

Abstract
Tumors are a serious threat to human health. The oncolytic virus is a kind of tumor killer virus which can infect and lyse cancer cells and spread through the tumor, while leaving normal cells largely unharmed. Mathematical models can help us to understand the tumor-virus dynamics and find better treatment strategies. This paper gives a new mathematical model of tumor therapy with oncolytic virus and MEK inhibitor. Stable analysis was given. Because mitogen-activated protein kinase (MEK) can not only lead to greater oncolytic virus infection into cancer cells, but also limit the replication of the virus, in order to provide the best dosage of MEK inhibitors and balance the positive and negative effect of the inhibitors, we put forward an optimal control problem of the inhibitor. The optimal strategies are given by theory and simulation.
1. Introduction
Tumors are a serious threat to human health, and chemotherapy and radiotherapy may not only kill cancer cells, but also damage human body normal cells at the same time [1]. The oncolytic virus is a kind of tumor killer virus which can infect and lyse cancer cells and spread through the tumor, while leaving normal cells largely unharmed [2]. When oncolytic viruses are inoculated into a cancer patient or directly injected into a tumor, these viruses will spread throughout the tumor and infect tumor cells. The viruses can be replicated in the infected tumor cells. When an infected tumor cell is lysed, it can burst out a mass of new oncolytic viruses. Then, these new viruses can infect much more neighboring tumor cells [3].
Experiments using oncolytic viruses such as adenovirus, CN706 [4], and ONYX-15 [5] in animal tumors show that these viruses are nontoxic and infect tumor cells specifically. Now, treatment of cancer with oncolytic virus has been clinically tested [6–8]. This treatment of cancer with oncolytic viruses has been explored by clinicians [9–11].
In recent years, in order to understand the cancer-virus dynamics and find better treatment strategies, some mathematical models have been set up [12–19]. Tian proposed a mathematical model to describe the development of a growing tumor and an oncolytic virus population as follows [18]:
| (1) |
where variables x, y, and v stand for the population of uninfected cells, infected tumor cells, and oncolytic viruses, respectively. The coefficient β represents the infection of the virus. The tumor growth is modeled by logistic growth, and K is the maximal tumor size. λ is the per capita tumor growth rate. δ means the lysis rate of the infected tumor cells. b represents the burst size of new viruses coming out from the lysis of an infected tumor cell. γ represents the death rate of the virus.
It was shown that when the threshold b < 1 + γ/(Kβ), the equilibrium solution (K, 0,0) is globally asymptotically stable [18], indicating that the oncolytic virus therapy finally has no effect. Obviously, the smaller the value of K, the more easily b < 1 + γ/(Kβ) holds. Since K represents the total number of tumor cells, smaller tumors may be more resistant to the treatment by oncolytic virus than large ones, which should be a contradiction. In [19], by replacing βxv with xv/(x + y + ε), we proposed the model
| (2) |
The meanings of variables x, y, and v and parameters λ, β, δ, γ, b, and K are the same as those in model (1), and ε is positive and sufficiently small. The threshold obtained by our model is b < 1 + (γ/β)(1 + ε/K), which is almost independent of K when ε is sufficiently small.
On the other hand, all the above papers did not consider coxsackie-adenovirus receptor (CAR). In fact, CAR is a main receptor when oncolytic viruses enter into tumor cells [20–22]. The successful entry of viruses into cancer cells is related to the presence of CAR. When oncolytic viruses infect the tumor cells, firstly, they combine with the CAR and are absorbed into the cells.
Mitogen-activated protein kinase (MEK) inhibitors have been shown to promote CAR expression and could increase oncolytic viruses infection into tumor cells. But MEK inhibitors may also limit the replication of viruses [23–25], which will affect the treatment by oncolytic virus. With the function of MEK, [25] gave a model:
| (3) |
The variables x, y, and v have the same meanings as those in model (2); z represents the average expression level of CAR on the surface of the cells. The intensity of MEK inhibitor application is captured in the parameter u, u ∈ [0,1]. If u = 0, there is no MEK inhibitor application, and the CAR expression level will gradually decline. If u = 1, the MEK inhibitor has the maximum possible effect. The model assumes that exponential growth can be slowed down by the inhibitor with expression 1 − u. CAR grow at the rate of g(p − z) and become extinct at the rate of c.
Based on models (2) and (3), we establish the following mathematical model:
| (4) |
The variables x, y, v, and z have the same meanings as those in model (3). The parameters λ, β, δ, γ, b, and K are the same as those of (1). The parameter u has the same meaning as that in model (3). All the parameters are strictly positive.
Since the use of MEK inhibitors not only results in enhanced oncolytic virus entry into the tumor cells, but also renders infected cells temporarily unable to produce viruses, the maximum dosage of MEK use may not result in the best treatment effect, so the optimal control-based schedules of MEK inhibitor application should be studied. The optimal MEK inhibitor application strategy can increase the efficacy of this treatment in an economical fashion. So, first, in this paper, we let the control variable u be a constant; a stability analysis of our model is conducted, and then the optimal control strategy is discussed; we also compare the optimal control with constant control by simulation.
2. Materials and Methods
2.1. Stability Analysis
System (4) always has two equilibrium points:
| (5) |
If ε is sufficiently small, when b > 1 + (α(K + ε)/βK)((gu + c)/gup), the third steady state E 2 = (x 2, y 2, v 2, z 2) exists in which
| (6) |
Here,
| (7) |
It should be noted that b > 1 + (α(K + ε)/βK)((gu + c)/gup) is equivalent to (b − 1)βz 2/α − 1 > 0 to ensure that y 2 > 0 when ε is sufficiently small.
The Jacobi matrix at point E 0 is
| (8) |
Obviously, μ 1 = r(1 − u) > 0 is one eigenvalue of J|E0 which means E 0 is unstable. The unstable result of E 0 seems consistent with the biological meaning that, without viruses and infected tumor cells, the tumor will grow from an initial small value around E 0.
As for equilibrium point E 1, we have the following theorem.
Theorem 1 . —
When b < 1 + (α(K + ε)/βK) · ((gu + c)/gup), E 1 is locally asymptotically stable. When b > 1 + (α(K + ε)/βK)((gu + c)/gup), E 1 is unstable.
Proof —
At the equilibrium point E 1, the Jacobi matrix is
(9) where H = βKgup/(K + ε)(gu + c). The eigenvalues of J|E1 are μ 1 = −(1 − u)r, μ 2 = −gu − c,
(10) Here, in which Δ2 = βKugp/(K + ε)(gu + c).
When b < 1 + (α(K + ε)/βK)((gu + c)/gup), we have
(11) which ensure that μ 3 and μ 4 are negative, so E 1 is locally asymptotically stable.
Similarly, when b > 1 + α(K + ε)(gu + c)/βKgup, μ 3 is positive, and E 1 is unstable.
Actually, we can prove that the equilibrium solution E 1 is globally asymptotically stable when b < 1 + α(K + ε)(gu + c)/βKgup. But we need to show the boundness of system (4). From the first two equations, we obtain
(12) By the comparison principle, we can obtain limt→∞sup(x(t) + y(t)) ≤ K.
From the third equation of (4), we can have
(13) It is easily shown that v(t) ≤ (1 − u)bδK/α.
Similarly, from
(14) we can get that z(t) ≤ gup/(gu + c) holds.
Theorem 2 . —
When b < 1 + (α(K + ε)/βK) · ((gu + c)/gup), E 1 is globally asymptotically stable.
Proof —
Consider the Lyapunov function V = y + (1/b)v; the derivative along a solution is given by
(15) Since 0 < x ≤ K, 0 < x + y ≤ K, we have xK + xε ≤ xK + Kε + Ky, which implies
(16) and because z ≤ gup/(gu + c), therefore,
(17) When b < 1 + (α(K + ε)/βK) · ((gu + c)/gup), we can have .
Let ; it is clear that E ⊂ {(x, y, v, z)∣v = 0}. Let M be the largest positively invariant subset of the set E; by the third equation of system (4), we can know that y(t) = 0, so M = {(x, y, v)∣y = 0, v = 0}. By LaSalle invariance principal [26], we know
(18) So, the limit equation of system (4) is
(19) Therefore, x(t) → K, z → gup/(gu + c) when t → ∞. So, E 1 is globally attractive; note that b < 1 + (α(K + ε)/βK)·((gu + c)/gup) can also ensure the local asymptotical stability of E 1, so we can know that E 1 of system (4) is globally asymptotically stable when b < 1 + (α(K + ε)/βK)·((gu + c)/gup).
Although we can prove the global asymptotical stability of E 1, we would not want this to happen, because the global asymptotical stability means the therapy does not have any effect. When b > 1 + (α(K + ε)/βK)((gu + c)/gup) holds, the coexistent steady state E 2 = (x 2, y 2, v 2, z 2) exists, but it is difficult to give the stable analysis of E 2, so we just give some simulations about it.
We choose β = 0.2 day−1, ε = 0.009, δ = 0.5 day−1, r = 6 cells day−1, α = 0.5 day−1, p = 10, g = 0.1 day−1, c = 0.5 day−1, and K = 9 × 108 cells.
The initial condition is x0 = (6 × 107, 0, 5 × 104, 4 × 102), where the unit of each is cells.
We choose b = 10 cell−1 day−1, b = 14 cell−1 day−1, respectively, and b > 1 + (α(K + ε)/βK)((gu + c)/gup) = 2.6389 all hold; the simulation results are shown in Figures 1 and 2.
The simulation results show that oncolytic virus therapy may keep the tumor stable at some level as shown in Figure 1 or keep oscillating at a certain range as shown in Figure 2. Since the cured equilibrium is always unstable, just from our model, we could not give the condition that ensures the tumor can be cured by oncolytic virus therapy, but if we choose appropriate u which satisfies
(20) the simulation shows that oncolytic virus therapy can prevent the tumor from getting worse and worse. Some other therapy methods should be combined to cure the tumor.
Figure 1.
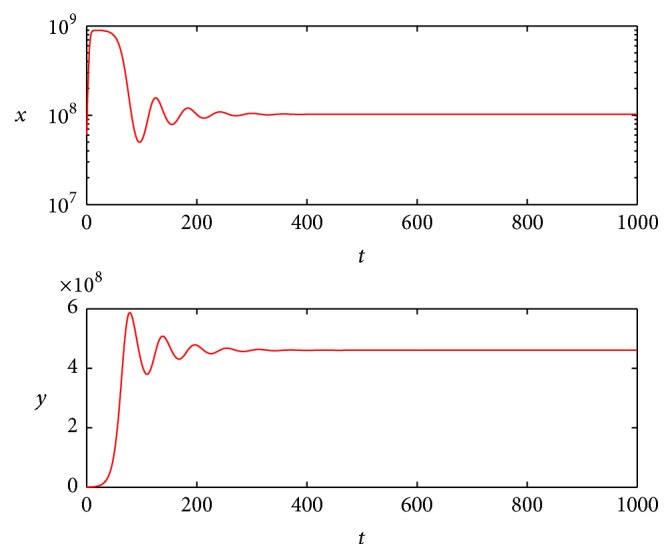
State dynamics for uninfected and infected tumor cells when b = 10.
Figure 2.
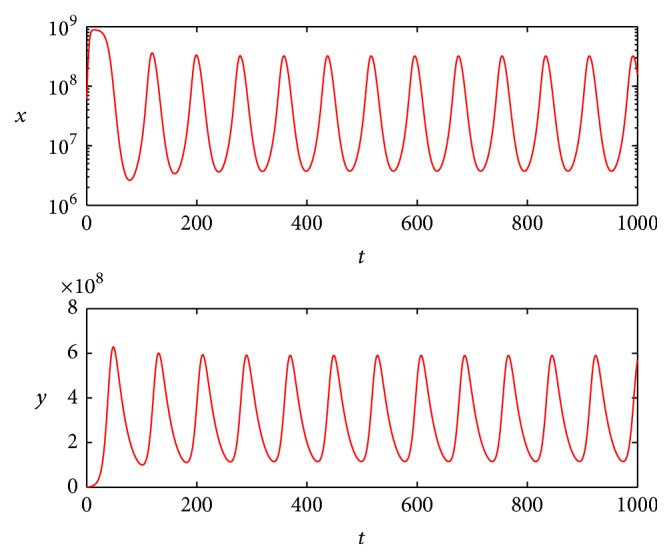
State dynamics for uninfected and infected tumor cells when b = 14.
2.2. The Optimal Control of MEK
In the simulation of Figures 1 and 2, we choose the control u as constant. Since the use of MEK inhibitors not only results in enhanced oncolytic virus entry into the cells, but also renders infected cells temporarily unable to produce viruses, how to use the MEK inhibitors optimally should be studied. In model (4), the function of MEK inhibitors was embodied by parameter u; we use it as the control variable. The control goal is not only to formulate an objective functional which lowers the levels of tumor cells during and at the end of therapy, but also to minimize the cost of MEK, so the objective function is defined as follows:
| (21) |
where t 0 represents the beginning time of the treatment and t f represents the terminal time of the treatment. a 11, a 22, b 11, b 22, b 33, and c 11 represent the cost coefficients for the variables, respectively.
For convenience, we define the state vector X = (x, y, v, z)T; system (4) can be written as
| (22) |
And the corresponding cost function is defined as follows:
| (23) |
Here,
| (24) |
Based on the dynamic constraint f(X(T), u(t), t) and the Lagrangian L(X(t), u(t), t), the Hamiltonian is as follows:
| (25) |
where L(X(t), u(t), t) = (1/2)(X T BX + c 11 u 2).
Using Pontryagin's minimum principle, the necessary conditions are given as follows:
-
(10)
(26) -
(20)
(27) -
(30)
(28)
where
| (29) |
-
(40)
(30) -
(50)
(31)
3. Results and Discussion
In this part, based on the minimum principle, we will give the simulation of optimal strategy by using the Runge–Kutta fourth-order scheme and the steepest gradient method [27].
We choose 100 days as the control time. The efficacy u can theoretically lie between 0 and 1, where 0 corresponds to no effectiveness of the MEK and 1 corresponds to full effectiveness of the MEK. However, the perfect efficacy of MEK is unlikely to be achieved totally, so we suppose that the maximum effect is 0.98.
It is difficult to choose the parameters exactly based on biological meaning without experiment data, since the global stability of E 1 means the therapy has no effect when
| (32) |
We choose β = 0.2 day−1, ε = 0.009, δ = 0.5 day−1, r = 6 cells day−1, α = 0.5 day−1, p = 10, g = 0.1 day−1, c = 0.5 day−1, K = 9 × 108 cells, b = 4 cell−1 day−1, a 11 = a 22 = 1, b 11 = b 22 = b 33 = 10−5, and c 11 = 8000.
Even if we use the maximum constant u = 0.98, b > 1 + (α(K + ε)/βK)((gu + c)/gup) = 2.5255 also holds.
We give and compare two control strategies with the same initial conditions:
| (33) |
The optimal control simulation results are shown in Figure 3. The corresponding state dynamics for uninfected and infected tumor cells under the optimal control are shown in Figure 4 with solid line.
Figure 3.
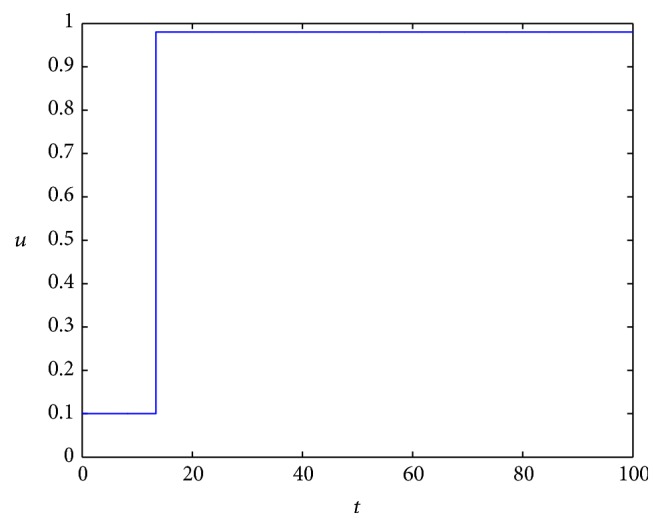
The optimal control of MEK.
Figure 4.
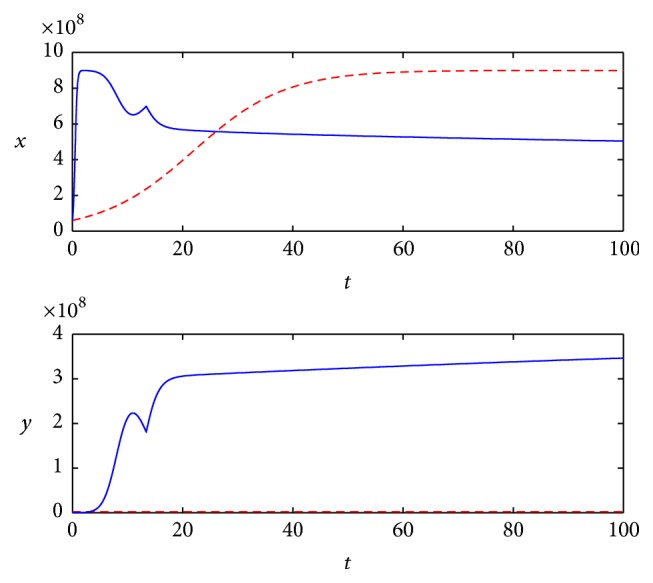
State dynamics for uninfected and infected tumor cells with different control strategies.
We choose constant control u = 0.98 to compare with the optimal control effect; the state dynamics for uninfected and infected tumor cells under the constant control are shown in Figure 4 with dotted line.
From the simulation, we can see that even if we use the maximum constant u = 0.98, the tumor cells still increase at the former stage and then keep stable at some level. But if we use the optimal control strategy as shown in Figure 3, that is to say, we need not use the maximum dosage of MEK all the time, though the tumor cells increase quickly with the lower dosage of MEK at the beginning stage, about 5 days later, it will begin to decrease and keep lower than that of constant control. As for the infected tumor, it is apparent that more tumor cells are infected with the optimal strategy; this in turn can help tumor cells keep at a contrarily lower level.
4. Conclusion
This paper introduces a new mathematical model of tumor therapy with oncolytic virus and MEK inhibitor. The stability of the equilibrium points is analyzed. Because inhibitors (MEK) can not only lead to greater oncolytic virus infection into cancer cells, but also cause cell cycle to stop, from theoretical analysis and numerical simulations, we compare optimal control strategy about the dosage of MEK inhibitor and constant control strategy with the same initial conditions. Simulations show that the optimal control has better control effect than constant control. But it should be pointed out that our model has no cure equilibrium point, so, just from our model, we could not say that the tumor can be cured only by oncolytic virus therapy. But the optimal control strategy can help to prevent the tumor from getting worse and worse. Some other therapy methods should be combined to cure the tumor.
As we cannot get the exact parameters based on biological meaning, more work should be done about the modeling and simulations.
Acknowledgments
This paper is supported by 2015 National Traditional Chinese Medicine Clinical Research Base Business Construction Special Topics (JDZX2015299).
Competing Interests
The authors declare that there are no competing interests regarding the publication of this paper.
References
- 1.Zhou L., He W. W., Zhu Z. N., et al. The clinical research progress for oncolytic adenovirus targeting cancer therapy. China Biotechnology. 2013;33(12):105–113. [Google Scholar]
- 2.Komarova N. L., Wodarz D. ODE models for oncolytic virus dynamics. Journal of Theoretical Biology. 2010;263(4):530–543. doi: 10.1016/j.jtbi.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelly E., Russell S. J. History of oncolytic viruses: genesis to genetic engineering. Molecular Therapy. 2007;15(4):651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez R., Schuur E. R., Lim H. Y., Henderson G. A., Simons J. W., Henderson D. R. Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Research. 1997;57(13):2559–2563. [PubMed] [Google Scholar]
- 5.Ganly I., Kirn D., Eckhardt G., et al. A phase I study of ONYX-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clinical Cancer Research. 2000;6(3):798–806. [PubMed] [Google Scholar]
- 6.Eager R. M., Nemunaitis J. Clinical development directions in oncolytic viral therapy. Cancer Gene Therapy. 2011;18(5):305–317. doi: 10.1038/cgt.2011.7. [DOI] [PubMed] [Google Scholar]
- 7.Donnelly O. G., Errington-Mais F., Prestwich R., et al. Recent clinical experience with oncolytic viruses. Current Pharmaceutical Biotechnology. 2012;13(9):1834–1841. doi: 10.2174/138920112800958904. [DOI] [PubMed] [Google Scholar]
- 8.Russell S. J., Peng K.-W., Bell J. C. Oncolytic virotherapy. Nature Biotechnology. 2012;30(7):658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miest T. S., Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nature Reviews Microbiology. 2014;12(1):23–34. doi: 10.1038/nrmicro3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel M. R., Kratzke R. A. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Translational Research. 2013;161(4):355–364. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 11.Aghi M., Martuza R. L. Oncolytic viral therapies—the clinical experience. Oncogene. 2005;24(52):7802–7816. doi: 10.1038/sj.onc.1209037. [DOI] [PubMed] [Google Scholar]
- 12.Si W., Zhang W. Control exponential growth of tumor cells with slow spread of oncolytic virus. Journal of Theoretical Biology. 2015;367:111–129. doi: 10.1016/j.jtbi.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 13.Wodarz D. Viruses as antitumor weapons: defining conditions for tumor remission. Cancer Research. 2001;61(8):3501–3507. [PubMed] [Google Scholar]
- 14.Choudhury B. S., Nasipuri B. Efficient virotherapy of cancer in the presence of immune response. International Journal of Dynamics and Control. 2014;2(3):314–325. doi: 10.1007/s40435-013-0035-8. [DOI] [Google Scholar]
- 15.Bajzer Z., Carr T., Josić K., Russell S. J., Dingli D. Modeling of cancer virotherapy with recombinant measles viruses. Journal of Theoretical Biology. 2008;252(1):109–122. doi: 10.1016/j.jtbi.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Novozhilov A. S., Berezovskaya F. S., Koonin E. V., Karev G. P. Mathematical modeling of tumor therapy with oncolytic viruses: regimes with complete tumor elimination within the framework of deterministic models. Biology Direct. 2006;1, article 6:1–18. doi: 10.1186/1745-6150-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y., Tian J. P., Wei J. Lytic cycle: a defining process in oncolytic virotherapy. Applied Mathematical Modelling. 2013;37(8):5962–5978. doi: 10.1016/j.apm.2012.12.004. [DOI] [Google Scholar]
- 18.Tian J. P. The replicability of oncolytic virus: defining conditions in tumor virotherapy. Mathematical Biosciences and Engineering. MBE. 2011;8(3):841–860. doi: 10.3934/mbe.2011.8.841. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y., Su Y. M. An improved model of tumor therapy with oncolytic virus. Journal of Henan University of Science & Technology. 2016;37(4):92–96. [Google Scholar]
- 20.Rauen K. A., Sudilovsky D., Le J. L., et al. Expression of the coxsackie adenovirus receptor in normal prostate and in primary and metastatic prostate carcinoma: potential relevance to gene therapy. Cancer Research. 2002;62(13):3812–3818. [PubMed] [Google Scholar]
- 21.Lacher M. D., Tiirikainen M. I., Saunier E. F., et al. Transforming growth factor-β receptor inhibition enhances adenoviral infectability of carcinoma cells via up-regulation of coxsackie and adenovirus receptor in conjunction with reversal of epithelial-mesenchymal transition. Cancer Research. 2006;66(3):1648–1657. doi: 10.1158/0008-5472.CAN-05-2328. [DOI] [PubMed] [Google Scholar]
- 22.Anders M., Christian C., McMahon M., McCormick F., Korn W. M. Inhibition of the Raf/MEK/ERK pathway up-regulates expression of the coxsackievirus and adenovirus receptor in cancer cells. Cancer Research. 2003;63(9):2088–2095. [PubMed] [Google Scholar]
- 23.Cherubini G., Petouchoff T., Grossi M., Piersanti S., Cundari E., Saggio I. E1B55K-deleted Adenovirus (ONYX-015) overrides G1/S and G 2/M checkpoints and causes mitotic catastrophe and endoreduplication in p53-proficient normal cells. Cell Cycle. 2006;5(19):2244–2252. doi: 10.4161/cc.5.19.3263. [DOI] [PubMed] [Google Scholar]
- 24.Bagheri N., Shiina M., Lauffenburger D. A., Korn W. M. A dynamical systems model for combinatorial cancer therapy enhances oncolytic adenovirus efficacy by MEK-inhibition. PLoS Computational Biology. 2011;7(2) doi: 10.1371/journal.pcbi.1001085.e1001085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zurakowski R., Wodarz D. Model-driven approaches for in vitro combination therapy using ONYX-015 replicating oncolytic adenovirus. Journal of Theoretical Biology. 2007;245(1):1–8. doi: 10.1016/j.jtbi.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaSalle J. P. The Stability of Dynamical Systems. Philadelphia, Pa, USA: SIAM; 1976. (Regional Conference Series in Applied Mathematics). [Google Scholar]
- 27.Pachpute G., Chakrabarty S. P. Dynamics of hepatitis C under optimal therapy and sampling based analysis. Communications in Nonlinear Science and Numerical Simulation. 2013;18(8):2202–2212. doi: 10.1016/j.cnsns.2012.12.032. [DOI] [Google Scholar]