

Abstract
Momelotinib, a small-molecule inhibitor of Janus kinase 1 and Janus kinase 2, has demonstrated efficacy in myelofibrosis patients with 300 mg, once-daily dosing. This open-label, non-randomized, phase 1/2 study evaluated the safety and therapeutic benefit of momelotinib with twice-daily dosing. A total of 61 subjects with primary myelofibrosis or post–polycythemia vera/post–essential thrombocythemia myelofibrosis with intermediate- or high-risk disease received momelotinib. A phase 1 dose escalation identified 200 mg twice daily as the optimal dose to be expanded in phase 2. The most frequent adverse events were diarrhea (45.9%), peripheral neuropathy (44.3%), thrombocytopenia (39.3%), and dizziness (36.1%), the latter primarily due to a first-dose effect. The response assessment according to the 2006 International Working Group criteria (≥8 weeks duration at any time point) demonstrated spleen response by palpation of 72% (36/50) and anemia response of 45% (18/40). Spleen response by magnetic resonance imaging obtained at 24 weeks was 45.8% (27/59) for all subjects and 54.0% (27/50) for those with palpable splenomegaly at baseline. The symptoms of myelofibrosis were improved in most subjects. Cytokine analysis showed a rapid decline in interleukin-6 with momelotinib treatment, and a slower reduction in other inflammatory cytokines. In the subgroup of subjects with the JAK2V617F mutation at baseline (n=41), momelotinib significantly reduced the allele burden by 21.1% (median) at 24 weeks. These results provide evidence of tolerability and a potential therapeutic activity of momelotinib for subjects that support further evaluation in ongoing, phase 3 randomized trials. (clinicaltrials. gov identifier:01423058).
Introduction
Primary myelofibrosis (PMF) is a chronic myeloproliferative neoplasm that originates at the level of the hematopoietic stem cell and is characterized by cytopenias, extramedullary hematopoiesis, megakaryocytic hyperplasia, marrow fibrosis, and systemic symptoms resulting from elevated levels of inflammatory and proangiogenic cytokines. A form of myelofibrosis, indistinguishable from PMF, can occur as part of the natural history of polycythemia vera (PV) and essential thrombocythemia (ET), referred to as post–PV myelofibrosis and post–ET myelofibrosis, respectively. The discovery of the JAK2V617F mutation contributed to the understanding of the role of the dysregulated Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway in myeloproliferative neoplasm, and paved the way for the development of small-molecule inhibitors of JAK1/2. The first-in-class JAK1/2 inhibitor ruxolitinib has been approved for myelofibrosis and is effective in the reduction of splenomegaly as well as the myelofibrosis-related symptom burden; however, anemia and thrombocytopenia are significant hematologic toxicities.1,2
Momelotinib (formerly known as CYT387) is a small-molecule, adenosine triphosphate-competitive inhibitor of JAK1 and JAK2. Kinase profiling of momelotinib indicated that this molecule has good selectivity over other JAK family kinases (JAK3, tyrosine kinase 2) and excellent selectivity over other tyrosine and serine/threonine kinases.3 Momelotinib has previously been evaluated in a phase 1/2 study in myelofibrosis patients (study CCL09101; clinicaltrials. gov identifier:00935987), in which the dose was escalated from a 100-mg once-daily (QD) capsule to a maximum of 400 mg QD.4 In that study, the maximum tolerated dose (MTD) was determined to be 300 mg QD, and the 400-mg QD dose met the definition of dose-limiting toxicity (DLT). Both the 150-mg QD and 300-mg QD doses demonstrated clinical responses and were expanded where both doses showed similar spleen responses.4 A dose expansion at 150 mg twice daily (BID) demonstrated similar efficacy and safety to the 150 mg QD dose and the MTD of 300 mg QD.5 This study was undertaken to investigate the possibility that additional therapeutic benefit may be achieved with a BID regimen with higher doses; therefore, further dose escalation was carried out in the current study. The use of a BID regimen was supported by the half-life of momelotinib, which ranged from 4 to 6 hours in the previous study.6
The primary objectives of this study were to determine the safety, tolerability, and pharmacokinetics (PK) of momelotinib for the BID dosing regimen, as well as to obtain information on the effectiveness of this dosing schema. The intention was to review the results from this study and those from the previous phase 1/2 study in order to select the appropriate dose for phase 3 trials.
Methods
Patient eligibility criteria
Subjects ≥18 years of age with PMF or post–ET/PV myelofibrosis diagnosis according to the revised World Health Organization criteria7 were eligible for enrollment if they had high-risk, intermediate-2 risk, or intermediate-1 risk myelofibrosis (defined by the International Prognostic Scoring System [IPSS]8) associated with symptomatic splenomegaly/hepatomegaly and/or were unresponsive to available therapy. Other eligibility criteria included life expectancy ≥12 weeks, Eastern Cooperative Oncology Group performance status ≤2, absolute neutrophil count ≥0.5×109/L, platelet count ≥50×109/L, and acceptable organ function within 7 days of initiating momelotinib. A washout period of 14 days was required for any previous systemic therapy. Key exclusion criteria were grade ≥2 peripheral neuropathy, acute active infection, or intercurrent illness that would jeopardize safety or compliance.
Study design
This multicenter, open-label phase 1/2 study (clinicaltrials. gov identifier:01423058) consisted of a dose-escalation phase (Part 1) to identify DLTs and/or MTD of momelotinib BID, and a dose-confirmation phase (Part 2), which was a cohort expansion at or below the MTD. In Part 1, the first cohort received 200 mg BID (doses approximately 12 hours apart) for a 28-day cycle. Higher dosing cohorts were initiated if ≤2 DLTs were experienced per 6 subjects in cycle 1 and following a review of the toxicity and efficacy of the lower dose. In Part 2, subjects were to receive the MTD or a lower dose shown to have significant clinical activity. Subjects in both phases were evaluated for 6 cycles. The study was approved by the institutional research and ethics boards of all of the participating institutions and was conducted in accordance with the principles of the Declaration of Helsinki.
Assessments
The primary safety endpoint was to determine the safety profile and MTD of momelotinib. Safety assessments included the characterization of DLTs, treatment-emergent adverse events (TEAEs), and adverse events (AEs) incidence and severity. The primary efficacy endpoint was the therapeutic response rate as defined by the number of subjects achieving complete remission, partial remission, or clinical improvement according to the 2006 International Working Group for Myeloproliferative Neoplasms Research and Treatment criteria (IWG-MRT).9
Spleen response by palpation: ≥50% decrease from baseline in palpable spleen length for baseline splenomegaly ≥10 cm that lasted ≥8 weeks, or resolution of palpable splenomegaly for baseline splenomegaly >5 cm and <10 cm that lasted ≥8 weeks.
Spleen response by magnetic resonance imaging (MRI): ≥35% reduction in spleen volume from baseline and, for the subgroup who had palpable splenomegaly, >5 cm below the left costal margin at baseline.
Anemia response: no red blood cell (RBC) transfusions for ≥8 weeks for transfusion-dependent subjects, or ≥2 g/dL increase in hemoglobin lasting ≥8 weeks for transfusion-independent subjects who had hemoglobin <10 g/dL at baseline. RBC transfusion-dependence was defined as ≥2 RBC units in the 30 days prior to the first dose of momelotinib. Two post hoc analyses were performed using a 12-week endpoint: one with the same transfusion-dependence definition as the 8-week analysis, and the other using a stricter definition for transfusion-dependence at baseline (i.e., 6 RBC units within the 12 weeks prior to the first dose with ≥1 unit administered in the 28 days prior to the first dose).
Myelofibrosis symptoms response: ≥50% reduction in total symptom score (TSS) using the Myelofibrosis Symptom Assessment Form lasting ≥12 weeks. TSS was defined as the sum of the scores for constitutional symptoms. See the Online Supplementary Methodology Information for the list.
For details of pharmacology and JAK2V617F allele burden analyses, see the Online Supplementary Methodology Information.
Results
A total of 61 subjects were enrolled between September 21, 2011, and July 30, 2012, and received at least one dose of momelotinib. Subject disposition and baseline characteristics are shown in Figure 1 and Table 1, respectively. The safety analysis and modified intention-to-treat analysis comprised of 61 and 60 subjects, respectively. Overall, 45 subjects (73.8%) completed at least six 28-day cycles of momelotinib BID treatment; 22 subjects continued until study closure in June 2014, at which time they were enrolled in an open-label maintenance study (clinicaltrials. gov identifier:02124746). Overall, 39 subjects discontinued, and the median duration of exposure to the study drug was 382 days (range 2–995 days) for the total safety cohort. The most common reason for study discontinuation was AEs, which occurred in 16 subjects (26.2%), followed by progressive disease (9 subjects [14.6%]) and subject withdrawal (8 subjects [13.1%] (Figure 1). Seven subjects (11.5%) died during the course of the study. None of the deaths were considered related to the study drug.
Figure 1.
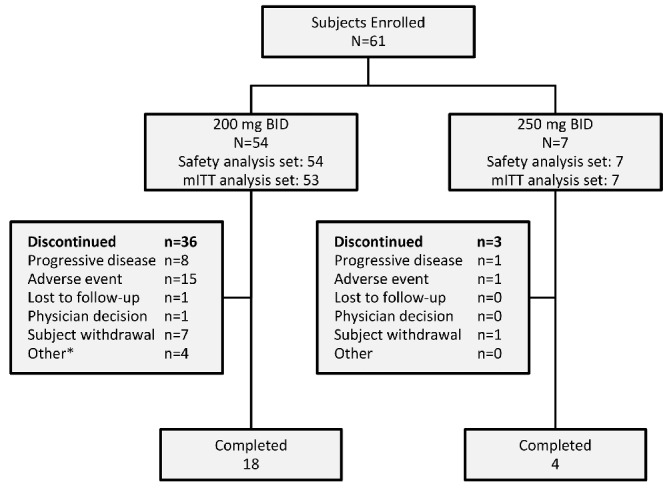
Subject disposition. *2 subjects with loss of response, 1 proceeded to bone marrow transplant, 1 lack of efficacy. BID: twice daily; mITT: modified intention-to-treat.
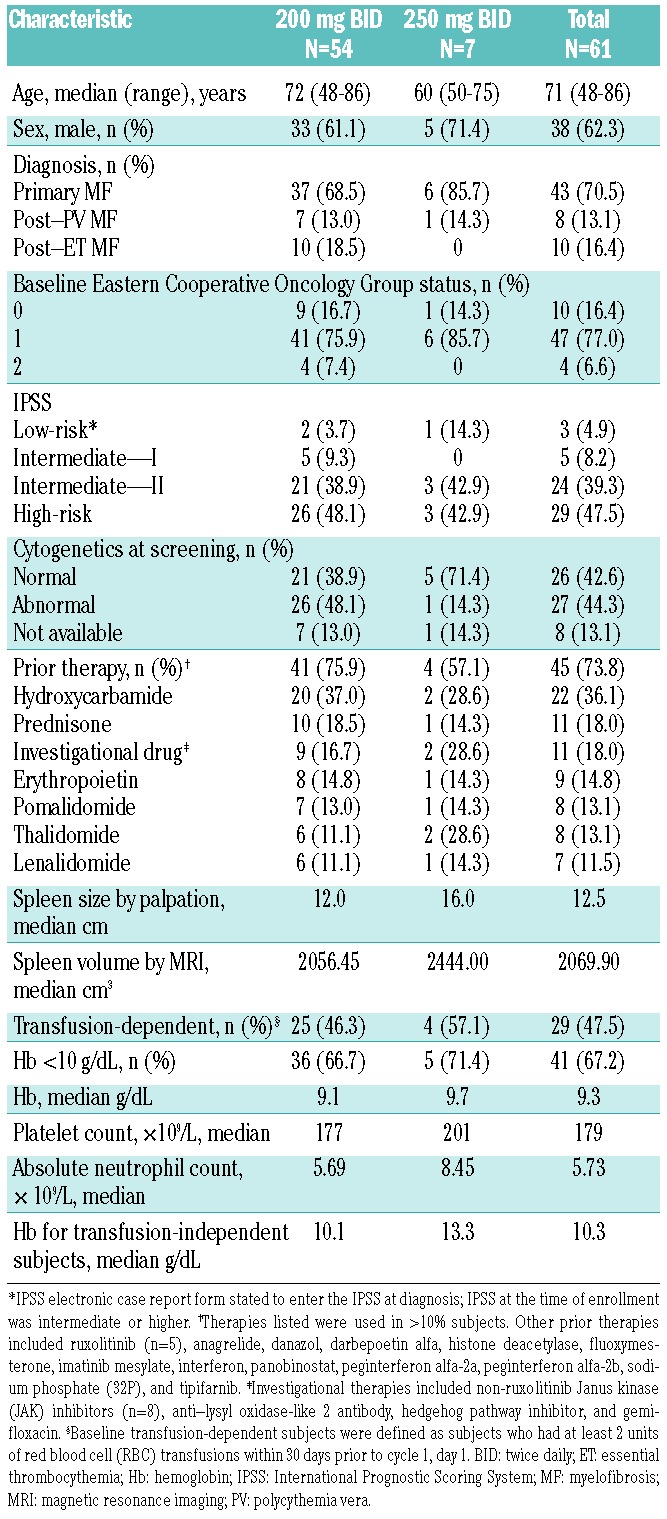
Table 1.
Baseline demographics and characteristics.
Safety analysis
Dose-escalation phase
In Part 1 of the study, 6 subjects were enrolled at the 200 mg BID dose level and 7 subjects were enrolled at the next dose level of 250 mg BID. Although no protocol-defined DLTs were identified, all but 1 of the 7 subjects at the 250 mg BID dose level required treatment interruptions, resulting in dose reductions in 5 subjects (4 due to thrombocytopenia and 1 due to elevated lipase) and 1 discontinuation (due to dysesthesia and skin rash). Therefore, the safety review committee designated 200 mg BID as the dose level for expansion, and an additional 48 subjects were enrolled at this dose level.
Toxicities
All 61 subjects experienced at least one AE, with 58 subjects (95.1%) having events related to the study drug as determined by the investigator. A total of 33 subjects (54.1%) experienced at least one serious AE (SAE), with 14 subjects (23.0%) reporting a treatment-related SAE.
Nonhematologic toxicities
The most frequently reported nonhematologic TEAEs, regardless of drug attribution, were diarrhea (28 subjects, 45.9%), peripheral neuropathy (27 subjects, 44.3%), dizziness (22 subjects, 36.1%), and hypotension (15 subjects, 24.6%). These TEAEs are summarized in Figure 2A and Online Supplementary Table S1.
Figure 2.

(A) Nonhematologic treatment-emergent adverse events (>10% of total subjects) across all momelotinib dose levels. (B) Hematologic treatment-emergent adverse events (>10% of total subjects) across all momelotinib dose levels.
First-dose hypotension/dizziness
A total of 27 subjects (44.3%) reported 1 or more AE(s) after the first dose—primarily hypotension, dizziness, and/or lightheadedness (14 subjects each, 23.0%; all grade 1). The majority of these AEs lasted ≤1 hour, with 86.3% resolving prior to the subject leaving the clinic. There was a mean decline of systolic blood pressure of 13 mm Hg and 15 mm Hg at 1 and 2 hours post-dose, respectively. Systolic blood pressure returned to baseline within 12 to 24 hours without intervention. None of these events led to hospital admissions or discontinuation of the drug.
Peripheral neuropathy
Peripheral neuropathy was observed in 27 subjects (44.3%) and was mainly reported as sensory in nature. Neuropathy events were classified as grade 1 (n=10), grade 2 (n=15), and grade 3 (n=2). The median time to neuropathy onset was 227 days (range: 6–785 days). For the two grade 3 neuropathy events, 1 subject discontinued treatment on day 221; the other subject had a drug interruption on day 702 (due to a grade 3 hand neuropathy that resolved without sequelae) and resumed treatment on day 785.
Overall, 5 subjects discontinued from the study because of symptoms of peripheral neuropathy. As the study generally captured only the highest reported grade of neuropathy and did not follow subjects beyond a day 30 safety visit, the reversibility of neuropathy could not be determined.
Cytokine release syndrome or withdrawal effect
Among the 39 subjects (64%) who discontinued the study drug prior to the end of the study, there were no reported cases of cytokine release syndrome or withdrawal effect.
Hematologic toxicities
Thrombocytopenia was observed in 24 study subjects (39.3%) (Figure 2B and Online Supplementary Table S2), with 18 (29.5%) experiencing grade ≥3 toxicity. Thrombocytopenia was more common in subjects receiving the 250 mg BID dose (5/7 subjects; 71.4%) than the 200 mg BID dose (19/53 subjects; 35.2%). Although no subject discontinued the study due to thrombocytopenia, there were 3 SAEs of grade 4 thrombocytopenia reported as related to momelotinib, all of which were resolved with drug interruption. Two bleeding events were associated with platelet counts of <50×109/L; one was a fatal intracranial bleed in a subject who had a baseline platelet count of 50×109/L, which declined to 37×109/L.
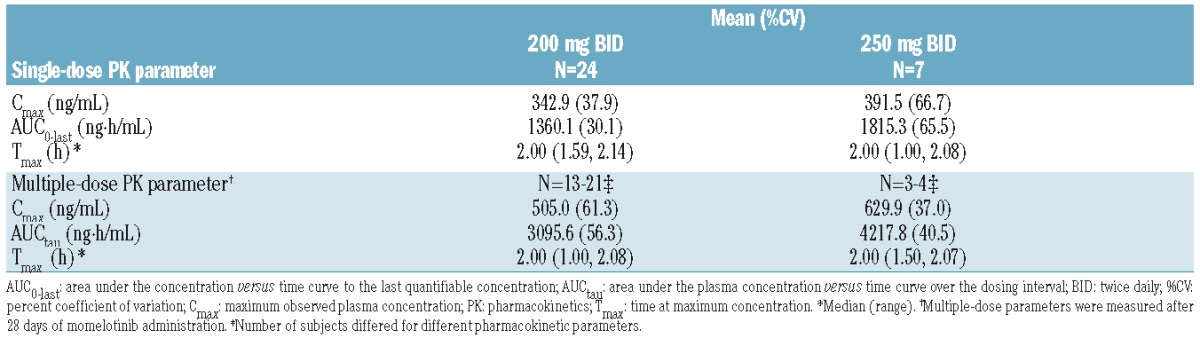
Pharmacokinetics
PK profiles were evaluated for 24 subjects from the 200 mg BID group and 7 subjects from the 250 mg BID group. Momelotinib concentrations peaked at around 2 hours post-dose for both the 200 mg and 250 mg BID cohorts. Momelotinib exposures were higher at the 250 mg BID dose than at the 200 mg BID dose (14% to 25% increase in maximum plasma concentration and 33% to 36% increase in the area under the curve; Table 2).
Table 2.
Pharmacokinetic parameters of momelotinib following single and multiple doses.
Efficacy evaluation
IWG-MRT response
Overall, 35 subjects (58.4%) showed a therapeutic response (clinical improvement: 34/60, 56.7%; partial remission 1/60, 1.7%) by investigator assessment during the study as per the 2006 IWG-MRT criteria. No subjects achieved a complete remission. Stable disease was seen in 21 subjects (35%).
Spleen response
Clinical improvement in splenomegaly was demonstrated using both palpation and MRI methods of measuring spleens (Figure 3). Only subjects who had an enlarged spleen at baseline demonstrated a spleen response by MRI. Of the 59 subjects with spleens who underwent a baseline MRI, 38 (64.4%) underwent repeat MRIs at 24 weeks, and 27 (45.8%) demonstrated a ≥35% reduction in spleen volume. Of the 50 subjects with baseline palpable splenomegaly >5 cm who underwent a baseline MRI, 27 (54.0%) demonstrated a ≥35% reduction in spleen volume (Table 3). Subjects who missed the 24-week MRI for any reason were classified as treatment failures. Spleen volume by MRI correlated with palpable spleen at the MRI designated time points, with the correlation coefficient ranging from 0.56 to 0.80.
Figure 3.
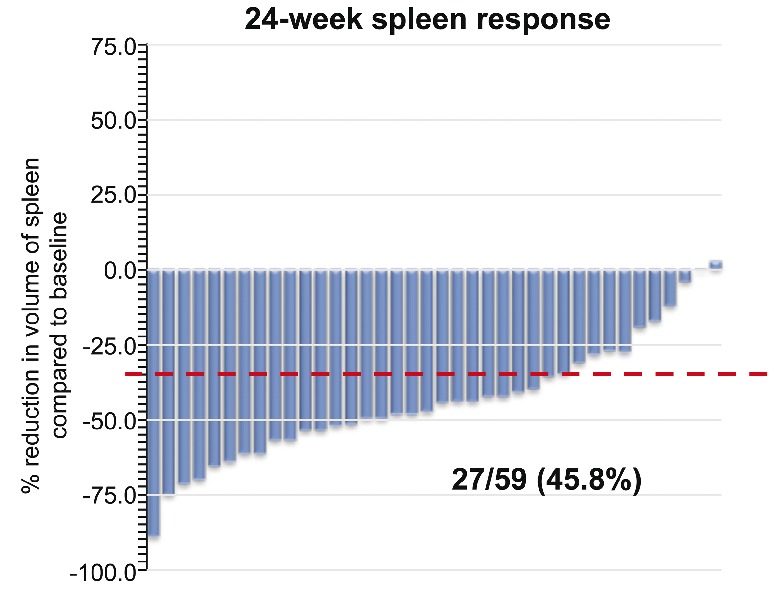
Spleen response showing percentage reduction from baseline in spleen volume by magnetic resonance imaging (MRI) for subjects with palpable splenomegaly at baseline (n=50). Baseline subjects included all subjects with spleens (n=59). At week 24, 38 subjects underwent MRI. Missed MRIs were due to adverse events (AEs) (9 subjects), withdrawal (5 subjects), missed appointments (4 subjects), progressive disease (2 subjects), and lost to follow-up (1 subject). Dashed red line indicates reduction of 35% from baseline.
Table 3.
Overall therapeutic response to momelotinib (modified intention-to-treat (mITT) population).

Anemia response
Subjects demonstrating an 8-week anemia response had either a transfusion-independent response (subjects who were transfusion-dependent at baseline) or a hemoglobin response (subjects who were transfusion-independent with hemoglobin <10 g/dL at baseline). Of the 29 subjects (48.3%) classified as transfusion-dependent at baseline, 15 (51.7%) achieved 8-week transfusion independence. Of the 11 baseline transfusion-independent subjects with a baseline hemoglobin <10 g/dL, 3 subjects (27.3%) had a rise in hemoglobin ≥2 g/dL lasting ≥8 weeks, for an overall anemia response seen in 18/40 subjects (45%; Table 3). A careful review of medications received prior to trial entry was done by the study authors (VG, SV, and CER) to ensure that these responses were not attributable to the discontinuation of myelosuppressive medications received prior to trial entry. Due to intervening changes in the accepted definitions of anemia response after the study had completed enrollment, a first post hoc evaluation of anemia response was performed using a 12-week rolling endpoint. The 12-week rolling anemia response was 25% (10/40), composed of a 31% (9/29) 12-week, transfusion-independent response combined with a 9% (1/11) hemoglobin response. A second post hoc anemia response evaluation defined transfusion-dependent subjects at baseline more stringently, as subjects who required at least 6 units of blood in the 12 weeks prior to the first dose and at least 1 of those units in the 28 days prior to study day 1. Using this stricter definition of transfusion dependence at baseline, the number of transfusion-dependent subjects decreased to 19, and the 12-week transfusion response in this heavily transfusion-dependent group was 21.1% (4/19 subjects; Table 3). While the 12-week response rates were lower than the 8-week response rates, the 12-week responses were more durable (Table 3).
Secondary efficacy endpoints
Overall, treatment with momelotinib resulted in a reduction of hepatomegaly and an improvement in almost all constitutional symptoms experienced by subjects at baseline (Table 3). Approximately one-third of subjects reported either a complete response (absence of symptoms [score=0]) or marked response (≥50% reduction in score from baseline) of their TSS at the 3-month or 6-month visit, and responses were durable up to the 24-month visit.
Progression-free survival and overall survival at 2 years were 74% (95% confidence interval [CI] 58–85) and 88% (95% CI 75–95), respectively.
In the 39 subjects who had both a baseline and an on-study bone marrow evaluation, fibrosis improved in 11 subjects (9 subjects improved by 1 grade, 1 subject improved by 2 grades, and 1 subject had complete resolution of a grade 3 fibrosis). In 3 subjects, fibrosis worsened by 1 grade. Eight of these 11 subjects also showed a clinical improvement according to IWG-MRT criteria.
In the study population, 41 subjects (68%) were positive for JAK2V617F at study entry. The median JAK2V617F allele burden significantly declined from baseline by 16.6% at week 12 (P=0.0063), and by 21.1% at week 24 (P=0.0019; Table 3, Figure 4). A complete molecular response was achieved by one subject whose JAK2V617F allele burden decreased from a baseline level of 26.4% to undetectable at week 12 and week 16, when the subject went off study.
Figure 4.
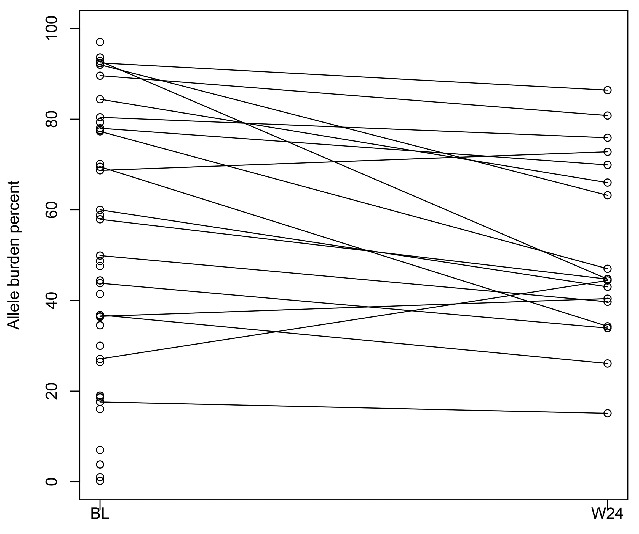
JAK2V617F allele burden. Plot shows baseline levels (n=41) and individual trajectories of allele burden to week 24 (n=18). BL: baseline; W24: week 24.
Cytokine assessments
Blood samples were collected pre-dose, at 6 and 24 hours post–first dose, and at weeks 8 and 20 for exploratory assessments of the effect of momelotinib on cytokines. At baseline, epidermal growth factor, transforming growth factor beta (TGF-β), and vascular endothelial growth factor-A (VEGF-A) were positively correlated with each other, as were tumor necrosis factor-alpha (TNF-α), insulin-like growth factor binding protein-1, and TNF receptor type II (TNFRII) (Spearman correlation coefficients >0.5; data not shown). Additional positive correlations were observed between interleukin (IL)-6 and C-reactive protein (CRP), and between IL-6 and IL-10 (Spearman correlation coefficients >0.5; data not shown). By week 8, CRP, erythropoietin, IL-6, IL-10, IL-12p40, TNFRII, and TNF-α were significantly decreased; all of these except erythropoietin remained low to week 20 (Figure 5). IL-6 showed the most rapid response, with a reduction of 30.3% within 6 hours after the first dose of momelotinib.
Figure 5.
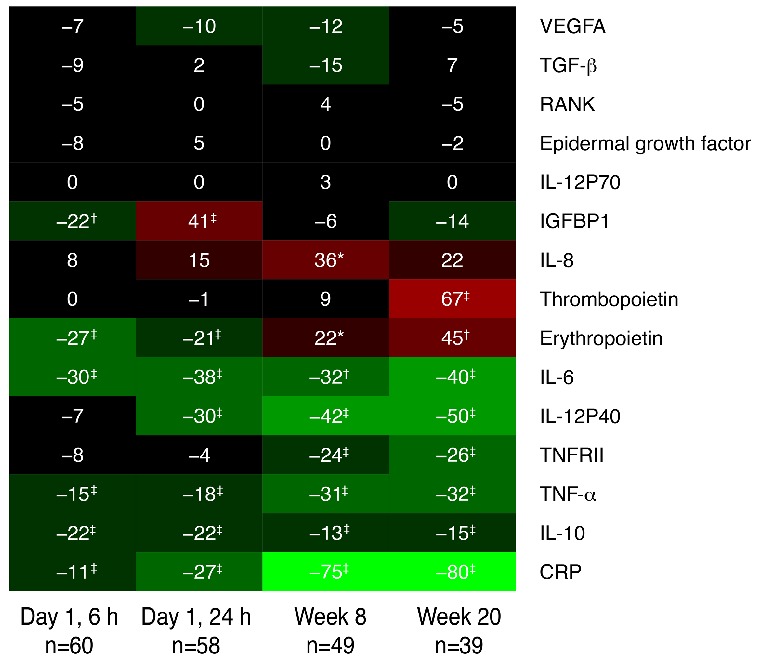
Heat map showing median percentage change in cytokine levels at each time point relative to pre-dose. Green and red represent decreased and increased levels from baseline, respectively. Changes from baseline tested using Wilcoxon signed-rank test. P values are indicated for median percent changes ≥10%: *0.01–0.05; †0.001–0.01; ‡<0.001. CRP: C-reactive protein; IGFBP1: insulin-like growth factor binding protein 1; IL: interleukin; IL-12P70: interleukin-12 subunit P70; RANK: receptor activator of nuclear factor kappa B; TGF-β: transforming growth factor beta; TNF-α: tumor necrosis factor alpha; TNFRII: tumor necrosis factor receptor type II; VEGFA: vascular endothelial growth factor A; lL-12P40: interleukin-12 subunit P40.
Discussion
This phase 1/2 study was designed to evaluate the safety and tolerability, to determine DLTs, MTD, and PK, and to obtain preliminary efficacy results of BID momelotinib in subjects with PMF or post–PV/ET myelofibrosis. BID dosing allowed for testing a higher total daily dose of momelotinib than QD dosing. No additional safety signals were observed at the higher doses. The discontinuation rate (59%) reflects, in part, the prolonged study duration for subjects and the wider availability of alternative treatment options during the conduct of the study, including ruxolitinib as a marketed therapy, and an increase in myelofibrosis clinical trials. It is important to note that the majority of subjects (73.8%) did successfully complete the first 6 cycles of treatment with momelotinib. It is difficult to compare discontinuation rates with previously reported studies of ruxolitinib and other JAK inhibitors because ruxolitinib studies enrolled JAK-naïve subjects, whereas this trial included 21% of subjects previously treated with JAK inhibitors. Direct comparisons can be truly assessed only in a head-to-head trial. The most common AEs observed were diarrhea, peripheral neuropathy, thrombocytopenia, and dizziness. The dizziness was primarily associated with a first-dose effect—sometimes associated with low-grade hypotension and flushing—and these events were generally grade 1 and self-limiting. This first-dose effect appeared to be more common with momelotinib than has been reported in other JAK inhibitors,1,2,10,11 but did not lead to dose interruption or study discontinuation. Diarrhea was reported in 45.9% of subjects, with 31.1% reporting the diarrhea to be drug-related, although these events were generally grade 1 and self-limiting (23.0% were grade 1 and 8.2% were grade 2). Overall, the AE profile was favorable for continuing the evaluation of momelotinib as treatment for myelofibrosis.
The evaluation of the frequency and severity of thrombocytopenia was partly confounded by existing thrombocytopenia in some subjects at baseline. Thrombocytopenia was also dose-dependent, and a higher frequency of grade 3/4 thrombocytopenia was seen in subjects treated with the 250 mg BID dose of momelotinib, and appeared to be reversible upon drug interruption.
Peripheral sensory neuropathy was a frequent nonhematologic toxicity observed in this trial. The incidence (44%, with the onset at a median of 32 weeks) was similar to that reported in a prior momelotinib phase 1/2 study.12 However, in the aforementioned prior study a significant number of subjects (49%) had already reported numbness and tingling at baseline in the Myelofibrosis Symptom Assessment Form. In the present study, grade 2 neuropathy was reported in 24% of subjects, and 2 subjects (3.3%) reported grade 3 neuropathy; this is higher than the rates observed in the previous momelotinib study, where a lower total daily dose of momelotinib was used.12 Five subjects discontinued therapy due to neuropathy, although the reversibility of peripheral neuropathy could not be determined because of the study design. The mechanism by which momelotinib might contribute to sensory neuropathy has not been identified. One should therefore remain vigilant about neurologic complications, as the development of three JAK inhibitors (XL019 [Exelixis, South San Francisco, CA, USA], fedratinib [Sanofi Aventis, Gentilly, France], and AZD1480 [AstraZeneca, London, UK]) have been halted due to neurologic complications primarily affecting the central nervous system.10,13,14
Spleen response rate by MRI at 24 weeks was similar to that which has been described for the JAK1/2 inhibitor ruxolitinib.1,2 Additionally, momelotinib demonstrated evidence of improvement in clinically important, anemia-related endpoints of transfusion independence and increased hemoglobin, using both 8-week criteria and the more clinically relevant 12-week criteria. A potential mechanism of the anemia benefit observed with momelotinib has been hypothesized to be due to the targeting of ACVR1/ALK-2, a key mediator in the hepcidin/inflammation pathway.15 If supported with further data, this characteristic could distinguish momelotinib compared with other JAK inhibitors. Data from ongoing, blinded, randomized trials with prespecified, anemia-related endpoints are anticipated in the near future.
Baseline cytokines were consistent with the inflammatory nature of myelofibrosis. Momelotinib treatment resulted in a rapid decrease in the inflammatory cytokine IL-6, with slower decreases in other inflammatory cytokines. After prolonged treatment, erythropoietin was increased relative to baseline. These changes are similar to those reported with the JAK1/2 inhibitor ruxolitinib.1
The current results provide evidence that momelotinib has therapeutic activity in the majority of subjects with myelofibrosis, as evidenced by the reduction of splenomegaly, improvements in anemia-related endpoints, and symptom response. This study confirms the findings of a previously conducted phase 1/2 study4 in a multicenter setting, and the results of these two studies helped in the selection of the dose used for phase 3 trials. Momelotinib is now under phase 3 evaluation for treatment of myelofibrosis in two ongoing randomized trials.
Supplementary Material
Acknowledgments
The authors thank Impact Communication Partners, Inc., for editorial assistance in preparing the manuscript.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/1/94
Funding
This study was supported by Gilead Sciences, Inc.
Previously presented in part at the European Hematology Association Meeting, Milan, Italy, June 2014. Cytokine data were presented at the American Society of Hematology Meeting, Orlando, FL, USA, December 2015.
References
- 1.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. [DOI] [PubMed] [Google Scholar]
- 2.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Höfener M, Pachl F, Kuster B, Sewald N. Inhibitor-based affinity probes for the investigation of JAK signaling pathways. Proteomics. 2015;15(17):3066–3074. [DOI] [PubMed] [Google Scholar]
- 4.Pardanani A, Laborde RR, Lasho TL, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia. 2013;27(6):1322–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pardanani A, Gotlib J, Gupta V, et al. Update on the long-term efficacy and safety of momelotinib, a JAK1 and JAK2 inhibitor, for the treatment of myelofibrosis. Blood. 2013;122(21):108. [DOI] [PubMed] [Google Scholar]
- 6.Xin Y, Shao L, Jun S, et al. The relative bioavailability, food effect and drug interaction with omeprazole of momelotinib (GS-0387, CYT387) tablet formulation in healthy subjects. Presented at: American Association of Pharmaceutical Scientists; November 2–6, 2014; San Diego, CA Abstract M1320. [Google Scholar]
- 7.Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110(4):1092–1097. [DOI] [PubMed] [Google Scholar]
- 8.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. [DOI] [PubMed] [Google Scholar]
- 9.Tefferi A, Barosi G, Mesa RA, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT). Blood. 2006;108(5): 1497–1503. [DOI] [PubMed] [Google Scholar]
- 10.Verstovsek S, Hoffman R, Mascarenhas J, et al. A phase I, open-label, multi-center study of the JAK2 inhibitor AZD1480 in patients with myelofibrosis. Leukemia Res. 2015; 39(2):157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komrokji RS, Seymour JF, Roberts AW, et al. Results of a phase 2 study of pacritinib (SB1518), a JAK2/JAK2(V617F) inhibitor, in patients with myelofibrosis. Blood. 2015;125(17):2649–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdelrahman RA, Begna KH, Al-Kali A, et al. Momelotinib treatment-emergent neuropathy: prevalence, risk factors and outcome in 100 patients with myelofibrosis. Br J Haematol. 2015;169(1):77–80. [DOI] [PubMed] [Google Scholar]
- 13.Verstovsek S, Tam CS, Wadleigh M, et al. Phase I evaluation of XL019, an oral, potent, and selective JAK2 inhibitor. Leuk Res. 2014;38(3):316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1(5):643–651. [DOI] [PubMed] [Google Scholar]
- 15.Asshoff M, Warr M, Haschka D, et al. The Jak1/Jak2 inhibitor momelotinib inhibits Alk2, decreases hepcidin production and ameliorates anemia of chronic disease (ACD) in rodents. Blood. 2015;126 (23):538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.