

Evidence of the presence of abnormalities in the primitive hematopoietic stem cells, as well as functional analysis, has demonstrated that Myelodysplastic syndrome (MDS) is propagated by rare and distinct human MDS propagating cells (MDS-PCs) in 5q- and in refractory anemia with ring sideroblastic (RARS) MDS patients.1,2 Bone marrow from these patients harbour Long-Term and Short-Term Hematopoietic Stem Cell (LT- and ST-HSCs) phenotype (Lin− CD34+ CD38− CD90+ CD49f+ CD45RA−). Therefore, low-risk MDS has been proposed as a stem cell disorder with mutations in the mature progenitors, originating from the more immature hematopoietic compartment.3 Here, we are reporting on another class of MDS; more precisely, an overlap MDS/MPN syndrome with a significantly elevated blast count. The study was part of a larger patient cohort that has been used to decipher clonal evolution.
It concerns a case report from a 46-year old male who, after presenting with bruising, was diagnosed as MDS/MPN-U (Myelodysplastic/Myeloproliferative Neoplasm, Unclassifiable). This patient had 8% bone marrow blasts with trisomy 8 in all metaphases, later transforming to acute myeloid leukemia (AML) (Online Supplementary Table S1). We performed whole-exome sequencing (WES) in order to identify the somatic mutations present in the CD34+ population (Online Supplementary Table S1&S2, See Online Supplementary Methods). We then isolated HSCs, MPPs, myeloid-lymphoid progenitors (MLPs), common myeloid progenitors (CMPs), and granulocyte macrophage progenitors (GMPs) (Online Supplementary Figure S1A), and performed targeted sequencing for gene mutations (EZH2, PHAC-TR3, WDR64, ASXL1, RUNX1 and ANGPTLP5) detected by the WES in the total CD34+ population (Figure 1A). For all targeted sequencing experiments, ≥500x depth (Q-score > q25) was achieved and, for many, it was much higher (average across all amplicons ≥1500x) (sensitivity between 2–5%) revealing that ASXL1 and ANGPTL5 gene mutations were not present at the stage of HSC and had arisen from the MPP compartment. However, all the other mutations, including EZH2, originated from HSCs. Similar scenarios where mutations were acquired in the mature compartments have also been reported in a recent study by Woll et al.1 One of the limitations of our sequencing analysis is that any mutation present at <2% mutant allele burden (MAB) will not have been detected. Thus, we could not exclude the possibility that ASXL1 and ANGPTL5 gene mutations were acquired in the HSC fraction, but remained at low level (below our detection level) until the cells reached the MPP stage and expanded.
Figure 1.
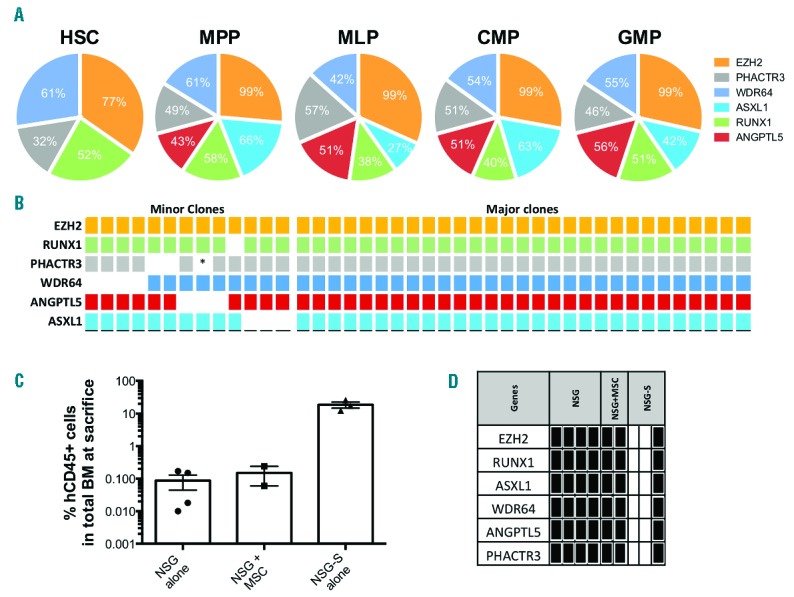
(A) Pie chart representing the percentage of mutant allele burden (MAB) of each mutation in the different hematopoietic compartments. Sequencing experiments were repeated twice for both HSC (total: 448 cells) and MPP (total: 1,608 cells) on independent reactions of the total HSC/MPP fractionated pools divided into two, one with a WGA step and one where we performed PCR directly on cells split 3 ways for EZH2, ASXL1 and RUNX1 mutations. For the other progenitors (>5,000 cells), targeted sequencing experiments were also repeated at least twice in the same fashion, once via WGA and once directly on remaining cells (non-WGA) split into relevant amplicon reactions (all 6). Mutant allele burdens were highly similar between experiments and averages are shown here. (B) Table showing the different mutations present in single colonies grown at day 0 from CD34+ cells. (*data not available). For all 42 colonies, targeted sequencing was performed directly on all the cells (without whole genome amplification); no repeat could thus be performed. (C) Bar chart representing the percentage of human CD45+ cells within the bone marrow retrieved from the different mouse models. (D) Table generated from sequencing analysis of the different mutations present in the patient, and whether these mutations were present in the human engrafted cells after transplantation in NSG or NSG-S model. Black square indicates that the specific mutation was detected; white square is for mutation not detected.
To delineate the clonal architecture of the patients’ bone marrow, we performed targeted sequencing of single colonies generated from the CD34+ population (Figure 1B). Our analysis revealed that the EZH2 gene mutation was the first aberration to occur in HSCs (42/42 colonies). The other mutations were present in fewer colonies, therefore indicating that these mutations were most likely to follow EZH2. A major clone harbouring all six mutations emerged from the colonies (29/42). Furthermore, we also found a clonal population, namely “minor” (13/42) clones (Figure 1B). These results are clearly suggestive of a founding genomic instability in the clonal MDS cell population, perhaps underpinned through mutations in epigenetic modifiers such as EZH2, leading to subsequent acquisition of additional compounding mutations that drive the progression of the disease. Notably, some of the EZH2 clones seem to have acquired mutations in a sequential order; others have done so in a random manner. It is possible, however, that some of these mutations were acquired and then lost, or that mutation drop-out occurred due to technical arte-facts, making it difficult to predict the evolutionary history of the mutations in this patient. Nevertheless, we confirmed the presence of the malignant clone(s) in cells isolated from long-term culture via FISH (Online Supplementary Figure S2).
Acknowledging the fact that bone marrow cells from MDS patients often fail to reconstitute in immunodeficient mice,4 we systematically injected, via intra-bone, primary patient CD34+ bone marrow cells into NOD-SCID/IL2Rgnull (NSG) mice, with or without autologous MSC. In addition, we also used the NSG-SGM3 (NSG-S) mouse model as these mice have previously been suggested5 to improve human engraftment (Figure 1C, See Online Supplementary Methods). The engraftment kinetics was assessed by bone marrow aspiration at different time points. As predicted in a previous work by A. Trump’s team, the NSG-S model showed a better engraftment than that observed in the other models5 (Online Supplementary Figure S3A). Although we confirmed the presence and maintenance of the malignant clone from cells retrieved from NSG mice (4/5 injected with CD34+ alone, and 2/2 when CD34+/MSC were co-injected), the data obtained from NSG-S mice was different. Three out of four NSG-S transplanted with MDS patient cells were engrafted. Interestingly, two out of the three engrafted NSG-S mice were reconstituted with wild-type patient (WT) cells (Figure 1D) (Online Supplementary Figure S3B&C) with a high percentage of human B-lymphoid cells (CD45+CD19+ 42% and 46%) in both cases. Only one NSG-S mouse displayed a high myeloid reconstitution from the MDS malignant clone (Figure 1D) (Online Supplementary Figure S3D). In agreement with previously published work,6 we observed an exhaustion of the normal HSPC compartment in NSG-S, which could explain why cells seen in these mice at week-6 and -8 failed to maintain after 12 weeks (Online Supplementary Figure S3C). For instance, engraftment assessed by bone marrow aspiration in NSG-S (mouse 1) showed 76% of human cells present at week-6, and 83.5% at week-8. From this last time point (from bone marrow aspiration) and at sacrifice (12 weeks), we sorted cells by FACS based on myeloid and lymphoid markers. Interestingly, none of the initial gene mutations were detected in these cells (Online Supplementary Figure S3C). Together, our results demonstrate that NSG mice, with or without MSC co-injection, engrafted consistently with MDS malignant clones. On the other hand, higher engraftment of human cells was observed in NSG-S mice, but none of the MDS patient related mutations were seen in 2 out of the 3 engrafted mice; reinforcing, therefore, the importance of genetic screening following xenotransplantation.
Next we evaluated the nature of the MDS-PCs. For this propose, HSCs (Lin−CD34+CD38−CD90+CD45RA−), MPPs (Lin−CD34+CD38-CD90−CD45RA−), and GMPs (Lin-CD34+CD38−CD123+CD45RA+) were sorted by FACS and injected via intra-bone into NSG and NSG-S mice (100 HSCs, 13000 MPPs, and 75000 GMPs cells were injected per mice). Engraftment was assessed at week 7, 12, 24 and 32 (Figure 2A). Interestingly, only the MPP fraction engrafted consistently in both the NSG and NSG-S mice. We did not detect any engraftment in mice injected with the HSC fraction. Although it is possible that a higher number of HSCs might be necessary to reveal the presence of MDS-PC in this cell fraction, the acquisition of the ASXL1 gene mutation within the MPP fraction, and not in HSCs (Figure 1A), could confer an advantage to the MPPs and their progenitors. Indeed, ASXL1 gene mutations have previously been reported to be associated with poor overall survival in MDS and CMML.7–9 Another potential interpretation is that the ASXL1 mutation might have initially been acquired in some HSCs but, because of the mutation, these HSCs down-regulated CD90 expression and, thus, these cells appear as MPPs.
Figure 2.
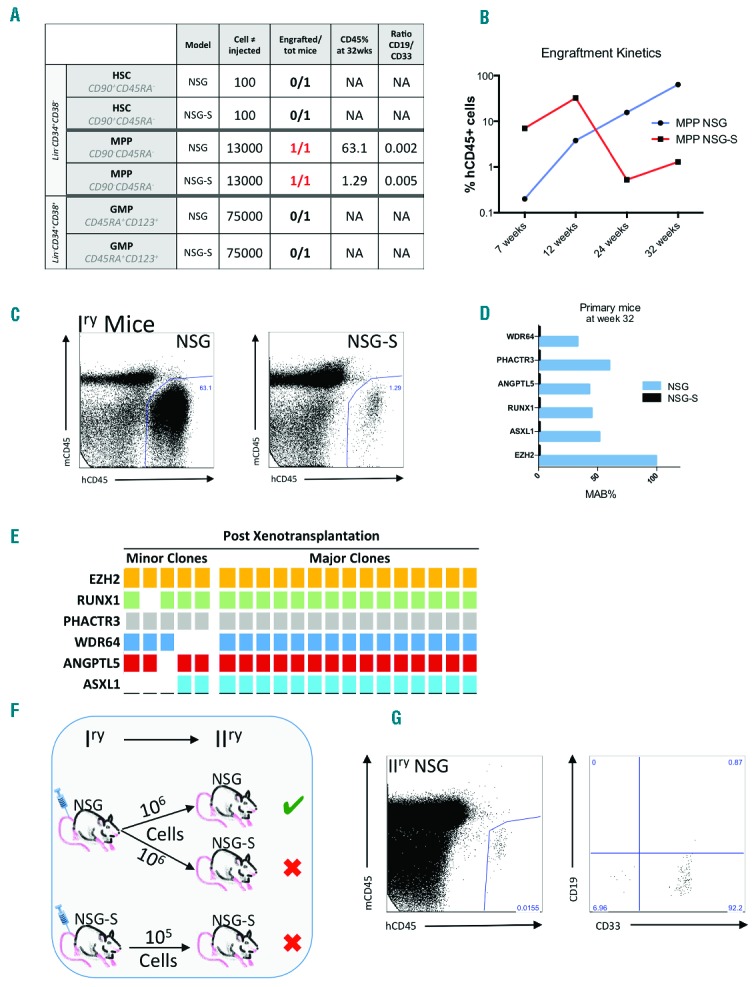
(A) Table recapitulating the different populations injected, the mouse model used, the number of cells injected, and the percentage of human CD45+ cells as well as the ratio lymphoid/myeloid at sacrifice. (B) Kinetics of engraftment of the MPP fraction in NSG and NSG-S mice. (C) FACS plots showing the percentage of human cells at sacrifice (32 weeks post-transplant) in NSG and NSG-S. (D) Bar chart representing the percentage of MAB of the different mutations present in HEC after passage into the mice. (E) Table showing the different mutations present in single colonies grown outside of transplanted mice F) Schematic of secondary mice engraftment. (G) FACS plot showing the percentage of human cells and myeloid cells at sacrifice (17 weeks post-transplant) in NSG.
Mice injected with MPPs displayed an elevated percentage of lymphoid cells within the human compartment at week 12 (47.7% in NSG and 66.9% in the NSG-S), suggesting the presence of WT MPPs cells, which were exhausted by week 24 and found only at low levels at week 32 (data not shown). Over the 32-week period, the human cell engraftment level in NSG increased to 63%, whilst it decreased in NSG-S to 1.3% (Figure 2A, B and C). Furthermore, the NSG-S model failed to maintain the MDS malignant clones, as determined by the lack of mutations in engrafted human CD45+ cells. Only cells recovered from the NSG mice displayed the same mutational landscape and mutant allele burden (MAB) as the original injected sample (day 0) (Figure 2D).
In order to decipher the type of clonal expansion observed in the mice, cells retrieved following the NSG xenotransplantation (after 32 weeks) were used for in vitro single cell colony assay. Our data showed that 75% of the human cells engrafted in NSG mice contained all the mutations as compared to 69% of the colonies from the day 0 sample (Figure 2E). Next, we injected cells from the primary NSG mice into NSG and NSG-S mice to confirm the presence of MDS-PCs in secondary animals (Figure 2F). Interestingly, human cell engraftment was observed only in the secondary NSG mice, but not in the NSG-S mice (Figure 2G). In conclusion, using two currently available mouse models, this report reveals, for the first time, the presence of MDS-PCs in the MPP compartment of MDS. This data also demonstrates that, despite the acquisition of mutation(s) in HSCs, even at the MDS stage, the propagating cells might not always be restricted to the HSC compartment. Similarly, we recently reported, as have others, that additional mutations could also be acquired outside the HSC compartment, compatible with MDS-PC activity outside the HSC compartment upon disease progression.1,2
We demonstrate here that combining the genetic mutation(s) analysis in sorted HSPCs with functional transplantation assays is an effective strategy in deciphering the nature and dynamics of the MDS-propagating cells. The linear or branching evolution of the disease progression remains a major challenge of MDS therapeutic strategy, evidenced in a recent study showing a complex and highly dynamic clonality upon lenalidomide treatment.10 Therefore, our study highlights the importance of discriminating between disease-initiating cells and disease–propagating cells, which is likely to be clinically relevant in devising a therapy regimen to target multiple clonal compartments and to monitor disease remission and/or relapse.
Supplementary Material
Acknowledgments
We would like to acknowledge the leukaemia and Lymphoma Research Fund (UK) for supporting K.R.P and A.E.S. We would also like to thank King’s College London and Kings College Hospital NHS trust for funding the Kings College Hemato-oncology Tissue Bank and Francis Crick core funds to DB. We would like to acknowledge Nigel B. Westwood, Rajani Chelliah and Swaibu Mugambwa for assisting with sample processing, tissue separation and clinical data. We would also like to thank Biological Resource Units, and the Flow Cytometry core facility members at FCI.
Footnotes
The online version of this letter has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Woll PS, Kjallquist U, Chowdhury O, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. 2014;25(6):794–808. [DOI] [PubMed] [Google Scholar]
- 2.Mian SA, Rouault-Pierre K, Smith AE, et al. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun. 2015;6:10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaiswal S, Ebert BL. MDS is a stem cell disorder after all. Cancer Cell. 2014;25(6):713–714. [DOI] [PubMed] [Google Scholar]
- 4.Nilsson L, Astrand-Grundstrom I, Anderson K, et al. Involvement and functional impairment of the CD34(+)CD38(−)Thy-1(+) hematopoietic stem cell pool in myelodysplastic syndromes with trisomy 8. Blood. 2002;100(1):259–267. [DOI] [PubMed] [Google Scholar]
- 5.Medyouf H, Mossner M, Jann JC, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14(6):824–837. [DOI] [PubMed] [Google Scholar]
- 6.Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood. 2015;125(17):2630–2640. [DOI] [PubMed] [Google Scholar]
- 7.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gelsi-Boyer V, Trouplin V, Roquain J, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010;151(4):365–375. [DOI] [PubMed] [Google Scholar]
- 9.Macedo LC, Silvestre AP, Rodrigues C, et al. Genetics factors associated with myelodysplastic syndromes. Blood Cells Mol Dis. 2015;55(1):76–81. [DOI] [PubMed] [Google Scholar]
- 10.Mossner M, Jann JC, Wittig J, et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood. 2016;128(9):1246–1259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.