

Abstract
Hematocrit control below 45% is associated with a lower rate of thrombosis in polycythemia vera. In patients receiving hydroxyurea, this target can be achieved with hydroxyurea alone or with the combination of hydroxyurea plus phlebotomies. However, the clinical implications of phlebotomy requirement under hydroxyurea therapy are unknown. The aim of this study was to evaluate the need for additional phlebotomies during the first five years of hydroxyurea therapy in 533 patients with polycythemia vera. Patients requiring 3 or more phlebotomies per year (n=85, 16%) showed a worse hematocrit control than those requiring 2 or less phlebotomies per year (n=448, 84%). There were no significant differences between the two study groups regarding leukocyte and platelet counts. Patients requiring 3 or more phlebotomies per year received significantly higher doses of hydroxyurea than the remaining patients. A significant higher rate of thrombosis was found in patients treated with hydroxyurea plus 3 or more phlebotomies per year compared to hydroxyurea with 0–2 phlebotomies per year (20.5% vs. 5.3% at 3 years; P<0.0001). In multivariate analysis, independent risk factors for thrombosis were phlebotomy dependency (HR: 3.3, 95%CI: 1.5–6.9; P=0.002) and thrombosis at diagnosis (HR: 4.7, 95%CI: 2.3–9.8; P<0.0001). The proportion of patients fulfilling the European LeukemiaNet criteria of resistance/intolerance to hydroxyurea was significantly higher in the group requiring 3 or more phlebotomies per year (18.7% vs. 7.1%; P=0.001) mainly due to extrahematologic toxicity. In conclusion, phlebotomy requirement under hydroxyurea therapy identifies a subset of patients with increased proliferation of polycythemia vera and higher risk of thrombosis.
Introduction
Polycythemia vera (PV) is a myeloproliferative neoplasm characterized by a high rate of thrombosis and bleeding.1,2 In the majority of patients, the disease is caused by the acquisition of mutations in the JAK2 gene resulting in an increased red cell mass and, frequently, in concomitant leukocytosis and thrombocytosis.3 The hyperviscosity resulting from red cell expansion has a central role in the pathogenesis of thrombosis in PV, whereas functional abnormalities of platelets and leukocytes have more recently been proposed as potential contributing factors.4–6
Control of symptoms and prevention of thrombosis and bleeding are the main objectives of treatment in PV.7 In order to achieve this, management with phlebotomies (PHL) and/or cytoreductive therapy is adopted according to risk of thrombosis and patient characteristics. When cytoreduction is indicated, hydroxyurea (HU) is the first-line therapy most commonly employed. Patients receiving HU are targeted to maintain the hematocrit (Hct) below 45%, since Hct control below 45% has been associated with a lower rate of thrombosis in both observational studies and randomized clinical trials.4,8 However, in daily clinical practice, a proportion of patients cannot adequately control the Hct with HU alone due to treatment side-effects or lack of response, and therefore require the concomitant use of PHL to achieve this.
The aim of the present study was to assess if PV patients treated with HU requiring frequent PHL have the same risk of thrombosis than those managed mainly with HU alone.
Methods
Study design
The Spanish Registry of Polycythemia Vera is a ‘real-life’ observational study which, by February 2016, included 1353 patients for whom baseline characteristics, therapies and complications during follow up are periodically up-dated. From this cohort, a total of 533 patients treated with HU with available data regarding hematologic values, PHL requirements, and HU dose were included for the present study. All patients included in the study were diagnosed after year 2000. In every case, the diagnosis of PV was reassessed using the criteria of the World Health Organization.9 The indication of HU was decided according to the criterion of the attending hematologist on the basis of the clinical guidelines and prevailing recommendations at that time. The treatment objective was to achieve Hct control below 45% without the need for PHL to achieve this, HU dose titration was performed according to individual clinical practice and patient characteristics. Supplemental PHL were performed in those patients in whom the Hct was not controlled with Hu alone. In general, the policy of the different centers was to increase the HU dose to achieve Hct control reserving PHL as a complementary therapy. The study was approved by the Ethics Committee of the Hospital del Mar, Spain. Informed consent for the inclusion in the registry and the scientific use of the patients’ clinicohematologic data was obtained in accordance with the requirements of the local ethics committees.
Data from the first 60 months of therapy with HU were retrospectively recovered. Hematocrit, leukocyte count, platelet count, number of PHL, and dose of HU were assessed at months 6, 12, 18, 24, 36, 48 and 60 of HU therapy. The requirement of PHL in each patient was calculated as the total number of PHL/time of follow up and expressed as the number of PHL per year.
Hematocrit response was defined as Hct less than 45%, regardless of any prior use of PHL. Complete hematologic response (CHR) was defined as the presence of Hct less than 45%, leukocyte count less than 10×109/L and platelet count less than 400×109. Months in Hct response or in CHR were calculated taken into consideration the response status at the different time points. Time in response was calculated as months in response / total months of follow up ×100 and expressed as percentage of follow up in response. Sustained response was defined as a response lasting more than 50% of the follow-up period. Intermittent response was defined as a response lasting less than 50% of follow-up period. The occurrence of resistance/intolerance to HU was recorded in those patients fulfilling at least one of the European LeukemiaNet (ELN) criteria.10 Different definitions of PHL requirement (> 2 PHL per year, > 3 PHL per year, and > 4 PHL per year) were explored for any possible association with the rate of thrombosis. Requirement of 3 or more PHL was selected to categorize the study groups due to its prognostic value and for its clinical relevance, since patients requiring 3 or more PHL per year require PHL at the majority of visits.
The primary outcome of the study was time to first thrombotic event from HU start. Study duration was 60 months after HU start. Patients were censored at last visit, at time of HU discontinuation, or at 60 months if they completed the study period. Secondary end points included probability of bleeding (major or minor) while on treatment with HU, Hct response, CHR, and probability of resistance/intolerance to HU. Thrombosis was defined according to the International Classification of Diseases (9th revision) including superficial thrombophlebitis. Severe hemorrhage was defined as a symptomatic bleeding in a critical organ or an overt hemorrhage requiring transfusion or associated with an Hb decrease of more than 20 g/L without transfusion.
Time-to-event curves were drawn up by the Kaplan-Meier method with the log-rank test for comparisons. Multivariate analysis was performed by Cox regression. All statistical analyses were performed with SPSS, v.22.
Results
Patients’ characteristics, treatment and response
Baseline characteristics at time of HU start are shown in Table 1. Median interval between PV diagnosis and HU start was 35 days, with 75% of patients starting on HU within the first year after diagnosis. Reasons for initiating HU were: age over 60 years n=334 (62.7%), previous thrombosis n=81 (15.2%), extreme thrombocytosis n=45 (8.4%), microvascular symptoms n=26 (4.9%), bleeding n=5 (0.9%), other n=20 (3.8%), not determined n=22 (4.1%). Median follow up under HU therapy was 36 months (range 6–60 months). Median starting dose of HU was 5 g per week (range 1–14 g). Complete data regarding antiplatelet and anticoagulant therapy were available in 448 and 516 patients, respectively, with 358 (80%) patients having received antiplatelet therapy and 48 (9%) anticoagulation therapy while on treatment with HU. There were no statistically significant differences in the proportion of patients receiving antiplatelet therapy or oral anticoagulants among the two study groups.
Table 1.
Main clinical and hematologic characteristics at time of hydroxyurea start in 533 patients with polycythemia vera.
The majority of patients achieved a stable Hct less than 45% during the study period (Figure 1A). Overall, 304 (57%) patients required one or more PHL at any time during the study. A total of 85 (16%) patients received 3 or more PHL per year (median 4, range 3–23). The remaining patients were included in the HU with 0–2 PHL per year group, in which the PHL requirements were significantly lower (median number of PHL per year 0, range 0–2). PHL requirements during follow up in the two study groups are shown in Table 2. The group of patients requiring 3 or more PHL per year had an inadequate Hct control, with the Hct levels being significantly higher than those requiring 0–2 PHL per year (Figure 1B). Leukocyte counts were slightly higher in the HU with 3 and more PHL patients, but the differences were not statistically significant (Figure 1C). Regarding platelet counts, both groups of patients presented similar values, mostly within the normal range (Figure 1D).
Figure 1.
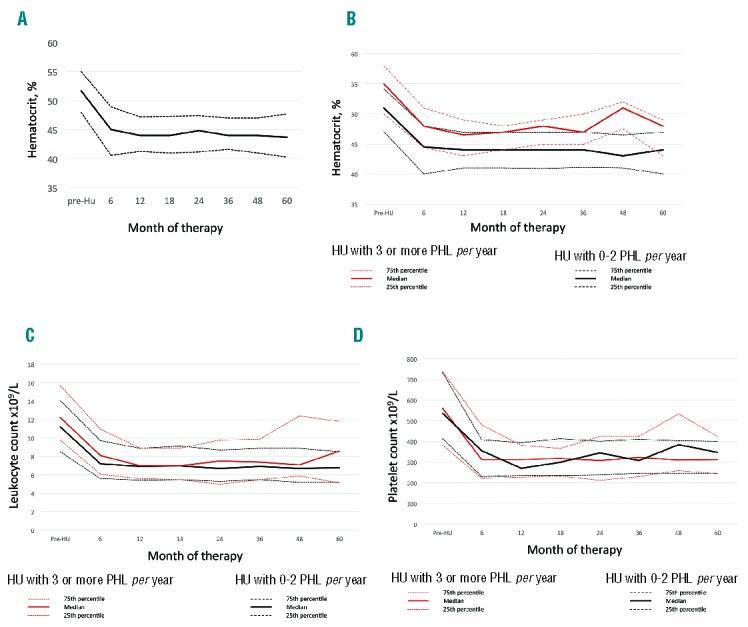
Hematocrit, leukocyte and platelet counts under hydroxyurea (HU) therapy. (A) Hematocrit in the whole cohort of patients. (B) Hematocrit in the two study groups (month 6, P<0.001; month 12, P=0.003; month 18, P=0.03; month 24, P=0.007; month 36, P=0.007; month 48, P<0.0001; month 60, P=0.1). (C) Leukocyte count in the two study groups (P=not significant). (D) Platelet count in the two study groups (P=not significant). 25th, 50th (median) and 75th percentiles are shown. HU: hydroxyurea; PHL: phlebotomies.
Table 2.
Frequency of hematocrit response, complete hematologic response, number of phlebotomies and hydroxyurea dose at different time points of treatment according to phlebotomy requirement in polycythemia vera.
Hematocrit response and CHR at any time point was achieved in 69% and 55% of patients, respectively. However, only 51% and 34% of the total patients had a sustained response in hematocrit and CHR, respectively. Hematocrit response and CHR at different time points in the two study groups are shown in Table 2. The proportion of patients achieving either Hct response or CHR was significantly lower in the group of patients requiring HU and 3 or more PHL per year. Patients requiring 3 or more PHL per year were treated with significantly higher doses of HU than the remainder. There was a trend to a progressive increase of the HU dose through follow up in patients with frequent PHL requirements (Table 2).
A total of 108 (20%) patients stopped HU during the study period. Reported reasons for discontinuation were: toxicity n=63, absence of response n=11, bleeding n=4, myeloid transformation n=1, chemotherapy for second neoplasia n= 4, other n=13, not available n=12. Resistance/intolerance to HU according to ELN criteria was observed in 51 (10%) patients. The proportion of patients fulfilling each of the definition criteria were: need for PHL despite 2 g/day of HU n=8 (1.5%), uncontrolled myeloproliferation n=3 (0.6%), failure to reduce massive splenomegaly n=0, cytopenia at the lowest dose of HU to achieve a response n=6 (1.1%), extrahematologic toxicity n=35 (6.6%). The 3-year probability of resistance/intolerance to HU was significantly higher in patients requiring 3 or more PHL per year than in those with 0–2 PHL per year (18.7% vs. 7.1%; P=0.001) (Figure 2).
Figure 2.
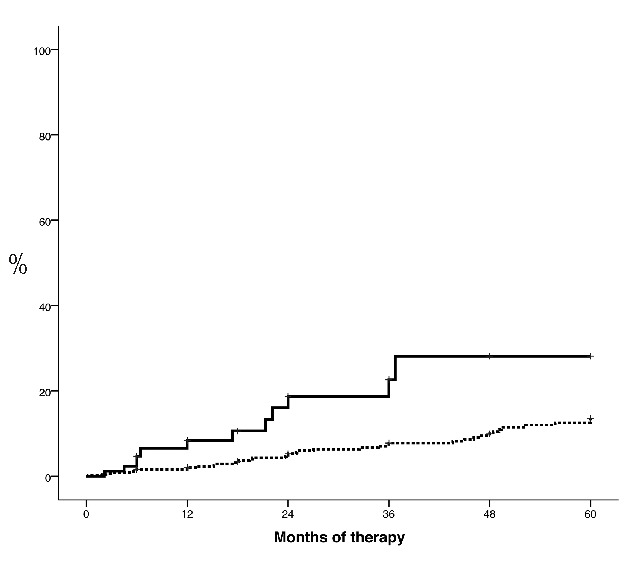
Time to resistance/intolerance to hydroxyurea according to European LeukemiaNet (ELN) criteria in patients with polycythemia vera treated with hydroxyurea (HU) and 3 or more phlebotomies per year (solid line) or with HU and 0–2 phlebotomies per year (dotted line). P=0.0001.
Thrombosis and bleeding
A total of 36 thrombotic events (22 arterial, 14 venous) were recorded resulting in a 3- and 5-year probability of thrombosis of 6.9% and 11%, respectively. Type of thrombotic events according to study groups are shown in Table 3. The probability of thrombosis was significantly higher in patients treated with HU and 3 or more PHL per year than in those treated with HU and 0–2 PHL per year (20.5% vs. 5.3% at 3 years; P<0.0001) (Figure 3). Hematocrit response and CHR status at month 6, 12, 18, 24, 36, 48 or 60 was not associated with a different rate of thrombosis. Patients with sustained hematocrit response or sustained CHR experienced similar rate of thrombosis than those with intermittent or absence of response. Other variables associated with a higher or a tendency towards a higher probability of thrombosis were: male sex (P=0.05), presence of either diabetes mellitus, active smoking, arterial hypertension or hypercholesterolemia (P=0.06), thrombosis prior to PV diagnosis (P=0.07), thrombosis at PV diagnosis (P<0.0001), and leukocyte count at time of HU start more than 10×109/L (P=0.09). Age and platelet count at time of HU start were not associated with a higher probability of thrombosis.
Table 3.
Type of thrombotic events under therapy with hydroxyurea according to phlebotomy requirements.
Figure 3.
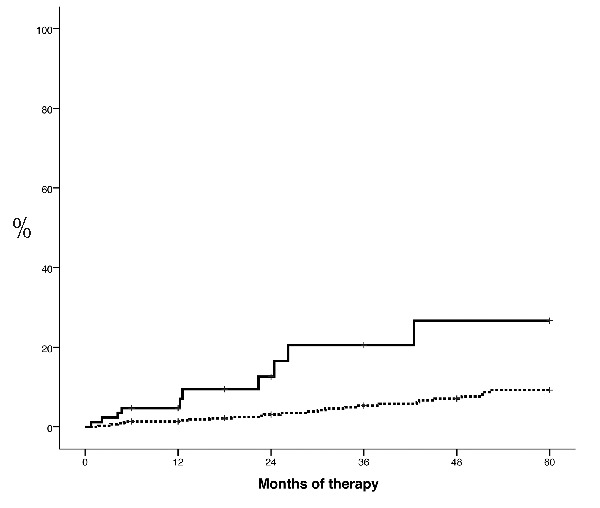
Time to thrombosis in patients with polycythemia vera treated with hydroxyurea (HU) and 3 or more phlebotomies per year (solid line) or with HU and 0–2 phlebotomies per year (dotted line). P<0.0001.
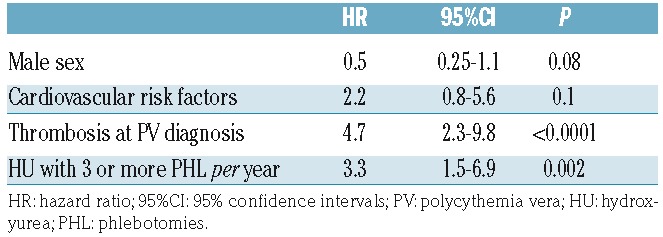
Multivariate analysis including sex, cardiovascular risk factors, thrombosis at PV diagnosis and need for PHL is shown in Table 4. Patients treated with HU and 3 or more PHL per year had a 3.3 fold increase (95% CI: 1.5–6.9) in the risk of thrombosis. In addition, patients with thrombosis at PV diagnosis showed the highest risk of developing thrombosis under HU therapy. When sustained hematocrit response or sustained CHR were included in the multivariate model, PHL requirement retained its prognostic value while hematocrit response or CHR were not associated with the risk of thrombosis.
Table 4.
Multivariate analysis of factors predicting thrombosis in 533 patients with polycythemia vera treated with hydroxyurea.
Twenty-five bleeding events (6 major, 19 minor) were registered, resulting in a 3- and 5-year probability of 4.4% and 6.7%, respectively. The 3-year probability of bleeding was higher in the group of patients requiring 3 or more PHL per year than in the remaining patients, but the difference was not statistically significant (7.4% vs. 4.2%, respectively; P=0.4) (Figure 4). In multivariate analysis, therapy with HU and 3 or more PHL per year was not associated with a higher risk of bleeding (HR: 5.5, 95% CI: 0.5–5.1; P=0.4) after adjusting for treatment with antiplatelet agents or oral anticoagulants.
Figure 4.
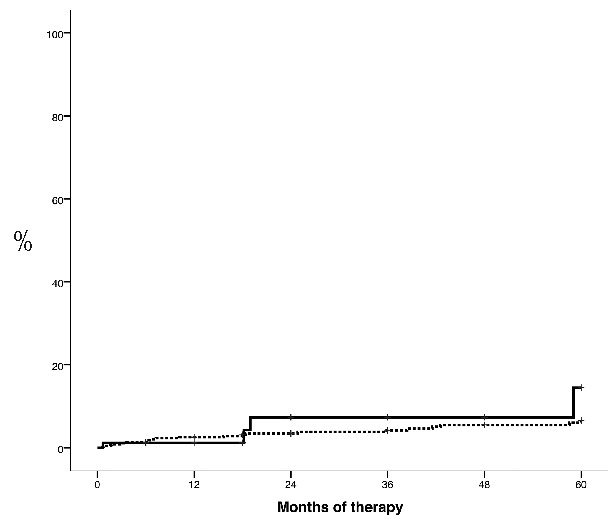
Time to bleeding (major or minor) in patients with polycythemia vera treated with hydroxyurea (HU) and 3 or more phlebotomies per year (solid line) or with HU and 0–2 phlebotomies per year (dotted line) (P=0.4).
Discussion
Hydroxyurea is the cytoreductive therapy most often used in PV. However, few studies have evaluated in detail the optimal management of this agent in clinical practice. In particular, no studies have examined whether the need of PHL under treatment with HU has any impact on the major complications of the disease. In the present study, we have shown that patients with PV treated with HU requiring 3 or more PHL per year have an increased risk of thrombosis and more frequently develop resistance/intolerance to HU.
According to current recommendations, PV patients under cytoreduction are targeted to maintain the Hct less than 45%.7,11 In this regard, most patients in the present study were able to keep the hematocrit below 45%, with the values observed being superimposable on to those reported in the higher intensity group of the CytoPV study.8 Moreover, our cohort of patients received HU dosages that were comparable or even higher than those previously reported by others.8,12 However, we identified a subgroup of patients requiring a higher intensity of treatment consisting of both higher HU doses and higher number of PHL. These patients, representing 16% of the total series, experienced a higher rate of thrombosis and, on multivariate analysis, PHL requirement while on HU therapy was an independent risk factor for thrombosis. This finding emphasizes the importance of an adequate HU dose adjustment to maintain the Hct below 45% without significant fluctuations. Alternatively, if HU dose cannot be increased, more frequent PHL or change to second-line therapy is advised.
It could be argued that the need for PHL could result from a lower intensity of cytoreductive treatment. In our series, however, the situation was just the opposite since patients with high PHL requirements received significantly higher doses of HU than those treated with HU alone. This finding suggests that this subgroup of patients have a disease with an increased proliferative capacity requiring a higher treatment intensity to achieve Hct control. This is supported by the observation that the hematocrit values prior to HU start were also significantly higher in the group of patients treated with HU and 3 or more PHL per year. An alternative explanation could be a lower individual sensitivity to treatment with HU that could result in poorer control of the disease when conventional doses of HU are used.
Although up to 16% of patients required 3 or more PHL per year to control the disease, the majority of these patients could not be classified as resistant to HU since the dose intensity of 2 g per day was not reached. In fact, only 1.5% of patients met the Hct resistance criterion defined by ELN as reported in previous studies.12,13 Nevertheless, a higher rate of resistance/intolerance to HU was observed in patients requiring 3 or more PHL per year mainly due to more frequent extrahematologic toxicity; this could be explained by the higher HU doses employed in the group requiring supplemental PHL. These features illustrate how difficult it is to apply ELN resistance criteria in daily clinical practice. In this regard, the concept of maximum tolerated dose of HU to maintain the Hct below 45% instead of a dose 2 g or more per day may be more appropriate when evaluating whether a patient is resistant to HU.14
An intriguing finding of the present work was the absence of a clear association between the Hct response and the risk of thrombosis. This does not in any way mean that patients should not be controlled according to the well-established criteria of response. In fact, patients treated with HU plus 3 or more PHL per year had poorer hematocrit control throughout the study and, therefore, a lower response rate. However, classification of patients based on the PHL requirement seems to better discriminate those patients at high risk of thrombosis than the categorization of responders/non-responders. Although our results suggest that the thrombotic risk of patients requiring frequent PHL is related to an inadequate Hct control, a detrimental effect of frequent phlebotomies PHL should still be taken into consideration. On the other hand, we have observed no significant differences in the leukocyte and platelet counts during treatment with HU between the two study groups, suggesting a predominant role of the increased red cell mass in the thrombotic risk of PV.
The main limitation of the present study is its retrospective design. The absence of a protocol that includes a standardized titration of the HU dose or the indication for PHL can result in significant bias that could affect the validity of the observed findings. On the other hand, the definition of study groups according to requirement of 3 or more PHL per year may also be criticized. The only way to solve these biases would be to conduct a prospective study with a pre-established definition of study groups, including a precise protocol of both dose titration and indication for PHL. Despite the aforementioned limitations, the detailed analysis of the treatment received by patients included in registries such as the present one is an excellent opportunity to evaluate and improve clinical practice in a ‘real-life’ scenario, which is often difficult to carry out within clinical trials.
In conclusion, PV patients treated with HU requiring 3 or more PHL per year have a higher risk of thrombotic complications. These findings highlight the importance of timely dose adjustment adapted to the proliferative activity of the disease.
Supplementary Material
Acknowledgments
We are indebted to all members of GEMFIN participating in the Spanish Registry of Polycythemia Vera.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/1/103
Funding
This work was supported by a grant from the Instituto de Salud Carlos III, Spanish Health Ministry, PI13/00557, PI1300393. The GEMFIN received a grant from Novartis for the development of the Spanish Registry of Polycythemia Vera and for conducting the present project.
References
- 1.Gruppo Italiano Studio Policitemia. Polycythemia vera: the natural history of 1213 patients followed for 20 years. Gruppo Italiano Studio Policitemia. Ann Intern Med. 1995;123(9):656–664. [DOI] [PubMed] [Google Scholar]
- 2.Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224–2232. [DOI] [PubMed] [Google Scholar]
- 3.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005; 434(7037): 1144–1148. [DOI] [PubMed] [Google Scholar]
- 4.Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous hematocrit in primary proliferative polycythaemia. Lancet. 1978;2(8102):1219–1222. [DOI] [PubMed] [Google Scholar]
- 5.Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood. 2007;109(6):2446–2452. [DOI] [PubMed] [Google Scholar]
- 6.Cervantes F, Arellano-Rodrigo E, Alvarez-Larrán A. Blood cell activation in myeloproliferative neoplasms. Haematologica. 2009; 94(11):1484–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33. [DOI] [PubMed] [Google Scholar]
- 9.Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110(4):1092–1097. [DOI] [PubMed] [Google Scholar]
- 10.Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol. 2010; 148(6):961–963. [DOI] [PubMed] [Google Scholar]
- 11.Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood. 2002;100(13):4272–4290. [DOI] [PubMed] [Google Scholar]
- 12.Alvarez-Larrán A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119(6):1363–1369. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez-Larrán A, Kerguelen A, Hernández-Boluda JC, et al. Frequency and prognostic value of resistance/intolerance to hydroxyurea in 890 patients with polycythemia vera. Br J Haematol. 2016;172 (5):786–793. [DOI] [PubMed] [Google Scholar]
- 14.McMullin MF, Wilkins BS, Harrison CN. Management of polycythaemia vera: a critical review of current data. Br J Haematol. 2016;172(3):337–349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.