

Abstract
Fusion genes involving ZNF384 have recently been identified in B-cell precursor acute lymphoblastic leukemia, and 7 fusion partners have been reported. We further characterized this type of fusion gene by whole transcriptome sequencing and/or polymerase chain reaction. In addition to previously reported genes, we identified BMP2K as a novel fusion partner for ZNF384. Including the EP300-ZNF384 that we reported recently, the total frequency of ZNF384-related fusion genes was 4.1% in 291 B-cell precursor acute lymphoblastic leukemia patients enrolled in a single clinical trial, and TCF3-ZNF384 was the most recurrent, with a frequency of 2.4%. The characteristic immunophenotype of weak CD10 and aberrant CD13 and/or CD33 expression was revealed to be a common feature of the leukemic cells harboring ZNF384-related fusion genes. The signature gene expression profile in TCF3-ZNF384-positive patients was enriched in hematopoietic stem cell features and related to that of EP300-ZNF384-positive patients, but was significantly distinct from that of TCF3-PBX1-positive and ZNF384-fusion-negative patients. However, clinical features of TCF3-ZNF384-positive patients are markedly different from those of EP300-ZNF384-positive patients, exhibiting higher cell counts and a younger age at presentation. TCF3-ZNF384-positive patients revealed a significantly poorer steroid response and a higher frequency of relapse, and the additional activating mutations in RAS signaling pathway genes were detected by whole exome analysis in some of the cases. Our observations indicate that ZNF384-related fusion genes consist of a distinct subgroup of B-cell precursor acute lymphoblastic leukemia with a characteristic immunophenotype, while the clinical features depend on the functional properties of individual fusion partners.
Introduction
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is a heterogeneous disease that can be subdivided according to primary recurrent genetic abnormalities that are strongly associated with characteristic biological and clinical features.1 The detection of these abnormalities can facilitate diagnosis, risk stratification, and targeted therapy. In approximately two-thirds of pediatric patients with BCP-ALL, well-characterized genetic abnormalities, including t(9;22) BCR-ABL1, the rearrangement of MLL at 11q23, t(1;19) TCF3-PBX1, t(17;19) TCF3-HLF, t(12;21) ETV6-RUNX1, hyperdiploidy, and hypodiploidy, can be detected by standard genetic analyses.1 In the remaining BCP-ALL patients, major pathogenic or driver cytogenetic abnormalities have yet to be clarified, and they are called “B-others”.
Recent studies using advanced analytical approaches have stratified a variety of subgroups harboring novel genetic abnormalities. For example, a high-risk subtype in B-others with a number of fusion genes involving tyrosine kinases, so-called “Ph-like acute lymphoblastic leukemia (Ph-like ALL)”, was recently discovered.2–3 Besides Ph-like ALL, other subgroups such as iAMP21, dic(9;20), and patients with PAX5 abnormalities have also been identified in B-others.4–7 However, a certain portion of B-others still remain genetically unclassified.
The zinc-finger protein 384 (ZNF384) gene encodes a transcription factor that regulates promoters of the extracellular matrix genes8 and is known to be involved in ALL through fusion with the TET family genes, such as the Ewing sarcoma breakpoint region 1 gene {EWSR1, t(12;22)}, TATA box binding protein-associated factor {TAF15, t(12;17)}, and transcription factor 3 {TCF3 or E2A, t(12;19)}.9–10 In addition to those previously reported ZNF384-related fusion genes, we identified EP300-ZNF384, t(12;22), as a novel recurrent fusion gene with an incidence of approximately 1% in B-others in a Japanese cohort.11 The recurrence of EP300-ZNF384 in pediatric BCP-ALL patients as well as a higher incidence in adolescent and young adult (AYA) BCP-ALL patients was confirmed by other groups.12–15 Furthermore, the CREB binding protein gene {CREBBP, t(12;16)(p13;p13)} has been most recently identified as another novel fusion partner for ZNF384.13 So far, however, patients harboring ZNF384-related fusion genes have been considered to be a minor subgroup in pediatric BCP-ALL.8–15 The BCP-ALL patients with EP300-ZNF384 possess a characteristic immunophenotype of weak CD10 and aberrant CD13 and/or CD33 expression.11 Although certain immunophenotypic features were still observed in some of the remaining B-others patients, cytogenetic abnormalities in these cases have yet to be clarified.
In order to further investigate unknown cytogenetic alterations in the remaining B-others, we employed whole transcriptome sequencing (WTS). Herein, we report the identification of an unexpectedly high incidence of fusion genes involving ZNF384 genes in BCP-ALL in our cohort. The immunophenotypic and gene-expression characteristics, accompanying genetic abnormalities as well as the clinical features of BCP-ALL harboring the ZNF384-related fusion gene are evaluated and discussed.
Methods
Patient selection
Following previous publication,11 91 ribonucleic acid (RNA) samples obtained from pediatric BCP-ALL patients classified as B-others were selected from the Tokyo Children’s Cancer Study Group (TCCSG) biobank as a discovery cohort for WTS to screen for unknown fusion genes. We then extended analysis to another 122 B-others’ RNA samples that were available in our biobank as a validation cohort by employing reverse transcription polymerase chain reaction (RT-PCR) (detailed information is presented in Figure 1, Online Supplementary Information and Online Supplementary Table S1).
Figure 1.

Flow chart of the analysis of patients. The respective numbers of patients and cohorts that were investigated are presented in a hierarchical fashion. See also the Online Supplementary Information and Online Supplementary Table S1. ALL: acute lymphoblastic leukemia; BCP-ALL: B-cell precursor ALL; B-others: BCP-ALL without conventional genetic abnormalities; RNA: ribonucleic acid; RNA-seq-Ex1 and Ex2: RNA-sequencing experiment 1 and 2; RT-PCR-Ex1 and Ex2: reverse transcription polymerase chain reaction experiment 1 and 2; Ex: experiment.
Diagnoses were made on the basis of the morphology and routine examinations, including the immunophenotype, cytogenetic analysis, DNA content analysis, and the RT-PCR detection of 7 conventional fusion transcripts, as described previously.16 The investigations were approved by the institutional review boards of all participating institutions. Informed consent was obtained from parents or guardians, and informed assent was obtained from the patients when appropriate, based on their age and level of understanding. In the TCCSG study, we first assigned patients to standard- (SR), intermediate- (IR) and high-risk (HR) groups based on their age and leukocyte count, and risk assessment was revised on day 8 based on the response of the patients to steroid monotherapy.17
Whole transcriptome sequencing and RT-PCR
Using total RNA extracted from the bone marrow or peripheral blood samples of the patients, WTS was performed and the fusion transcripts were investigated by employing deFuse, an algorithm for gene fusion discovery, as described in the Online Supplementary Information. For the confirmation of the whole transcriptome screening results and screening for additional cases in the validation cohort, RT-PCR was performed as described previously,18 using the primers listed in the Online Supplementary Table S2, followed by Sanger sequencing. The details of the data analyses of WTS to identify genetic mutations and deletions are also described in the Online Supplementary Information.
Whole exome sequencing (WES)
Exome libraries were prepared from 100 ng of genomic DNA using the Nextera Rapid Capture Exome Kit (Illumina, Inc., San Diego, CA, USA), and sequenced using SBS v4 reagents with the HiSeq2500 sequencing system (Illumina). More than 10 Gb of sequence was obtained for each library by paired-end sequencing (126 bp ×2). The details of the whole exome data analyses are described in the Online Supplementary Information.
Microarray and data analyses
The complementary DNAs (cDNAs) were amplified and labeled using the Ovation® Pico WTA System and Encore™ Biotin Module (NuGEN Technologies Inc., San Carlos, CA, USA), as instructed by the manufacturer. The labeled probes were hybridized to Human Genome U133 Plus 2.0 Arrays (Affymetrix, Santa Clara, CA, USA). The arrays were analyzed using GeneChip Operating Software 1.2 (Affymetrix, Santa Clara, CA, USA) and GeneSpring GX 13.1 software (Agilent Technologies, Santa Clara, CA, USA), and the details of data analyses, including gene set enrichment analysis (GSEA), are described in the Online Supplementary Information.
Statistical analysis
An unpaired t-test with Welch’s correction was performed for the positivity of cell surface antigens.
Mutual univariable analysis of characteristics were conducted using the Fisher’s exact test or χ2 test for qualitative variables. Overall survival (OS) and event-free survival (EFS) were estimated by the Kaplan-Meier method and compared with the log-rank test. Analyses were performed with the use of Prism software, version 6.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Detection of the fusion genes involving ZNF384 in pediatric BCP-ALL patients
The 213 selected RNA samples from B-others patients were analyzed by WTS and/or RT-PCR followed by Sanger sequencing (Figure 1 and Online Supplementary Tables S1 and S3). We identified a total of 15, 3 and 3 patients with TCF3-ZNF384, TAF15-ZNF384 and CREBBP-ZNF384, respectively. Three fusion genes were reported previously.9,10,12–15,19–22 In addition, we identified a novel ZNF384-related fusion gene, namely BMP2K-ZNF384 (1 patient). Although we recently reported 6 patients with EP300-ZNF384,11 another 3 patients with this fusion gene were further identified in this study (Figure 1 and Online Supplementary Tables S1 and S4).
Structure of the fusion genes involving ZNF384
The structure and sequences of the fusion points of each fusion gene as well as a schematic representation of the predicted fusion proteins are depicted in Figure 2. In 3 out of 15 TCF3-ZNF384 patients, the fusion point of ZNF384 was the same as those previously reported, and exon 11 of TCF3 was fused to exon 3 of ZNF384 (presented in Figure 2A as Type (a)).10 In addition to this known fusion point, we identified several novel fusion points of this fusion gene. Most frequently, for example, exon 13 of TCF3 was fused to exon 3 of ZNF384 with in-frame joining in 7 patients (Figure 2A Type (c)). The predicted protein from all of the fusions does not have the principal functional domains of wild-type TCF3, whereas it retains almost all of the part of the ZNF384 protein.
Figure 2.

Structure of the ZNF384-related fusions. Structures of fusion proteins and nucleotide sequences of fusion points of (A) TCF3-ZNF384, (B) TAF15-ZNF384, (C) CREBBP-ZNF384, and (D) BMP2K-ZNF384 are presented. Exon numbers and boundaries are marked below the protein structures. The black arrowhead shows the donor site fusion point. The white arrowhead shows the acceptor site fusion point. Five and four different in-frame fusions for TCF3-ZNF384 and CREBBP-ZNF384, respectively, are depicted with the nucleotide sequence, predicted amino acids, and chromatogram. The patients in whom a particular fusion isoform was found are indicated on the right. Ex: exon; TAZ2: transcription adaptor putative zinc finger; KIX: kinase-inducible domain interacting; RING: really interesting new gene; PHD: plant homeodomain; CREB: cyclic adenosine monophosphate (AMP) response element binding protein; RNA: ribonucleic acid; ZN: zinc finger; RAN: Ras-related nuclear proteins; BMP-2: bone morphogenetic protein 2.
The structure and sequences of the fusion point of TAF15-ZNF384 are depicted in Figure 2B. Although several isoforms have been reported in this fusion gene,16 only one isoform joining exon 6 of TAF15 to exon 3 of ZNF384 was observed in 3 of our patients.
In the case of CREBBP-ZNF384, we identified 5 isoforms in 3 patients (Figure 2C). The exons 4, 5, and 6 of CREBBP fused in-frame to exon 3 or 2 of ZNF384 (Figure 2C Type (a–d)). Among all of the fusion points, most functional domains of CREBBP were lacking in the resulting fusion proteins, while the complete ZNF384 protein was retained. In contrast, exon 7 of CREBBP was fused out-of-frame to exon 3 of ZNF384 in 1 patient (Figure 2C Type (e)).
In the case of BMP2K-ZNF384, 2 isoforms were identified in one patient and nucleotide 2228 (exon 15) and nucleotide 2117 (exon 14) of BMP2K were fused in-frame to nucleotide 266 (exon 3) of ZNF384 (Figure 2D, Type (a, b)), and the predicted protein retained the kinase domain of BMP2K but lacked the c-terminus portion. The 3 patients with EP300-ZNF384 exhibited identical split sequences, as we reported previously (data not shown).11
Frequency of ZNF384-related fusion gene-positive patients
A consecutive series of 333 ALL patients enrolled in the TCCSG L0416/0616 study11 included 130 B-others patients and 161 BCP-ALL patients with conventional genetic abnormalities (291 BCP-ALL patients, Figure 1 and Online Supplementary Table S1). Since the majority of these B-others patients of the L0416/0616 cohort (121 patients, 93.1% of B-others) were studied in the present analysis, we estimated the frequency of ZNF384-related fusion genes in childhood ALL based on this cohort. As we reported previously, 3 patients with EP300-ZNF384 were included in the L0416/0616 cohort.11 In the study herein, we identified another 9 patients with ZNF384-related fusion genes in this cohort. Therefore, the frequency of the total of 12 patients with ZNF384-related fusion genes in B-others was estimated as 9.2% (4.1% in BCP-ALL, Figure 1 and Online Supplementary Table S1). Of those fusion genes, TCF3-ZNF384 was the most recurrent (7 patients), with a frequency of 5.4% in B-others (2.4% in BCP-ALL).
Immunophenotypic characteristics of BCP-ALL patients with ZNF384-related fusion genes
As we reported previously, all BCP-ALL patients with EP300-ZNF384 showed a dull or negative expression of CD10 and aberrant expression of one or more myeloid antigens based on immunophenotypic examination.11,13 Similar to EP300-ZNF384-positive patients, all of the other BCP-ALL patients with ZNF384-related fusion genes were found to have a weak or negative expression of CD10, ranging from 0.39% to 67.34% (mean: 19.44±18.23%, Figure 3 and Online Supplementary Table S5), and there was a significant difference compared with TCF3-PBX1-positive patients and B-others in which ZNF384-related fusion genes were not identified (wild-type ZNF384), for the most part strongly expressing CD10 (mean: 98.66±0.89%, P<0.001 and mean: 77.71±25.79%, P<0.001, respectively). In addition, 7 and 17 patients out of 22 patients with ZNF384-related fusion genes (31.82% and 77.27%, respectively) exhibited more than 20% expression of CD13 (mean: 10.70±10.37%) and CD33 (mean: 59.86±34.01%), significantly distinct from BCP-ALL patients with TCF3-PBX1 and wild-type ZNF384 expressing neither CD13 (mean: 0.26±0.33%, P<0.001 and mean: 2.76±7.9%, P<0.0015, respectively), nor CD33 (mean: 0.59±0.76%, P<0.001 and mean: 6.63±1.72%, P<0.0001, respectively).
Figure 3.

Immunophenotypic characteristics of B-cell precursor acute lymphoblastic leukemia (BCP-ALL) patients with ZNF384-related fusion genes. (A) Typical histograms of CD19, CD10, aberrant myeloid antigens (CD13 and CD33), and CD66c of ZNF384-related fusion gene-positive patients and TCF3-PBX1 patients are indicated with a positive rate (%). X-axis, fluorescence intensity; Y-axis, relative cell number. (B) The positivity (percentage) of CD10, 13, and 33 of ZNF384-related fusion gene-positive BCP-ALL patients (22 cases, excepting EP300-ZNF384) and TCF3-PBX1-positive or wild-type ZNF384 BCP-ALL patients was plotted on a scattergram. The detailed list of positivity for each immunophenotypic marker of the patients is presented in the Online Supplementary Table S5. ZNF384-WT indicates cases without ZNF384 rearrangement.
Additional genetic abnormalities in ZNF384-related fusion gene-positive patients
To detect the additional genetic abnormalities in BCP-ALL with ZNF384-related fusion genes, we performed multiplex ligation-dependent probe amplification (MLPA) analysis. However, MLPA analysis did not reveal that ZNF384-translocated cases had deleted or amplified regions in IKZF1, PAX5, ETV6, BTG1, EBF1, CRLF2, and ERG, and only 2 patients with CDKN2A/B deletion and 1 patient with RB1 deletion out of 15 were detected (Table 1 and Online Supplementary Figure S1).
Table 1A.
Clinical characteristics of acute lymphoblastic leukemia (ALL) with ZNF384-related fusion genes.

Table 1B.
Additional genetic characteristics of acute lymphoblastic leukemia (ALL) with ZNF384-related fusion genes.

We preliminarily identified 126 genes as candidates for recurrent genetic abnormalities occurring in BCP-ALL by performing somatic point mutation calling from matched leukemia-normal patient samples based on the results from WES data from 100 patients with pediatric BCP-ALL. We therefore subjected 7 DNA samples of patients with ZNF384-related fusion genes to WES to filtrate the 126 genes by multi-sample calling for single-nucleotide variants and short insertions/deletions (Online Supplementary Information). Together with the data of WTS performed for 9 patients (6 patients were subjected to both WES and WTS), we analyzed them to identify somatic mutations impacting coding sequences. As a consequence, we identified 7 recurrently mutated genes which had been recognized earlier as abnormalities involved in leukemia, including NRAS, KRAS, PTPN11, EZH2, MLL2, and ASH1L (Table 1 and Online Supplementary Figure S1). Principally, activating mutations in RAS signaling pathway genes, including NRAS, KRAS, and PTPN11, were detected in 4 out of 10 ZNF384-translocated patients subjected to WES and/or WTS. On the other hand, mutations in B-cell developmental genes, such as PAX5, VPREB1, BTG1, and IKZF1, were not detected at all in DNA samples from BCP-ALL with ZNF384-related fusion genes.
Gene expression profile of TCF3-ZNF384-positive patients
To assess the functional aspects of TCF3-ZNF384, we performed DNA microarray-based expression profiling. Hierarchical clustering analysis using the selected gene probe sets2,3 showed that the major subtypes of BCP-ALL patients with conventional genetic abnormalities separated into distinct clusters. Remarkably, ZNF384-related fusion gene positive patients were located in the same cluster and clearly separated from those of other conventional genetic subtypes (Online Supplementary Figure S2A) as well as TCF3-PBX1-positive patients (Online Supplementary Figure S2B). The data indicate that the gene expression profile of TCF3-ZNF384-positive ALL is related to EP300-ZNF384- and CREBBP-ZNF384-positive ALL, but not to other conventional genetic subtypes.
Therefore, we proceeded to directly compare the gene expression of the most frequent TCF3-ZNF384-positive ALL to that of TCF3-PBX1-positive ALL possessing the same fusion partner, and with both translocations disrupting one allele of TCF3 that drives the B-cell differentiation program. As shown in the Online Supplementary Figure S2C and Figure 4A, TCF3-ZNF384-positive patients were clearly separated in a distinct cluster from TCF3-PBX1-positive patients by hierarchical clustering, and the differential expression of 3,698 genes (up: 1,984, down: 1,714, >2.0) were identified (Online Supplementary Table S6), indicating that two TCF3-translocated subtypes were significantly distinct. Interestingly, the B-cell developmental genes VPREB1, PAX5, and IKZF1-3 were listed as more highly expressed genes in TCF3-PBX1-positive ALL than in TCF3-ZNF384-positive ALL (Online Supplementary Table S6 and Figure 4B). The transcriptional regulators, such as BACH2, HIST1H3A, and LEF,1 were also upregulated in TCF3-PBX1-positive ALL, while GATA3, ERG, NCOR1, and TOX were upregulated in TCF3-ZNF384-positive ALL. It is noteworthy that gene set enrichment analysis revealed a significant enrichment for hematopoietic stem cell (HSC) signatures in TCF3-ZNF384-positive ALL while lymphoid features, such as Pro-B cells and early T-cell precursors (ETP), were more prominent in TCF3-PBX1-positive ALL (Figure 4C and Online Supplementary Table S7). The signatures of granulocyte-macrophage progenitor (GMP)/multi-lymphoid progenitor (MLP) and megakaryocyte-erythroid progenitor (MEP) were enriched in TCF3-ZNF384- and TCF3-PBX1-positive ALL, respectively, whereas statistical significance was not observed.
Figure 4.

Characteristics of gene expression profile in TCF3-ZNF384-positive acute lymphoblastic leukemia (ALL). (A) Two-way hierarchical clustering was performed on filtered microarray probes, those up- or downregulated by 2-fold or more in TCF3-ZNF384- (red, n=10), in comparison with TCF3-PBX1-positive (blue, n=19) ALL. The results are displayed using a heat map as a dendrogram. (B) Among the genes that exhibited a 2 times higher or lower expression in either TCF3-ZNF384- and TCF3-PBX1-positive ALL (listed in the Online Supplementary Table S6), the expressions of PAX5 and VPREB genes measured by microarray analysis were plotted on a scatter diagram, and bars representing the mean±SD are presented. The data of wild-type ZNF384 B-cell precursor acute lymphoblastic leukemia (BCP-ALL) patients are also presented. (C) Gene set enrichment analysis (GSEA) for curated gene sets of hematopoietic precursors was performed on the differentially expressed genes between TCF3-ZNF384- (red) and TCF3-PBX1-positive (blue) ALL. Enrichment plots for the hematopoietic stem cells (HSCs), multi-lymphoid progenitor (MLP), pro-B cell (Pro-B), early T-cell precursors (ETP), common myeloid progenitors (CMP), granulocyte-monocyte progenitor (GMP), and megakaryocyte-erythroid progenitor cell (MEP) signatures are presented. Bold lines represent significant enrichments {false discovery rate (FDR) q-value<0.25 and/or nominal (NOM) P-value<0.05)}. NES: normalized enrichment score.
The above analyses could just as well detect the lack of specific TCF3-PBX1 features rather than the common features among cases with TCF3-ZNF384. Thus we subsequently compared the gene expression of TCF3-ZNF384-positive ALL to that of wild-type ZNF384. There is the possibility that the wild-type ZNF384 patients still include a variety of subgroups harboring novel genetic abnormalities, and some of them might be related to ZNF384-related fusion genes by their gene expression. Indeed, hierarchical clustering analysis showed that wild-type ZNF384 patients were not necessarily located in the same cluster, and some of the cases were dispersed in the clusters of the major subtypes of BCP-ALL with conventional genetic abnormalities (Online Supplementary Figure S2D), indicating the heterogeneity of wild-type ZNF384 patients. Furthermore, hierarchical clustering of ZNF384-related fusion gene positive patients and wild-type ZNF384 patients indicated that 21 out of 104 wild-type ZNF384 patients located in the same cluster as ZNF384-related fusion gene positive patients (Online Supplementary Figure S2E). However, when we compared TCF3-ZNF384-positive patients and wild-type ZNF384 patients; excepting those located in the ZNF384-related fusion gene positive cluster; they were clearly separated into a distinct cluster by hierarchical clustering (Online Supplementary Figure S2F), and the differential expression of 4,515 genes (up: 2,485, down: 2,030, >2.0) were identified (Online Supplementary Table S8). The B-cell developmental genes VPREB1 and IKZF1, but not IKZF2 or IKZF3, were listed as more highly expressed genes in wild-type ZNF384 ALL than in TCF3-ZNF384-positive ALL, and PAX5 also listed as a relatively highly expressed gene in wild-type ZNF384 ALL (1.83-fold). The transcriptional regulators such as BACH2, HIST1H3A, and LEF1 were also upregulated in wild-type ZNF384 ALL, while GATA3 and NCOR1, but not ERG and TOX, were upregulated in TCF3-ZNF384-positive ALL. The gene set enrichment analysis also revealed an enrichment for HSC, GMP and MLP signatures in TCF3-ZNF384-positive ALL while Pro-B, ETP and MEP were more prominent in wild-type ZNF384 ALL, whereas statistical significance was not obvious (Online Supplementary Figure S2G and Online Supplementary Table S7).
Clinical characteristics and outcomes of ZNF384-related fusion gene positive patients
The clinical findings and outcomes of BCP-ALL patients with ZNF384-related fusion genes, other than EP300-ZNF384, are summarized in Table 1. They were aged between 1 and 12 years (median: 3) at presentation and comprised of 12 males and 10 females. Their initial white blood cell (WBC) counts at presentation ranged from 1,220 to 150,200 (median: 26,975). The analysis of fluids obtained by lumbar puncture revealed no indication of central nervous system involvement, with the exception of one patient with TCF3-ZNF384 (Case-15). Thirteen out of 22 patients (59.1%) were classified into an IR at the initial diagnosis, based on an advanced age (4 patients), elevated WBC counts (7 patients), or both (2 patients). In addition, 5 patients were classified as HR (22.7%) based on markedly elevated WBC counts, but not an advanced age, and thus the SR included only 4 patients (18.2%). Five out of 15 (33.3%) patients with TCF3-ZNF384 had a relapse in the bone marrow, all of the patients received stem-cell transplantation, and 3 patients died. One patient each of TAF15- and CREBBP-ZNF384-positive patients had a relapse in the bone marrow, but both patients were salvaged by chemotherapy. It should be emphasized that no patient died in those with ZNF384-related fusion genes, other than TCF3-ZNF384.
Importantly, 21 out of 22 patients showed a normal karyotype on cytogenetic analysis by conventional G-banding, but the predicted chromosomal translocations, including t(12;19)(p13;p13) for TCF3-ZNF384, t(12;17)(p13;q11) for TAF15-ZNF384, t(12;16)(p13;p13) for CREBBP-ZNF384, and t(4;12)(q21;p13) for BMP2K-ZNF384 were not detected at all (Table 1). The remaining patient with CREBBP-ZNF384 (Case-19) showed 47,X,add(X)(p22),t(12;12)(p13:q13),+21 at the initial diagnosis, but this was not consistent with the predicted chromosomal translocation.
Since TCF3-ZNF384 was the most recurrent among ZNF384-related fusion genes detected in the pediatric BCP-ALL patients, we next compared the characteristics of TCF3-ZNF384-positive patients with those of wild-type ZNF384, as well as EP300-ZNF384- and TCF3-PBX1-positive patients (Table 2). When compared with wild-type ZNF384, initial WBC counts and a trend to be classified in higher risk groups (IR and HR) were significantly higher in TCF3-ZNF384-positive patients (P<0.05 and P<0.01, respectively). In contrast, the age at diagnosis was not significantly different between TCF3-ZNF384-positive patients and those with wild-type ZNF384. The response to steroid monotherapy using the cut-off of 1,000/μL for the blast count in peripheral blood on day 8 was significantly poorer in TCF3-ZNF384-positive patients than in those with wild-type ZNF384 (P<0.01).
Table 2.
Statistical comparison of clinical characteristics between TCF3-ZNF384-, EP300-ZNF384-, TCF3-PBX1-positive, and ZNF384-related fusion gene-negative B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
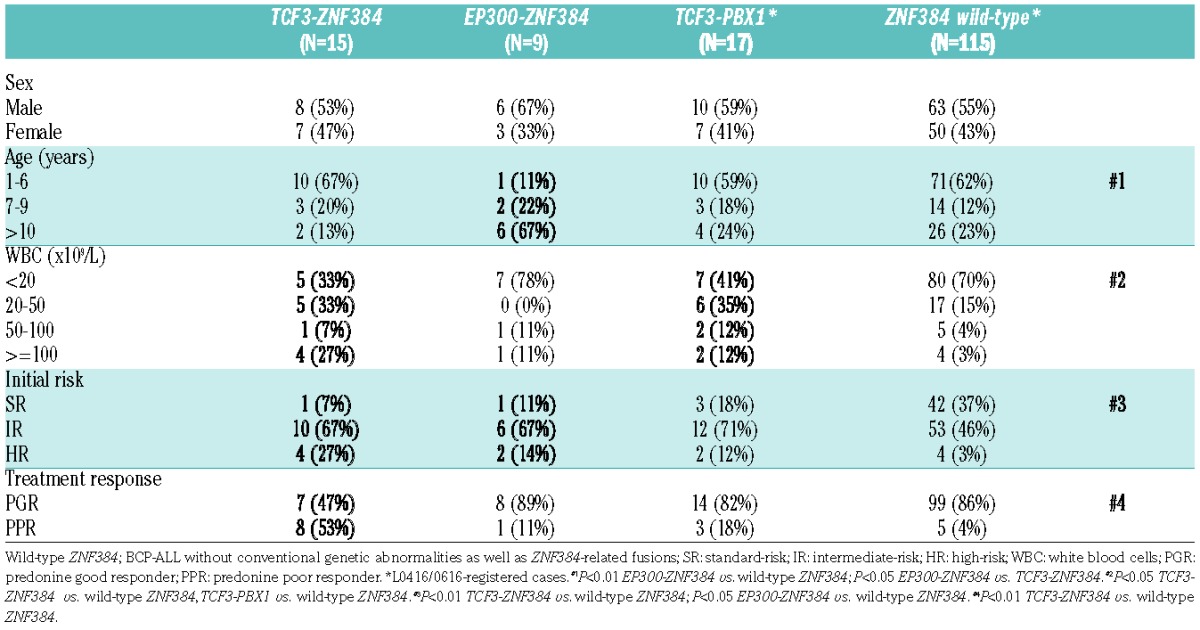
As we reported previously, BCP-ALL patients with EP300-ZNF384 exhibit an advanced age but not significantly elevated WBC counts.11 Indeed, compared to the patients with wild-type ZNF384, those with EP300-ZNF384 exhibit a significantly older age at presentation (P<0.01) but not significantly elevated WBC counts (P=0.37), as presented in Table 2. Therefore, the clinical features of TCF3-ZNF384-positive patients were clearly distinct from those of EP300-ZNF384-positive patients. In the case of TCF3-PBX1-positive ALL, initial WBC counts were significantly higher than in wild-type ZNF384, whereas other features were not significantly different.
The 5-year EFS among TCF3-ZNF384-positive patients and those with wild-type ZNF384 were 50.0% and 73.3% (P=0.35) and the 5-year OS rates were 72.2% and 89.8% (P=0.15), respectively (Figure 5). Although there was no statistic significance, the clinical outcomes of TCF3-ZNF384-positive patients were more unfavorable than in wild-type ZNF384 patients. In contrast, the 5-year EFS and the 5-year OS rates of EP300-ZNF384-positive patients were 83.3% and 100%, respectively, thus being more favorable than in TCF3-ZNF384-positive and wild-type ZNF384 patients.
Figure 5.

Outcomes of patients with ZNF384-related fusion genes. (A) Kaplan-Meier estimates of event-free survival (EFS) for patients with TCF3-ZNF384, EP300-ZNF384, and wild-type ZNF384 (log-rank P=0.35 in wild-type ZNF384 vs. TCF3-ZNF384, log-rank P=0.17 in TCF3-ZNF384 vs. EP300-ZNF384. (B) Overall survival (OS) for the same as above (log-rank P=0.15 in wild-type ZNF384 vs. TCF3-ZNF384, log-rank P=0.14 in TCF3-ZNF384 vs. EP300-ZNF384.
Discussion
We identified a novel fusion gene, BMP2K-ZNF384, in addition to the previously reported ZNF384-related fusion genes, including TCF3-, TAF15-, EWSR-, EP300-, “CREBBP-”, “SYNRG-”, and ARID1B-ZNF384, and thus 8 fusion partners for the ZNF384 gene have been identified so far.9–15,19–22 Considering the current condition, whereby the frequency of B-others in which specific cytogenetic abnormalities are not present are markedly decreasing, the incidence of ZNF384-related fusion genes in our cohort was unexpectedly high. In the literature, 8 ALL patients with TAF15-ZNF384 have been reported.9,19–21 Similarly, 2 and 3 ALL patients with EWSR1- and TCF3-ZNF384, respectively, have also been described previously.9–10,20–22 Because 6 out of 13 patients in the literature are adults (>21 years), this is the first report on the high frequency of the recurrence of ZNF384-related fusion genes in childhood BCP-ALL in a single nation. It is possible that the differences in the outbreak frequency of ZNF384-related fusion genes in BCP-ALL depending on racial differences are present, and this type of fusion might be common in Asians but not in Caucasians. Alternatively, since ZNF384-related fusion genes are difficult to detect with conventional G-banding, as we presented in this report, ZNF384-related fusion genes may be found in Caucasians at a similar frequency to that in our cohort upon using more sensitive methods such as PCR or fluorescence in situ hybridization (FISH).
As we presented herein, BCP-ALL patients with ZNF384-related fusion genes possess a characteristic immunophenotype. All of the BCP-ALL patients with ZNF384-related fusion genes, including EP300-ZNF384 that we reported recently, exhibit the dull or negative expression of CD10 and for the most part express CD13 and/or CD33. Our observations are consistent with previous reports because at least 8 among 13 patients with ZNF384-related fusion genes were CD10-negative and expressed CD13 and/or CD33 based on the literature.10,20–22 Since ZNF384 is a commonality in all of these fusion genes, the aberrant function of the ZNF384 protein may be responsible for the characteristics of the immunophenotype. The fact that BCP-ALL patients with TCF3-PBX1 are clearly positive for CD10 and have no aberrant myeloid antigens should support the above notion. A correlation between the high expression of the GATA3 gene and CD13/CD33 expression on primary BCP-ALL blasts with ZNF384-related fusion genes was suggested,13 whereas the role of ZNF384-fusion proteins in the expression characteristics of the antigen remains largely unknown. BCP-ALL harboring MLL rearrangement also reveals the negative expression of CD10 and aberrant expression of myeloid antigens, while such patients frequently express CD65 and CD15 as aberrant myeloid antigens and are mainly accompanied by NG2 (7.1) expression.23 Thus, the presence of ZNF384-related fusion genes should be predictable, based on the characteristic immunophenotype of dull or negative CD10 expression accompanied by the aberrant expression of CD13 and/or CD33.
In contrast to the similarity of the immunophenotypic characteristics, the clinical features of BCP-ALL with the ZNF384-related fusion gene are not uniform, and are thought to depend on the fusion partner gene of ZNF384. In the TCF3-ZNF384 fusion gene-positive ALL patients, more than half of the patients were classified into an IR or HR group because of an elevated WBC count at presentation, while their onset age was not advanced and there was no significant difference compared with wild-type ZNF384 patients. In addition, approximately half of the patients exhibited a poor response to prednisolone, and one-third relapsed, indicating that BCP-ALL patients with the TCF3-ZNF384 fusion gene include a certain proportion of patients with a poor response to conventional chemotherapy. It is noteworthy that stem cell transplantation is effective as salvage therapy for a part of the relapsed TCF3-ZNF384-positive patients. On the other hand, we previously reported that EP300-ZNF384 fusion gene-positive patients exhibited a relatively advanced age but no significant elevation of the WBC count at presentation, and showed a good response to prednisolone as well as a favorable response to conventional chemotherapy.11 Moreover, although one patient has relapsed, the OS rate of 9 patients with EP300-ZNF384 is currently 100%. Therefore, the clinical features are significantly different between TCF3-ZNF384-positive and EP300-ZNF384-positive patients.
As the fusion genes involving the TCF3 gene, TCF3-PBX1, TCF3-ZNF384, and TCF3-HLF are known, recent reports have indicated that the genomic and transcriptomic landscape of TCF3-HLF-positive ALL differs markedly from TCF3-PBX1-positive ALL, and that TCF3-HLF-positive ALL exhibited enriched stem cell and myeloid features, while lymphoid features were more prominent in TCF3-PBX1–positive ALL.24 Moreover, the recurrent intragenic deletions of PAX5 or VPREB1 as well as activating mutations in the RAS signaling pathway genes (NRAS, KRAS, and PTPN11) were common in TCF3-HLF-positive ALL but rare in TCF3-PBX1-positive ALL. In the case of TCF3-ZNF384-positive ALL, abnormalities in such B-cell developmental genes were also revealed to be rare. However, the gene expression analysis of TCF3-ZNF384-positive ALL revealed that PAX5 as well as VPREB1 expressions were lower compared with TCF3-PBX1-positive and wild-type ZNF384 ALL, and the gene expression profile of TCF3-ZNF384-positive ALL was markedly distinct from that of TCF3-PBX1-positive and wild-type ZNF384 ALL. In addition, the signature of TCF3-ZNF384-positive ALL exhibited enrichment of the stem cell signature, similar to that of TCF3-HLF-positive ALL. The low expression of the gene regulating the pro- to pre-B-cell transition and expression of a HSC/myeloid-signature could be features of TCF3-ZNF384-positive ALL. Our data also indicate the activating mutations in RAS signaling pathway genes and CDKN2A/B-deletion in a part of the patients with TCF3-ZNF384. Further studies involving a large series of patients would be required to elucidate the prognostic impact of these gene abnormalities in a part of TCF3-ZNF384-positive patients.
It has been shown that TAF15-, EWSR1-, and TCF3-ZNF384 induce 3T3 fibroblast transformation.9,25 Since ZNF384 is a commonality in 8 related fusion genes identified in BCP-ALL, the aberrant function of the ZNF384 protein may participate in the development of BCP-ALL, whereas the precise function of ZNF384 in cellular transformation has yet to be clarified. Furthermore, the role of the defects in the function of TET family partner molecules in the development of ALL is still controversial. On the other hand, the loss of functions of both EP300 and CREBBP has been reported to participate in tumorigenesis by means of epigenetic alterations mediated by reduced histone acetylation activities.26–29 Recent reports also described the marked involvement of CREBBP mutations in the relapse of high hyperdiploid BCP-ALL.27–28 Therefore, both the loss of function in CREBBP or EP300 and the gain of aberrant ZNF384 function could cooperatively affect leukemogenesis in ALL harboring these two fusion genes. In contrast, the role of BMP2K abnormality in leukemogenesis is unclear. Further studies to investigate the molecular basis of ZNF384-related fusion proteins are required to clarify the precise role of the abnormalities of ZNF384 as well as its fusion partner genes in leukemogenesis.
In conclusion, we clarified that ALL patients harboring ZNF384-related fusion genes constitute a subgroup with a characteristic immunophenotype in B-others of a Japanese cohort with an unexpectedly high incidence, and that the TCF3-ZNF384 fusion gene is the most recurrent. The clinical features of BCP-ALL with ZNF384-related fusion genes, however, depend on the functional defect of the fusion partner gene of ZNF384. Additional studies to elucidate the biological function of the ZNF384-related fusion protein should shed light on the molecular basis of the development of BCP-ALL.
Supplementary Material
Acknowledgments
We thank K. Itagaki, H. Yagi, Y. Katayama, A. Tamura, K. Takeda, K. Hayashi, and the staff of the Laboratory for Genotyping Development, Riken Center for Integrative Medical Sciences for their excellent data management and experimental assistance. We thank all members of the Committees of ALL and of Research and Diagnosis of TCCSG.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/1/118
Funding
This work was supported in part by a Health and Labour Sciences Research Grant (3rd-term comprehensive 10-year strategy for cancer control H22-011), the Grant of the National Center for Child Health and Development (26–20), and the Advanced Research for Medical Products Mining Programme of the National Institute of Biomedical Innovation (NIBIO, 10–41, –42, –43, –44, –45), and Biobank Japan project funded by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Japan Agency for Medical Research and Development (AMED), and the Practical Research for Innovative Cancer Control from AMED.
These funding sources played no role in the collection, analysis, or interpretation of the results, or in the writing of the manuscript and decision to submit it.
References
- 1.Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541–1552. [DOI] [PubMed] [Google Scholar]
- 2.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2): 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rieder H, Schnittger S, Bodenstein H, et al. dic(9;20): a new recurrent chromosome abnormality in adult acute lymphoblastic leukemia. Genes Chromosomes Cancer. 1995;13(1):54–61. [DOI] [PubMed] [Google Scholar]
- 5.Moorman AV, Richards SM, Robinson HM, et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21). Blood. 2007;109(6):2327–2330. [DOI] [PubMed] [Google Scholar]
- 6.Stasevich I, Inglott S, Austin N, et al. PAX5 alterations in genetically unclassified childhood Precursor B-cell acute lymphoblastic leukaemia. Br J Haematol. 2015; June 26 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Marincevic-Zuniga Y, Zachariadis V, Cavelier L, et al. PAX5-ESRRB is a recurrent fusion gene in B-cell precursor pediatric acute lymphoblastic leukemia. Haematologica. 2016;101(1):e20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bidwell JP, Torrungruang K, Alvarez M, et al. Involvement of the nuclear matrix in the control of skeletal genes: the NMP1 (YY1), NMP2 (Cbfa1), and NMP4 (Nmp4/CIZ) transcription factors. Crit Rev Eukaryot Gene Expr. 2001;11(4):279–297. [PubMed] [Google Scholar]
- 9.Martini A, La Starza R, Janssen H, et al. Recurrent rearrangement of the Ewing’s sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res. 2002;62(19):5408–5412. [PubMed] [Google Scholar]
- 10.Zhong CH, Prima V, Liang X, et al. E2A-ZNF384 and NOL1-E2A fusion created by a cryptic t(12;19)(p13.3; p13.3) in acute leukemia. Leukemia. 2008;22(4):723–729. [DOI] [PubMed] [Google Scholar]
- 11.Gocho Y, Kiyokawa N, Ichikawa H, et al. A novel recurrent EP300-ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia. 2015;29(12):2445–2448. [DOI] [PubMed] [Google Scholar]
- 12.Yasuda T, Tsuzuki S, Kawazu M, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48(5):569–574. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y-F, Wang B-Y, Zhang W-N, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lilljebjorn H, Henningsson R, Hyrenius-Wittsten A, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7:11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shago M, Abla O, Hitzler J, Weitzman S, Abdelhaleem M. Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer. (in press) [DOI] [PubMed] [Google Scholar]
- 16.Kiyokawa N, Iijima K, Tomita O, et al. Significance of CD66c expression in childhood acute lymphoblastic leukemia. Leuk Res. 2014;38(1):42–48. [DOI] [PubMed] [Google Scholar]
- 17.Manabe A, Ohara A, Hasegawa D, et al. Significance of the complete clearance of peripheral blasts after 7 days of prednisolone treatment in children with acute lymphoblastic leukemia: the Tokyo Children’s Cancer Study Group Study L99-15. Haematologica. 2008;93(8):1155–1160. [DOI] [PubMed] [Google Scholar]
- 18.Tomita O, Iijima K, Ishibashi T, et al. Sensitivity of SNX2-ABL1 toward tyrosine kinase inhibitors distinct from that of BCR-ABL1. Leuk Res. 2014;38(3):361–370. [DOI] [PubMed] [Google Scholar]
- 19.Grammatico S, Vitale A, La Starza R, et al. Lineage switch from pro-B acute lymphoid leukemia to acute myeloid leukemia in a case with t(12;17)(p13;q11)/TAF15-ZNF384 rearrangement. Leuk lymphoma. 2013;54(8):1802–1805. [DOI] [PubMed] [Google Scholar]
- 20.La Starza R, Aventin A, Crescenzi B, et al. CIZ gene rearrangements in acute leukemia: report of a diagnostic FISH assay and clinical features of nine patients. Leukemia. 2005;19(9):1696–1699. [DOI] [PubMed] [Google Scholar]
- 21.Nyquist KB, Thorsen J, Zeller B, et al. Identification of the TAF15-ZNF384 fusion gene in two new cases of acute lymphoblastic leukemia with a t(12;17)(p13;q12). Cancer Genet. 2011; 204(3):147–152. [DOI] [PubMed] [Google Scholar]
- 22.Barber KE, Harrison CJ, Broadfield ZJ, et al. Molecular cytogenetic characterization of TCF3 (E2A)/19p13.3 rearrangements in B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer 2007;46(5):478–486. [DOI] [PubMed] [Google Scholar]
- 23.Behm FG, Smith FO, Raimondi SC, Pui CH, Bernstein ID. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood. 1996;87(3):1134–1139. [PubMed] [Google Scholar]
- 24.Fischer U, Forster M, Rinaldi A, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet. 2015;47(9): 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corveleyn A, Janssen H, Martini A, Somers R, Cools J, Marynen P. Cellular transformation of NIH3T3 fibroblasts by CIZ/NMP4 fusions. J Cell Biochem. 2005;94(6):1112–1125. [DOI] [PubMed] [Google Scholar]
- 26.Pasqualucci L, Dominguez-Sola D, Chiarenza A, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pastore A, Jurinovic V, Kridel R, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16(9):1111–1122. [DOI] [PubMed] [Google Scholar]
- 28.Malinowska-Ozdowy K, Frech C, Schonegger A, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015;29(8):1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen C, Bartenhagen C, Gombert M, et al. Next-generation-sequencing of recurrent childhood high hyperdiploid acute lymphoblastic leukemia reveals mutations typically associated with high risk patients. Leuk Res. 2015;39(9):990–1001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.