 mutations by using quantitative PCR. Although the JAK2V617F allele burden is readily available, it is not routinely reported, although various assays yield similar quantification results.31
Here we present a prognostic model that is based on a relatively large cohort. The internal validation of this model was confirmed by bootstrap resampling. By using only 2 variables, we developed a simple, easily applied model with excellent discrimination power for survival outcome of patients with newly diagnosed PMF.
Although this prognostic model needs to be validated with a large, independent patient population, it has a larger log-likelihood ratio than that of the IPSS or DIPSS, suggesting that it has superior, clinically applicable value.
Supplementary Material
Acknowledgments
We thank Tamara Locke for editing our manuscript.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/1/79
Funding
This study was supported by the NIH/NCI under award number P30 CA016672.
References
- 1.Barosi G. Myelofibrosis with myeloid metaplasia: diagnostic definition and prognostic classification for clinical studies and treatment guidelines. J Clin Oncol. 1999;17(9):2954–2970. [DOI] [PubMed] [Google Scholar]
- 2.Dupriez B, Morel P, Demory JL, et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996;88(3): 1013–1018. [PubMed] [Google Scholar]
- 3.Elliott MA, Verstovsek S, Dingli D, et al. Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk Res. 2007;31(11):1503–1509. [DOI] [PubMed] [Google Scholar]
- 4.Tam CS, Kantarjian H, Cortes J, et al. Dynamic model for predicting death within 12 months in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. J Clin Oncol. 2009;27(33):5587–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. [DOI] [PubMed] [Google Scholar]
- 6.Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. [DOI] [PubMed] [Google Scholar]
- 7.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. [DOI] [PubMed] [Google Scholar]
- 8.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. [DOI] [PubMed] [Google Scholar]
- 9.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014;28(7):1472–1477. [DOI] [PubMed] [Google Scholar]
- 11.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. [DOI] [PubMed] [Google Scholar]
- 12.Jones AV, Kreil S, Zoi K, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106(6):2162–2168. [DOI] [PubMed] [Google Scholar]
- 13.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. [DOI] [PubMed] [Google Scholar]
- 14.Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–3476. [DOI] [PubMed] [Google Scholar]
- 15.Guglielmelli P, Barosi G, Specchia G, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009;114(8):1477–1483. [DOI] [PubMed] [Google Scholar]
- 16.Tefferi A, Lasho TL, Huang J, et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008;22(4):756–761. [DOI] [PubMed] [Google Scholar]
- 17.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. [DOI] [PubMed] [Google Scholar]
- 18.Nussenzveig RH, Swierczek SI, Jelinek J, et al. Polycythemia vera is not initiated by JAK2V617F mutation. Exp Hematol. 2007;35(1):32–38. [DOI] [PubMed] [Google Scholar]
- 19.Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10(21):7252–7259. [DOI] [PubMed] [Google Scholar]
- 20.Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barosi G, Bergamaschi G, Marchetti M, et al. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood. 2007;110(12):4030–4036. [DOI] [PubMed] [Google Scholar]
- 22.Koren-Michowitz M, Landman J, Cohen Y, et al. JAK2V617F allele burden is associated with transformation to myelofibrosis. Leuk Lymphoma. 2012;53(11):2210–2213. [DOI] [PubMed] [Google Scholar]
- 23.Tefferi A, Lasho TL, Finke C, et al. Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: differences in phenotype and prognostic impact. Leukemia. 2014;28(7):1568–1570. [DOI] [PubMed] [Google Scholar]
- 24.Rumi E, Pietra D, Pascutto C, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124(7):1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guglielmelli P, Rotunno G, Fanelli T, et al. Validation of the differential prognostic impact of type 1/type 1-like versus type 2/type 2-like CALR mutations in myelofibrosis. Blood Cancer J. 2015;5:e360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tenedini E, Bernardis I, Artusi V, et al. Targeted cancer exome sequencing reveals recurrent mutations in myeloproliferative neoplasms. Leukemia. 2014;28(5):1052–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tefferi A, Lasho TL, Abdel-Wahab O, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24(7):1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tefferi A, Pardanani A, Lim KH, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23(5):905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. [DOI] [PubMed] [Google Scholar]
- 30.Barbui T, Thiele J, Gisslinger H, Finazzi G, Vannucchi AM, Tefferi A. The 2016 revision of WHO classification of myeloproliferative neoplasms: Clinical and molecular advances. Blood Rev. 2016. June 11 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 31.Lippert E, Girodon F, Hammond E, et al. Concordance of assays designed for the quantification of JAK2V617F: a multicenter study. Haematologica. 2009;94(1):38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.