

T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive cancer of developing thymocytes, and remains fatal in 20% of pediatric and 50% of adult patients.1,2 Frequent application of multi-agent cytotoxic drugs leads to disease relapse and high toxicities, underscoring the need for targeted therapies. The suppression of aberrant NOTCH1 signaling in T-ALL cells by gamma secretase inhibitors (GSIs) has been met with much enthusiasm; however, the gastrointestinal toxicities and drug resistance of GSIs restrain their clinical applications.3 The proto-oncogene MYC is a transcriptional target of NOTCH1 and a dominant driver of T-ALL pathogenesis.3 Targeting MYC-mediated transcriptional programs through BET bromodomain inhibitor JQ1 exhibits anti-leukemic efficacy in vitro and in vivo.4 However, global repression of transcription is predicted to cause toxicities. Identification of drug(s) synergizing with JQ1 to kill T-ALL cells may enhance the efficacy while reducing toxicities. Protein kinase CK2 is a tetrameric serine-threonine kinase composed of two catalytic (α or α′) and regulatory (β) subunits that can phosphorylate NOTCH1.5 CK2 inhibition by CX-4945, a potent and specific inhibitor in clinical trials for treating breast cancer and multiple myeloma, significantly reduces growth and survival of human T-ALL cells,6 and down-regulates NOTCH1 in lung cancer cells.7 However, it remains unclear whether the cytotoxic effect of CX-4945 on T-ALL cells is associated with repression of NOTCH1 signaling. Here we show that CK2 inhibition by CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells, implicating an alternative strategy to target NOTCH1 signaling in refractory/relapsed T-ALL.
CK2 (α and β) was previously found up-regulated in human T-ALL cells;8 whether this upregulation is linked to the temporal regulation of CK2 during T-cell development is unknown. To address this question, we analyzed publicly available databases and cross-compared the expression of CK2 subunits among subsets of developing T cells and patient T-ALL cells that are arrested at different developmental stages.9 We found that the transcript levels of all CK2 subunits (α, α′ and β) were significantly higher in patient T-ALL cells, compared to normal T cells regardless of their developmental stages (Figure 1A and B and Online Supplementary Figure S1A). We next examined protein levels of CK2, cleaved-NOTCH1 and MYC by Western blots in a panel of primary T-ALL patient samples. Consistent with its elevated transcript levels, CK2α′ protein levels were up-regulated in patient T-ALL cells, compared with those in normal thymus (Figure 1C and D). While CK2α protein levels were not significantly elevated in the patient samples examined (Figure 1C and Online Supplementary Figure S1B), they significantly correlated with those of cleaved-NOTCH1 and MYC (Figure 1E and F). In addition, we observed significant correlation between protein levels of CK2α′ and cleaved-NOTCH1 (Figure 1G). Western blotting analysis of CK2α, CK2α′, CK2β, cleaved-NOTCH1 and MYC in a panel of human T-ALL cell lines (JURKAT, ALL-SIL, RPMI-8402 and MOLT-3) revealed that human T-ALL cells expressed significantly higher levels of CK2α′, NOTCH1 and MYC, compared with those in normal thymus (Online Supplementary Figure S2). These results demonstrate that CK2, the α′ subunit in particular, is aberrantly expressed in human T-ALL cells regardless of their stages of differentiation blockade, and its expression correlates with cleaved-NOTCH1 and MYC.
Figure 1.
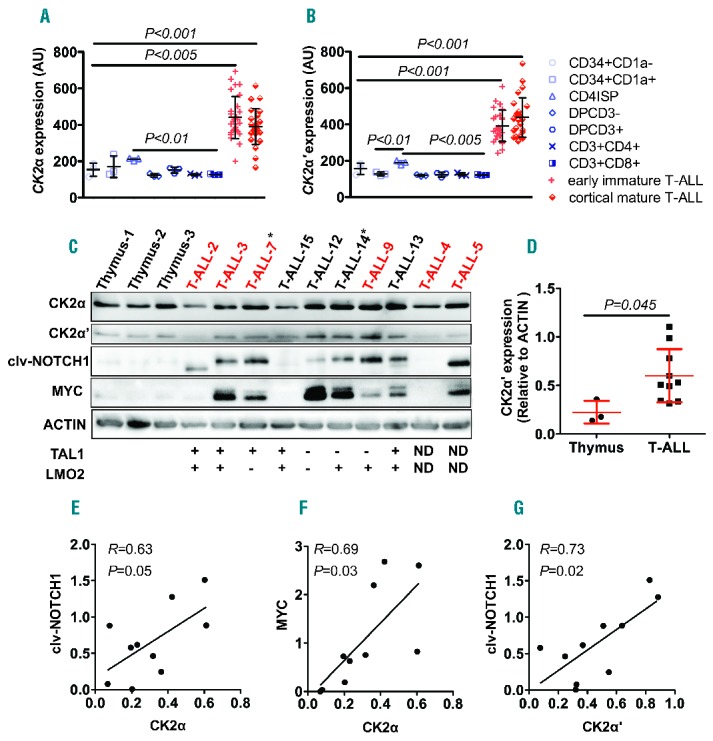
CK2 expression is elevated in human patient T-cell acute lymphoblastic leukemia (T-ALL) cells, and correlates with those of NOTCH1 and MYC. (A and B). Both CK2α (probe ID: ILMN_2386355) (A) and CK2α′ (probe ID: ILMN_1723843) (B) transcripts are elevated in early immature and mature T-ALL patient samples, compared with different subsets of T cells. Mean±SD of CK2α: 440.6±21.9 for early immature T-ALL and 391.1±19.6 for cortical/mature T-ALL versus 154±21.1 for CD34+CD1a−, 170.1±34.35 for CD34+CD1a+, 211.1±4.55 for CD4ISP, 122.3±5.49 for DPCD3−, 151.5±9.28 for DPCD3+, 126.3±3.19 for CD3+CD4+, 127.1±2 for CD3+CD8+; P<0.005 and P<0.001; and CK2α′: 392.2±16.3 for early immature T-ALL and 438.1±21.74 for cortical/mature T-ALL versus 156.8±17.89 for CD34+CD1a−, 128.1±5.5 for CD34+CD1a+, 190±6.81 for CD4ISP, 119.4±2.61 for DPCD3−, 124.1±7.54 for DPCD3+, 124.6±5.18 for CD3+CD4+, 122.4±1.94 for CD3+CD8+. P<0.001 for all comparisons; n=28 for early immature T-ALL, 25 for cortical/mature T-ALL and 3 for subsets of T cells, respectively. Among the different T-cell subsets, the expression of CK2α (A) and CK2α′ (B) is slightly but significantly higher in CD4ISP cells, compared to double-positive or single-positive subsets of T cells (P<0.01 and P<0.005, respectively). (C) Western blotting analysis of CK2α, CK2α′, cleaved-NOTCH1 (clv-NOTCH1) and MYC in patient T-ALL samples, compared with normal thymus. ACTIN serves as a loading control. Patient sample number marked in red indicates NOTCH1 mutations and asterisk (*) denotes FBW7 mutations. (D) CK2α′ versus ACTIN protein ratios demonstrating that CK2α′ levels are significantly higher in primary T-ALL patient samples, compared with those in control thymocytes (mean±SD of CK2α′ to ACTIN ratio: 0.60±0.09 versus 0.22± 0.07; P=0.045; n=10 and 3, respectively). (E–G) Pearson correlation tests reveal that CK2α (E and F) and CK2α′ (G) protein levels significantly correlate with those of NOTCH1 and MYC, or NOTCH1 alone (n=10; P=0.05, 0.03 and 0.02, respectively). AU: arbitrary unit. All human samples were collected and analyzed after informed consent and with approval of the Institutional Review Board and the Ethics Committee without linked identifiers.
To understand whether CX-4945 can modulate NOTCH1 signaling in the context of T-ALL cells, we treated JURKAT and ALL-SIL cells that up-regulate all three subunits of CK2, as well as RPMI-8402 cells expressing much less CK2, with CX-4945. CK2 enzymatic activity was efficiently inhibited by CX-4945 treatment in these cells, as demonstrated by decreased levels of phospho-AKT serine 129 (Figure 2A and Online Supplementary Figure S3B),10 as well as CK2 kinase assays (Online Supplementary Figure S3A). Interestingly, CK2 inhibition by CX-4945 led to a dose-dependent decrease of cleaved-NOTCH1 in JURKAT and ALL-SIL cells as early as 8 hours post treatment (Figure 2A). The effect of CX-4945 on cleaved-NOTCH1 was less obvious in RPMI-8402 cells, consistent with low expression of CK2 in this cell line (Online Supplementary Figure S2B). To understand why CX-4945 treatment reduced NOTCH1 levels, we blocked proteasome-mediated degradation in JURKAT and ALL-SIL cells with the proteasome inhibitor MG132. Because JURKAT cells are exquisitely sensitive to MG132 treatment, rapidly inducing apoptosis,11 we performed this experiment in JURKAT cells that over-express BCL-2 and thus are apoptotic-resistant. In the absence of CX-4945, MG132 treatment led to increased NOTCH1 levels in both T-ALL cell lines, indicating that NOTCH1 is degraded by the proteasome in these cells (Figure 2B). Importantly, MG132 treatment restored cleaved-NOTCH1 levels that declined upon CX-4945 treatment in both cell lines (Figure 2B), suggesting that CX-4945 treatment promotes the degradation of NOTCH1 in these cells. Pulse-chase analysis was subsequently performed to measure the half-life of cleaved-NOTCH1 in JURKAT and ALL-SIL T-ALL cells with and without CX-4945 treatment (Figure 2C), and revealed that NOTCH1 was degraded at least twice as fast in TALL cells upon CX-4945 treatment, compared to control DMSO-treated cells (Figure 2C).
Figure 2.
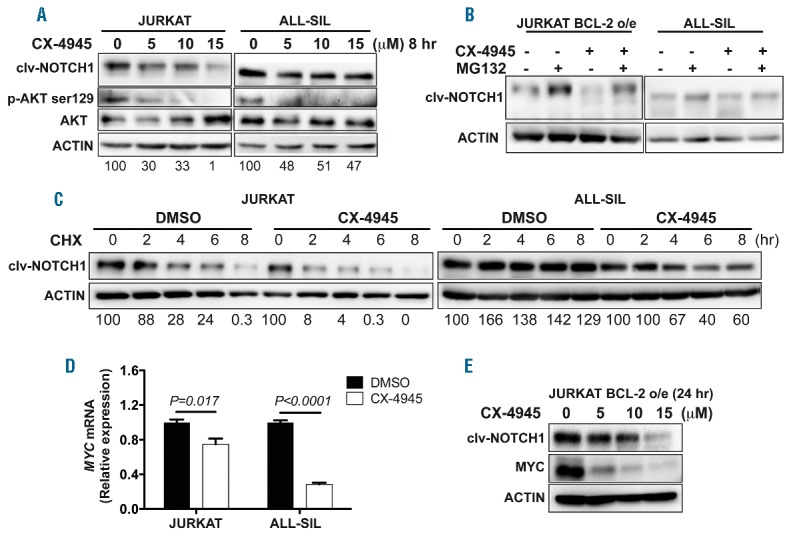
CK2 inhibition by CX-4945 decreases NOTCH1 and MYC levels in human T-cell acute lymphoblastic leukemia (T-ALL) cells by destabilizing NOTCH1. (A) CX-4945 treatment in both human JURKAT and ALL-SIL T-ALL cells for 8 hours leads to decreased expression of cleaved-NOTCH1 (clv-NOTCH1) and phospho-AKT at Serine 129 site (P-AKT ser129). (B) Blocking proteasome-mediated degradation by MG132 for 8 hours rescues the decreased levels of clv-NOTCH1 in JURKAT and ALL-SIL T-ALL cells upon CX-4945 treatment. (C) Pulse-chase analysis of the half-life for clv-NOTCH1 reveals a less stable NOTCH1 upon CX-4945 (5 μM) treatment, compared with those treated with DMSO (3.96±0.45 hours vs. 1.59±1.54 hours in JURKAT cells and more than 8 hours vs. 5.75±1.77 hours in ALL-SIL cells; n=3 per group). CHX: cycloheximide. Data from one out of three biological repeats are shown. (D) qRT-PCR analysis revealing significantly decreased transcript levels of MYC in both JURKAT (P=0.017) and ALL-SIL (P<0.0001) T-ALL cells treated with CX-4945, compared with those in DMSO-treated cells. Data from one out of two biological repeats are shown. (E) CX-4945 treatment for 24 hours leads to decreased protein levels of both clv-NOTCH1 and MYC in JURKAT-BCL-2 over-expressing (o/e) T-ALL cells that are resistant to apoptosis. Clv-NOTCH1 protein amounts (relative to ACTIN) are shown in the bottom of panels (A) and (C). hr: hours.
Because MYC is a direct transcriptional target of NOTCH1 and critical for T-ALL pathogenesis,3 we next examined the extent to which CX-4945 treatment decreases MYC transcript levels through quantitative RT-PCR analysis. Compared with DMSO-treated T-ALL cells, CX-4945 treatment led to a significant reduction of MYC transcripts as early as eight hours post treatment (Figure 2D and additional data not shown). To preclude the possible effects of apoptosis on overall protein levels, we treated JURKAT BCL-2-over-expressing cells that are apoptotic-resistant with CX-4945, and observed drastic reductions of both cleaved-NOTCH1 and MYC protein levels after 24 hours of treatment (Figure 2E). These data demonstrate that CX-4945 treatment destabilizes NOTCH1, leading to subsequent downregulation of MYC transcript and protein levels.
Because both JQ1 and CX-4945 exhibit anti-leukemic efficacy as single agents,4,6 and our results show that CX-4945 destabilizes NOTCH1 and down-regulates MYC in T-ALL cells, we next asked whether CX-4945 synergizes with JQ1 to kill human T-ALL cells. To this end, we treated JURKAT, ALL-SLL, RPMI-8402 and MOLT-3 cells with serial dilutions of CX-4945 and JQ1 in combination and analyzed relative cell viability. In both JURKAT and ALL-SIL cells that significantly up-regulate CK2 subunits (CK2α, CK2α′ and CK2β; see Online Supplementary Figure S2A), CX-4945 and JQ1 exhibited strong synergism, with an average combination index (CI) of 0.31 and 0.16, respectively (Figure 3A and B; where a CI of 1 indicates an additive effect, CI<1 is synergistic, and CI>1 antagonistic). However, for RPMI-8402 and MOLT-3 cells, which express moderate levels of CK2β (Online Supplementary Figure S2A), the synergism is weaker, with an average CI of 0.49 and 0.75, respectively (Figure 3A and B). Our data demonstrate that T-ALL cells, especially those with CK2 upregulation, are more sensitive to the combination treatment of CX-4945 and JQ1 than to single agents.
Figure 3.

JQ1 synergizes with CX-4945 in killing T-cell acute lymphoblastic leukemia (T-ALL) cells. (A) Combination treatment of CX-4945 and JQ1 depicted as normalized isobolograms shows strong synergism between the two drugs in JURKAT and ALL-SIL cell lines [combination index (CI)=0.31 and 0.16, respectively] but weaker synergism in RPMI-8402 and MOLT-3 cell lines (CI=0.49 and 0.75, respectively). CalcuSyn software was used to analyze combination data to produce the isobolograms normalized to the IC50 of each drug. The black diagonal line connects x- and y-axes of the normalized isobologram. Red dots on the black line represent additive dose combinations. Red dots below the black line represent synergistic drug combinations. Red dots above the black line represent antagonism. The T-ALL cell lines were treated with the following combination doses of CX-4945 and JQ1 for 72 hours, respectively: CX-4945 from 1.0 to 10 μM and JQ1 from 0.1 to 10 μM. (B) Cell viability upon combination treatment with CX-4945 2.5 μM and JQ1 1 μM is significantly reduced in all cell lines (except in MOLT-3 for JQ1 treatment vs. combination treatment), compared with those by single-agent treatment. Cell viability was determined with CellTiter-Blue after 72 hours of treatment for all four T-ALL cell lines treated with DMSO, CX-4945 (2.5 μM), JQ1 (1 μM) and both drugs in combination (CX-4945: 2.5 μM; JQ1: 1 μM). (C) Apoptosis analysis was performed on JURKAT, ALL-SIL, RPMI-8402 and MOLT-3 cells stained with Annexin V and PI after 48 hours of treatment with DMSO, CX-4945 (2.5 μM), JQ1 (1 μM) and both drugs in combination (CX-4945: 2.5 μM; JQ1: 1 μM). Cells were examined by flow cytometry to determine early apoptosis (PI-, Annexin V+) and late apoptosis (PI+, Annexin V+). Values in (B and C) are means±standard deviation (SD), and represent three biological replicates. Statistical significance was determined by two-tailed t-test.
To understand the cellular basis for the synergism of CX-4945 and JQ1 in T-ALL cells, we performed apoptosis and cell cycle analyses. We used Annexin V and propidium iodide (PI) staining to document that the combination treatment of CX-4945 (2.5 μM) and JQ1 (1 μM) in our tested four T-ALL cells significantly induced apoptosis, compared with either drug alone (Figure 3C and Online Supplementary Figure S4). Furthermore, Western blotting analysis showed much stronger expression of cleaved-PARP in human T-ALL cells subjected to combination treatment than those in single-agent-treated cells (Online Supplementary Figure S5). Finally, cell cycle analysis of T-ALL cells failed to reveal cell cycle arrest upon single or combination treatment (data not shown), indicating that apoptosis is the primary cellular basis of synergism.
To determine the possible toxicities of the combination treatment, we treated ALL-SIL T-ALL cells and normal peripheral blood monocytes (PBMC) with a higher dosage of CX-4945 (5 μM) and JQ1 (2 μM) than those used in Figure 3B and C, alone or in combination for 48 hours. Importantly, PBMC were less sensitive to CX-4945, JQ1, or combination treatment, compared with ALL-SIL T-ALL cells (Online Supplementary Figure S6A). Moreover, when we tested the combination treatment in PBMC, we observed an antagonistic effect (CI=1.11) (Online Supplementary Figure S6B).
In conclusion, our studies show that CK2 inhibitor CX-4945 destabilizes NOTCH1 and synergizes with JQ1 against human T-ALL cells. Both FBW7 and ITCH can degrade NOTCH1,12 and ITCH may promote NOTCH1 degradation in T-ALL cells (e.g. JURKAT) with FBW7 mutations.13 Importantly, CX-4945 exhibits striking synergy with JQ1 in T-ALL cells that up-regulate CK2, cleaved-NOTCH1 and MYC. CX-4945 induces proapoptotic unfolded protein response (UPR) in T-ALL cells,6 while JQ1 down-regulates MYC that normally activates prosurvival UPR.14 Hence, CX-4945 and JQ1 may synergistically kill T-ALL cells by enabling the switch of prosurvival to proapoptotic UPR. Although JQ1 can also synergize with the GSI to kill T-ALL cells,15 due to the toxicities and drug resistance of GSI, the combination of CX-4945 and JQ1 may offer a better approach to target NOTCH1 signaling in refractory/relapsed T-ALL. Both CX-4945 and JQ1 structural analogs are currently in clinical trials as single agents to treat solid and hematologic cancers (clinicaltrials.gov identifiers: 02128282 and 02157636). Our studies provide a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-ALL using pre-clinical in vivo models. Our data from TALL cell lines suggest that patient T-ALL cells with elevated CK2 expression could be more sensitive to the treatment than those with low CK2 expression. Given the wide involvement of CK2 and NOTCH1/MYC in cancers, the combination treatment of JQ1 and CX-4945 should be investigated in other cancer types.
Supplementary Material
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Ko RH, Ji L, Barnette P, et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol. 2010;28(4):648–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marks DI, Paietta EM, Moorman AV, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. 2009;114(25):5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: challenges and opportunities for anti-NOTCH1 therapy in T-cell acute lymphoblastic leukemias and lymphomas. Clin Cancer Res. 2008;14(17):5314–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roderick JE, Tesell J, Shultz LD, et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood. 2014;123(7):1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ranganathan P, Vasquez-Del Carpio R, Kaplan FM, et al. Hierarchical phosphorylation within the ankyrin repeat domain defines a phosphoregulatory loop that regulates Notch transcriptional activity. J Biol Chem. 2011;286(33):28844–28857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buontempo F, Orsini E, Martins LR, et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: targeting the unfolded protein response signaling. Leukemia. 2014;28(3):543–553. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, Long H, Yang YL, et al. Inhibition of CK2alpha down-regulates Notch1 signalling in lung cancer cells. J Cell Mol Med. 2013;17(7):854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118(11):3762–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K, et al. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood. 2013;122(1):74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Maira G, Salvi M, Arrigoni G, et al. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005;12(6):668–677. [DOI] [PubMed] [Google Scholar]
- 11.Park HS, Jun do Y, Han CR, Woo HJ, Kim YH. Proteasome inhibitor MG132-induced apoptosis via ER stress-mediated apoptotic pathway and its potentiation by protein tyrosine kinase p56lck in human Jurkat T cells. Biochem Pharmacol. 2011;82(9):1110–1125. [DOI] [PubMed] [Google Scholar]
- 12.Chastagner P, Israël A, Brou C. AIP4/Itch regulates Notch receptor degradation in the absence of ligand. PloS One. 2008;3(7):e2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204(8):1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hart LS, Cunningham JT, Datta T, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. 2012;122(12):4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knoechel B, Roderick JE, Williamson KE, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46(4):364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.