

Abstract
Background
We previously showed that the levels of both Giα‐2 and Giα‐3 proteins were augmented in spontaneously hypertensive rats (SHRs) before the onset of hypertension. In addition, intraperitoneal injection of pertussis toxin, which inactivates both Giα proteins, prevented the development of hypertension in SHRs. The aim of the present study was to determine the specific contributions of Giα‐2 and Giα‐3 proteins to the development of hypertension.
Methods and Results
Antisense oligodeoxynucleotide of Giα‐2 and Giα‐3 encapsulated in PEG/DOTAP/DOPE cationic liposomes were administrated intravenously into 3‐week‐old prehypertensive SHRs and Wistar Kyoto rats, whereas the control Wistar Kyoto rats and SHRs received PBS, empty liposomes, or sense. The knockdown of Giα‐2 but not Giα‐3 protein attenuated tachycardia and prevented the development of hypertension up to age 6 weeks; thereafter, blood pressure started increasing and reached the same level as that of untreated SHRs at 9 weeks. Furthermore, Giα‐2 and Giα‐3 antisense oligodeoxynucleotide treatments significantly decreased the enhanced levels of Giα‐2 and Giα‐3 proteins, respectively, and enhanced levels of superoxide anion and NADPH oxidase activity in heart, aorta, and kidney and hyperproliferation of vascular smooth muscle cells from SHRs aged 6 weeks. In addition, antisense oligodeoxynucleotide treatment with Giα‐2 but not Giα‐3 restored enhanced inhibition of adenylyl cyclase by oxotremorine to WKY levels.
Conclusions
These results suggested that the enhanced expression of Giα‐2 but not Giα‐3 protein plays an important role in the pathogenesis of hypertension and tachycardia in SHRs.
Keywords: antisense, Giα‐2 proteins, Giα‐3 proteins, hypertension, oxidative stress, tachycardia
Subject Categories: Animal Models of Human Disease, Cell Signalling/Signal Transduction, Oxidant Stress, Hypertension, Gene Therapy
Introduction
Guanine nucleotide binding proteins (G proteins) are a large family of guanosine triphosphate binding proteins that play a crucial regulatory role as transducers in a variety of cell‐signaling pathways. G proteins are heterotrimeric proteins composed of 3 distinct subunits; α, β, and γ. All α subunits possess intrinsic GTPase activity and are responsible for specificity of receptor–effector interaction.1 According to amino acid homology, G proteins are classified into 4 major subfamilies and are represented as Gsα, Giα, Gqα/α11, and Gα12/α13. G proteins exert their action via several signal transduction pathways including adenylyl cyclase and receptor‐mediated activation of phospholipase C and A2.2, 3, 4 The inhibition and stimulation of adenylyl cyclase is mediated via Giα and Gsα, respectively. Three distinct Giα forms (Giα‐1, Giα‐2, and Giα‐3) are encoded by 3 different genes. All isoforms of Giα proteins inhibit adenylyl cyclase and activate atrial K+ channels,4, 5 whereas the 4 different isoforms of Gsα that were revealed by molecular cloning result from different splicing of 1 gene. Gsα is associated with adenylyl cyclase stimulation and increased cyclic AMP (cAMP) production.6
Giα proteins and associated inhibition of adenylyl cyclase signaling have been shown to be implicated in a variety of cellular functions, including vascular permeability,7, 8 salt and water transport,9, 10 and catecholamine release,11 all of which play key roles in the regulation of blood pressure (BP). Alteration in Giα protein levels is associated with impairment of cellular function that results in various pathological states including hypertension12 and heart failure.13, 14 Enhanced expression of Giα‐2 and Giα‐3 proteins and their genes was also shown in cardiovascular tissues from several animal models of hypertension including spontaneously hypertensive rats (SHRs),15, 16, 17, 18, 19, 20, 21, 22, 23, 24 whereas the levels of G0α and Gsα were not altered in hypertension.15, 18, 19 Moreover, high salt intake in salt‐sensitive hypertensive Dahl rats was shown to enhance expression of Giα proteins in heart failure, hypertension, and cardiac hypertrophy,25 whereas central Giα‐2 protein‐gated pathways were reported to counter the development of salt‐sensitive hypertension.26 Enhanced expression of Giα proteins and the Gi protein–mediated pathway in cardiovascular tissues and kidney was shown to contribute to the maintenance of hypertension in SHRs.27, 28 In contrast, Giα‐1 that is present predominantly in the brain and neural tissue and absent in heart and aorta23, 29 does not appear to be implicated in the regulation of cardiovascular functions including BP in SHRs.
The augmented expression of Giα proteins has been reported to occur before the onset of hypertension in SHRs and DOCA–salt hypertensive rats30, 31 and suggests that the enhanced expression of Giα‐2 and Giα‐3 proteins is a factor contributing to the development of hypertension rather than a consequence of hypertension. This was further supported by our study showing that a single intraperitoneal injection of pertussis toxin, which inactivates both Giα‐2 and Giα‐3 proteins, prevented the development of hypertension in prehypertensive SHRs aged 2 weeks.32 It is not clear whether the enhanced expression of Giα‐2 or Giα‐3 or both contributes to the development of hypertension in SHRs.
The present study was undertaken to examine the specific contributions of Giα‐2 and Giα‐3 to the development of hypertension in SHRs by using an antisense oligodeoxynucleotide approach. Antisense oligodeoxynucleotide is composed of short fragments of single‐strand DNA (13–25 nucleotides). The main concept underlying antisense therapy is simple: Use of a certain DNA sequence that is complementary to a specific mRNA will inhibit its transcription/gene expression and consequently inhibit protein synthesis.33, 34 The antisense approach has been used extensively and successfully in hypertension research. Antisense targeting angiotensin receptor type 1, angiotensinogen, thyrotropin‐releasing hormone, epidermal growth factor receptor, and insulin‐like growth factor 1 receptor35, 36, 37, 38, 39 attenuated BP in SHRs. Moreover, antisense targeting β‐adrenergic receptor exhibited profound and prolonged reduction in BP.40 Because antisense oligodeoxynucleotide is not serum stable and can be degraded easily by exonuclease and endonuclease,41, 42 it was necessary to use the delivery system. The recent advancement in nanotechnology was positively reflected in the improvement of the liposomes as drug‐delivery vectors. Liposome‐based nanomedicines offer an interesting approach for delivery of gene therapeutic agents like antisense.
This study provided evidence that knockdown of Giα‐2 but not Giα‐3 protein using antisense oligodeoxynucleotide encapsulated in PEG/DOTAP/DOPE cationic liposomes attenuated the development of hypertension and tachycardia and suggested that overexpression of Giα‐2 but not Giα‐3 protein plays an important role in tachycardia and the development of hypertension in SHRs.
Methods
Antisense Oligodeoxynucleotide
Antisense and the inverted‐form sense oligomer targeting Giα‐2 and Giα‐3 proteins were purchased from Alpha DNA. These oligodeoxynucleotides were modified by phosphothioation of 3 nucleotides on both sides. The following sequences were used: Giα‐2 antisense, 5′‐C*T*T*GTCGATCATCTTA*G*A*3′; Giα‐3 antisense, 5′A*A*G*TTGCGGTCGATC*A*T*3′; Giα‐2 sense, 5′T*C*T*AAGATGATCGACA*A*G*3′; and Giα‐3 sense, 5′‐A*T*G*ATC‐GACCGCAAC*T*T*3′.
Liposomes
Cationic lipid DOTAP and helper lipid DOPE were purchased from Avanti Polar Lipids and mixed at a 1:1‐M ratio. In addition, 10% DSPE‐PEG(2000) (Avanti Polar Lipids) was incorporated in the lipid mixture to increase liposome stability, to prolong circulation time in the blood stream, and to prevent leakage and aggregation of liposomes, as described previously.43
Formulation of Lipoplexes
DOTAP, DOPE, and PEG were dissolved in chloroform. Dried lipid film was formulated by exposing the lipid mixture to a slow N2 stream for evaporation of chloroform and then to a vacuum system overnight to ensure complete removal of chloroform traces. The lipid film was hydrated using sterile PBS with pH 7.4 at room temperature. The suspension was left at room temperature for 1 hour to allow assembly and formulation of liposomes. Liposomes were exposed to freeze/thaw cycles (7–9 cycles) to facilitate size management using liquid nitrogen and warm tap water. The size was homogenized using manual graded extrusion with 800‐, 400‐, and 100‐nm polycarbonate membranes (LiposoFast; Avestin, Inc). The final size of the liposomes was 100 to 200 nm with a polydispersity index of 0.1 measured using a dynamic light scattering technique (Malvern Zetasizer; Malvern Instruments). Antisense or sense of Giα‐2 or Giα‐3 was added to the final concentration of 200 μg/mL in sterile PBS with pH 7.4 and an N:P ratio of 2 calculated according to the Felgner equation.44 Lipoplexes were left for 1 hour at room temperature to allow encapsulation of antisense or sense. To ensure that antisense was encapsulated inside the liposome, fluorescently labeled antisense was used.
Animals
Male SHRs aged 2 weeks and age‐matched normotensive Wistar Kyoto (WKY) rats were purchased from Charles River Laboratories International, Inc. Animals were maintained at room temperature with free access to water and regular rat chow in 12‐hour light/dark cycles. Rats were left for 1 week for adaptation. SHRs and WKY rats were divided into 6 groups (control, empty liposomes, Giα‐2 antisense, Giα‐2 sense, Giα‐3 antisense, Giα‐3 sense; 6–10 rats per group). Antisense or sense of Giα‐2 or Giα‐3 (1 mg/kg body weight) and empty liposomes were injected intravenously via tail vein once into prehypertensive SHRs aged 3 weeks and age‐matched WKY rats. BP was monitored every week by the tail‐cuff method without anesthesia, using the CODA standard noninvasive BP system (Kent Scientific Corp). After measurement of BP, heart rate, and body weight, groups of rats (WKY and SHRs with and without different treatments, as indicated above) aged 6 and 9 weeks were euthanized by decapitation after CO exposure. Thoracic aortas, hearts, and kidneys were removed by dissection and used for determination of Giα protein levels, oxidative stress, and other biochemical assays. All animal procedures used in the present study were approved by the Ethics Committee for experimentation on animals of the University of Montreal (approval 99050). The investigation conforms to the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health.
BP and Heart Rate Measurement
Rats were left for 1 week for adaptation. BP was measured weekly using the CODA noninvasive tail‐cuff method, according to the recommendation of American Heart Association.45 The CODA tail‐cuff BP system uses volume pressure recording sensor technology to measure tail BP. Volume pressure recording is clinically validated and provides 99% correlation with telemetry and direct BP and heart rate measurements. BP was expressed as milligrams of mercury and heart rate as beats per minute.
Cell Culture
Aortic vascular smooth muscle cells (VSMCs) from SHRs aged 6 or 9 weeks and age‐matched WKY rats (control group) and empty liposome–, sense‐, and antisense‐treated SHRs and WKY rats were cultured, as described previously.30, 31 The purity of the cells was checked with an immunofluorescence technique using α‐actin, as described previously.46 These cells were found to contain high levels of smooth muscle–specific actin. The cells were plated in 75‐cm2 flasks and incubated at 37°C in 95% air and 5% CO2 humidified atmosphere in DMEM (with glucose, l‐glutamine, and sodium bicarbonate) containing antibiotics and 10% heat‐inactivated FBS. Confluent cell cultures were starved by incubation for 3 hours in DMEM without FBS at 37°C to reduce the interference by growth factors present in the serum. The cell lysates were prepared and used for Western blotting and adenylyl cyclase activity determination.
Preparation of Heart, Aorta and Kidney Particulate Fraction
Heart, aorta, and kidney particulate fractions were prepared, as described previously.18, 31 After rats were sacrificed, hearts, aortas, and kidneys were removed by dissection, quickly frozen in liquid nitrogen, and then pulverized into fine powder using a mortar and pestle precooled in liquid nitrogen. The powder was stored at −80°C until assayed. The powder was homogenized (12 strokes) in a glass homogenizer with a buffer containing 25 mmol/L Tris‐HCl (pH 7.5), 25 mmol/L NaCl, 1 mmol/L sodium orthovanadate, 10 mmol/L sodium fluoride, 10 mmol/L sodium pyrophosphate, 2 mol/L benzamidine, 2 mmol/L ethylene‐bis(oxy‐ethylenenitrolo)tetraacetic acid, 2 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 1% Triton X‐100, 0.1% sodium dodecyl sulfate, and 0.5 μg/mL leupeptin. The homogenate was centrifuged at 12 000g for 15 minutes at 4°C. The supernatants were transferred to a fresh microcentrifuge tube without disturbing the pellet. Protein concentration was determined by Bradford assay. The supernatant was used for Western blotting.
Western Blotting
Western blotting of Gi proteins was performed using specific antibodies, as described previously.20 After SDS‐PAGE, the separated proteins were electrophoretically transferred to a nitrocellulose membrane with a semi–dry transfer blot apparatus (Bio‐Rad Laboratories) at 15 V for 45 minutes. After transfer, the membranes were washed twice in PBS and were incubated in PBS containing 5% skim milk at room temperature for 1 hour. The blots were then incubated with the following specific antibodies: Giα‐2 (L5), Giα‐3 (C‐10), and dynein (74‐1; Santa Cruz Biotechnology) incubated in PBS containing 0.1% Tween 20 overnight at 4°C. The antigen–antibody complexes were detected by incubating the blots with goat anti–rabbit immunoglobulin G (Bio‐Rad Laboratories) conjugated with horseradish peroxidase for 1 hour at room temperature. The blots were then washed 3 times with PBS before reacting with enhanced chemiluminescence Western blotting detection reagents (Santa Cruz Biotechnology). Quantitative analysis of the protein was performed by densitometric scanning of the autoradiographs using the enhanced laser densitometer LKB Ultroscan XL and quantified using the Gelscan XL evaluation software (version 2.1) from Pharmacia.
Determination of Adenylyl Cyclase Activity
Adenylyl cyclase activity was determined by measuring 32P‐labeled cAMP formation from α‐32P–labeled ATP, as described previously.47, 48 Briefly, the assay medium containing 50 mmol/L glycylglycine (pH 7.5), 0.5 mmol/L MgATP, α‐32P–labeled ATP ([1.5–10]×106 count per minute), 5 mmol/L MgCl2 (in excess of the ATP concentration), 100 mmol/L NaCl, 0.5 mmol/L cAMP, 1 mmol/L IBMX, 0.1 μmol/L EGTA, 10 μmol/L GTPμS, and an ATP‐regenerating system consisting 2 mmol/L phosphocreatine, 0.1 mg creatine kinase per milliliter, and 0.1 mg myokinase per milliliter in a final volume of 200 μL was preincubated for 2 minutes at 37°C. The reaction was initiated by the addition of the membrane proteins (20–30 μg) to the reaction mixture. The reactions were terminated after 10 minutes by the addition of 0.6 mL of 120 mmol/L zinc acetate. The cAMP was purified by coprecipitation of other nucleotides with ZnCO3, addition of 0.5 mL of 144 mmol/L Na2CO3, and subsequent chromatography by the double‐column system, as described by Salomon.49
Determination of Cell Proliferation
Cell proliferation was quantified by DNA synthesis, which was evaluated by incorporation of 3H‐labeled thymidine into cells, as described previously.50 Subconfluent VSMCs from control and antisense‐treated SHRs and WKY rats were plated in 6‐well plates for 24 hours and were serum deprived for 24 hours to induce cell quiescence. The 3H‐labeled thymidine (1 μCi/ml) was added and further incubated for 4 hours before the cells were harvested. The cells were rinsed twice with ice‐cold PBS and incubated with 5% trichloroacetic acid for 1 hour at 4°C. After being washed twice with ice‐cold water, the cells were incubated with 0.4 N sodium hydroxide solution for 30 minutes at room temperature, and radioactivity was determined using a liquid scintillation counter (Wallac 1409; Perkin Elmer Life Science). Cell viability was checked with the trypan blue exclusion technique and indicated that >90% to 95% of cells were viable.
Determination of Superoxide Anion Production and NADPH Oxidase Activity
Basal superoxide anion production and NADPH oxidase activity in heart, aorta, and kidney were measured using the lucigenin‐enhanced chemiluminescence method with a low concentration (5 μmol/L) of lucigenin, as described previously.20, 51 The heart, aorta, and kidney tissues from control and antisense‐treated SHRs and WKY rats were washed in oxygenated Krebs HEPES buffer and placed in scintillation vials containing lucigenin solution, and the emitted luminescence was measured with a liquid scintillation counter (Wallac 1409; Perkin Elmer Life Science) for 5 minutes. The average luminescence value was estimated, the background value was subtracted, and the result was divided by the total weight of tissue in each sample.
The NADPH oxidase activity in the samples was assessed by adding 10 to 4 mol/L NADH (Sigma‐Aldrich) in the vials before counting. Basal superoxide–induced luminescence was then subtracted from the luminescence value induced by NADH.
Statistical Analysis
Data are expressed as mean±SD and were analyzed using 1‐way ANOVA in conjunction with the Newman–Keuls test with GraphPad Prism 5 software (GraphPad Software). A mean difference between groups was considered statistically significant at P<0.05.
Results
Role of Liposome Quality on Antisense Transfection Efficiency
In the current study, we used freshly formulated liposomes composed of monocationic lipid DOTAP and neutral helper lipid DOPE at a 1:1‐M ratio. A novel upgrade in antisense/liposome‐based hypertension research was the incorporation of 10% DSPE‐PEG(2000) to increase the circulation time of the liposomes. Other important factors affecting circulation time and thus transfection efficiency are size and polydispersity index. Manual extrusion was used to ensure size control and homogeneity. Liposome size was 100 to 200 nm with a polydispersity index of 0.1 measured using dynamic light scattering (Figure 1). Figure 1 shows that antisense was efficiently encapsulated in liposomes, as determined using fluorescently labeled antisense (FITC). Figure 1A shows fluorescently labeled Giα‐2 antisense encapsulated inside liposomes that appeared as green circles under fluorescent microscopy. Figure 1B shows measurement of the size of the PEG‐cationic liposomes and the polydispersity index using dynamic light scattering. The size of the freshly formulated liposomes ranged from 100 to 200 nm with a polydispersity index of 0.1.
Figure 1.
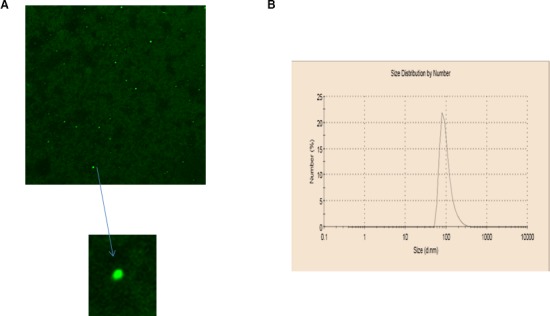
Role of liposome quality in antisense transfection efficiency. A, Antisense encapsulation inside the liposomes. Antisense oligodeoxynucleotide labeled with 5′ fluorescein (FITC) was used to ensure that antisense was encapsulated inside the liposomes. Antisense took the shape of liposomes and appeared as green spherical bodies under fluorescent microscopy; their size ranged from 100 to 200 nm. Measurement of the size of the PEG‐cationic liposomes and polydispersity index using dynamic light scattering. B, The size of the freshly formulated liposomes ranged from 100 to 200 nm with a polydispersity index of 0.1.
Effect of Giα‐2 and Giα‐3 Knockdown on BP and Heart Rate
To investigate the contribution of enhanced expression of Giα‐2 on BP, the effect of Giα‐2 knockdown on BP was examined by single intravenous injection of Giα‐2 antisense into prehypertensive rats aged 3 weeks, and the results are shown in Figure 2. Systolic BP (SBP) (Figure 2A) was not different in SHRs aged 3 weeks compared with age‐matched WKY rats and started to increase from the age of 4 weeks. Treatment of SHRs with Giα‐2 antisense and not with empty liposomes or Giα‐2 sense prevented the increase in SBP up to age 6 weeks (123±2.9 mm Hg); thereafter, SBP started to increase and reached the same level as that of SHR control groups at 9 weeks (190±5 mm Hg). A second injection of Giα‐2 antisense at that time point again decreased BP significantly but not to the level in control WKY rats. In contrast, Giα‐2 antisense treatment did not have any significant effect on the SBP in WKY rats (121.8±1.6 versus 123.8±2 mm Hg). In addition, diastolic BP (Figure 2B) and mean arterial BP (Figure 2C) were slightly higher in all SHR groups compared with WKY groups at the age of 3 weeks. The knockdown of Giα‐2 protein by antisense prevented any further increase in diastolic and mean BP up to age 6 weeks; however, at age 9 weeks, like SBP, diastolic and mean arterial BPs reached the same levels as those of SHR control groups.
Figure 2.
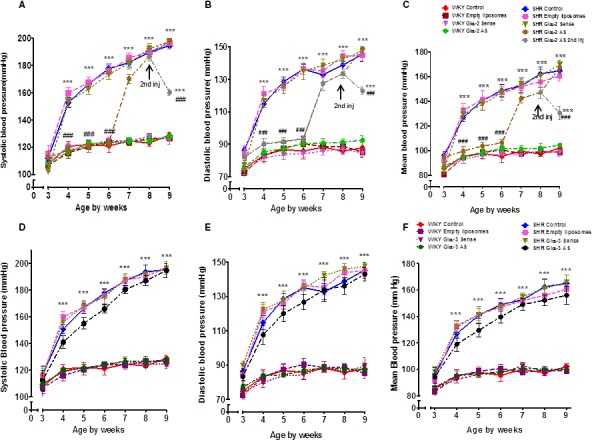
Effect of Giα‐2 and Giα‐3 knockdown on the development of high blood pressure (BP). Antisense (AS) or sense of Giα‐2 and Giα‐3 encapsulated in PEG‐cationic liposomes (1 mg/kg body weight) and empty liposomes were injected intravenously via tail vein once into prehypertensive spontaneously hypertensive rats (SHRs) aged 3 weeks and age‐matched Wistar Kyoto (WKY) rats. A second injection of AS Giα‐2 (1 mg/kg body weight) was given at 8 weeks to the AS‐treated SHR group. Systolic BP (A and D), diastolic BP (B and E), and mean arterial BP (C and F) were monitored weekly using the CODA system until rats were aged 9 weeks. Values are mean±SD of 6–10 rats in each group. ***P<0.001 vs WKY control, ### P<0.001 vs SHR control.
To investigate whether enhanced expression of Giα‐3 also contributes to the development of hypertension in SHRs, the effect of Giα‐3 knockdown on BP was examined using Giα‐3 antisense, and the BP profile is shown in Figure 2D through 2F. SBP (Figure 2D), diastolic BP (Figure 2E), and mean arterial BP (Figure 2F) started to increase at the age of 4 weeks in all groups of control SHRs (SHR control, empty liposome, and Giα‐3 sense group); however, BP was slightly decreased in the SHR group treated with Giα‐3 antisense compared with the control SHR groups, but this decrease was not significant (mean arterial BP: control SHRs 148.5±4.2 mm Hg; Giα‐3 antisense–treated SHRs, 139.7±4.5 mm Hg). At 9 weeks, the Giα‐3 antisense–treated SHR group had the same BP as that of SHR control groups. In contrast, Giα‐3 antisense treatment did not have any significant effect on the mean arterial BP in treated WKY rats (95.6±4.9 versus 98.2±2 mm Hg).
Both Giα‐2 and Giα‐3 antisense treatments did not show any adverse side effects on the health of rats because all rats treated with antisense gained weight during the study period (body weights were 142.5±4.5 g for WKY control rats, 144±3 g for Giα‐2 antisense–treated WKY rats, 141±5 g for Giα‐3 antisense–treated WKY rats, 135±3.5 g for control SHRs, 139.2±3.9 g for Giα‐2‐antisense–treated SHRs, and 136.0±6.0 g for Giα‐3 antisense–treated SHRs). In addition, the ratio of heart weight:body weight was not different in SHRs aged 6 and 9 weeks compared with age‐matched WKY rats, and it was not affected by the antisense treatment, as reported previously.31
Prehypertensive tachycardia has been reported in SHRs.52, 53 To further examine the contribution of enhanced expression of Giα‐2 and Giα‐3 proteins to the regulation of heart rate in SHRs, the effect of antisense of Giα‐2 and Giα‐3 treatment on heart rate was examined in SHRs and WKY rats at age 6 and 9 weeks (Figure 3. At age 6 weeks (Figure 3A), all SHR control groups had higher heart rates than age‐matched WKY rats by ≈25%. Treatment of SHRs with Giα‐2 antisense significantly attenuated increased heart rate by ≈15%. In contrast, Giα‐3 antisense treatment did not have any significant effect on the heart rate. Furthermore, treatment of rats with empty liposomes, Giα‐2 sense, and Giα‐3 sense had no impact on heart rate in SHR and WKY groups. Moreover, the increased heart rate in SHRs at age 9 weeks compared with age‐matched WKY rats was not attenuated by these treatments (Figure 3B).
Figure 3.
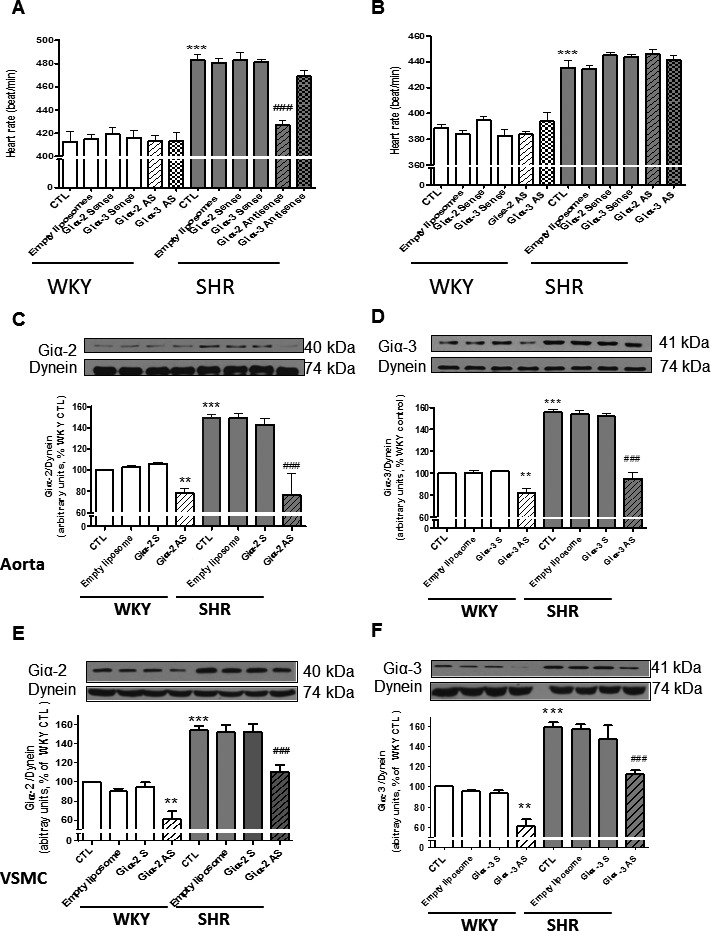
A and B, Effect of Giα‐2 and Giα‐3 knockdown on heart rate in spontaneously hypertensive rats (SHRs) and age‐matched Wistar Kyoto (WKY) rats. Antisense (AS) or sense of Giα‐2 and Giα‐3 encapsulated in PEG‐cationic liposomes (1 mg/kg body weight) and empty liposomes were injected intravenously via tail vein once into prehypertensive SHRs aged 3 weeks and age‐matched WKY rats. Heart rate was monitored weekly using the CODA system. A, Effect of Giα‐2 and Giα‐3 AS, sense, and empty liposomes on heart rate in 6‐week‐old SHRs and WKY rats. B, Effect of Giα‐2 and Giα‐3 AS, sense, and empty liposomes on heart rate in 9‐week‐old SHRs and WKY rats Values are mean±SD of 6–10 rats in each group. ***P<0.001 vs WKY control (CTL), ### P<0.001 vs SHR CTL. C through F, Effect of Giα‐2 and Giα‐3 knockdown on the expression of Giα‐2 or Giα‐3 proteins in aorta and vascular smooth muscle cells (VSMCs) from SHRs aged 6 weeks and age‐matched WKY rats. AS or sense of Giα‐2 and Giα‐3 encapsulated in PEG‐cationic liposomes (1 mg/kg body weight) and empty liposomes were injected intravenously via tail vein once into prehypertensive SHRs aged 3 weeks and age‐matched WKY rats. Aorta (C and D) and VSMC lysates (E and F) from 6‐week‐old SHRs and WKY rats with or without treatments were subjected to Western blotting using antibodies against Giα‐2 (C and E) and Giα‐3 (D and F). The protein bands were quantified by densitometric scanning. The results are expressed as ratio of Gi protein:dynein of WKY rats taken as 100%. Values are mean±SD of 4–6 separate experiments using different rats or cell populations in each group. **P<0.01, ***P<0.001 vs WKY CTL; ### P<0.001 vs SHR CTL.
Effect of Giα‐2 and Giα‐3 Knockdown on Giα Protein Levels in Aorta and Aortic VSMCs From SHRs and WKY Rats
To examine the relationship between BP and Giα protein expression, we determined the levels of Giα proteins by Western blotting in aortas and VSMCs from untreated control SHRs and age‐matched WKY rats and those treated with different interventions at age 6 weeks, when the increase in BP was prevented by Giα‐2 antisense treatment, and age 9 weeks, when BP in antisense‐treated SHRs was augmented to the same level as that of control SHRs. Results indicated that, as reported previously,15, 17 expression of Giα‐2 (Figure 3C and 3E) and Giα‐3 (Figure 3D and 3F) proteins was significantly enhanced in aortas (Figure 3C and 3D) and VSMCs (Figure 3E and 3F) from rats aged 6 weeks by ≈50% to 55% in all SHR groups compared with WKY rats. In addition, the treatment of rats with Giα‐2 antisense but not with sense or empty liposomes completely abolished enhanced expression of Giα‐2 in aorta (Figure 3C); inhibition of ≈70% to 80% was observed in VSMCs (Figure 3E). Similarly, Giα‐3 antisense but not sense or empty liposomes also attenuated enhanced expression of Giα‐3 protein in aortas (Figure 3D) and VSMCs (Figure 3F) from SHRs aged 6 weeks by ≈90% and 60%, respectively. In addition, Giα‐2 and Giα‐3 antisense also attenuated expression of Giα‐2 and Giα‐3, respectively, in aortas and VSMCs from WKY rats by ≈20% to 40%. In contrast, the enhanced expression of Giα‐2 or Giα‐3 was not attenuated by Giα‐2 or Giα‐3 antisense treatments in aortas from SHRs aged 9 weeks (data not shown).
Effect of Giα‐2 and Giα‐3 Knockdown on the Expression of Giα Proteins in Hearts and Kidney From SHRs and WKY Rats
To examine the relationship between heart rate and levels of Giα proteins, we determined the levels of Giα‐2 and Giα‐3 in hearts from SHRs and WKY rats with and without antisense treatment (Figure 4. As reported previously,15, 17 the expression of Giα‐2 (Figure 4A) and Giα‐3 (Figure 4B) was significantly enhanced by 75% and 55%, respectively, in hearts from SHRs compared with WKY rats. In addition, antisense of both Giα‐2 and Giα‐3 restored the enhanced expression of Giα‐2 and Giα‐3 proteins, respectively, to levels in WKY controls. Giα‐2 and Giα‐3 antisense also attenuated the expression of Giα‐2 and Giα‐3, respectively, in hearts from WKY rats by ≈50% and 25%, respectively. In contrast, levels of Giα‐2 (Figure 4C) and Giα‐3 (Figure 4D) proteins that were significantly enhanced by ≈50% in hearts from SHRs aged 9 weeks compared with WKY rats were not attenuated by Giα‐2 or Giα‐3 antisense treatments. These results suggested a role of enhanced levels of Giα‐2 but not Giα‐3 protein in increased heart rate in SHRs.
Figure 4.
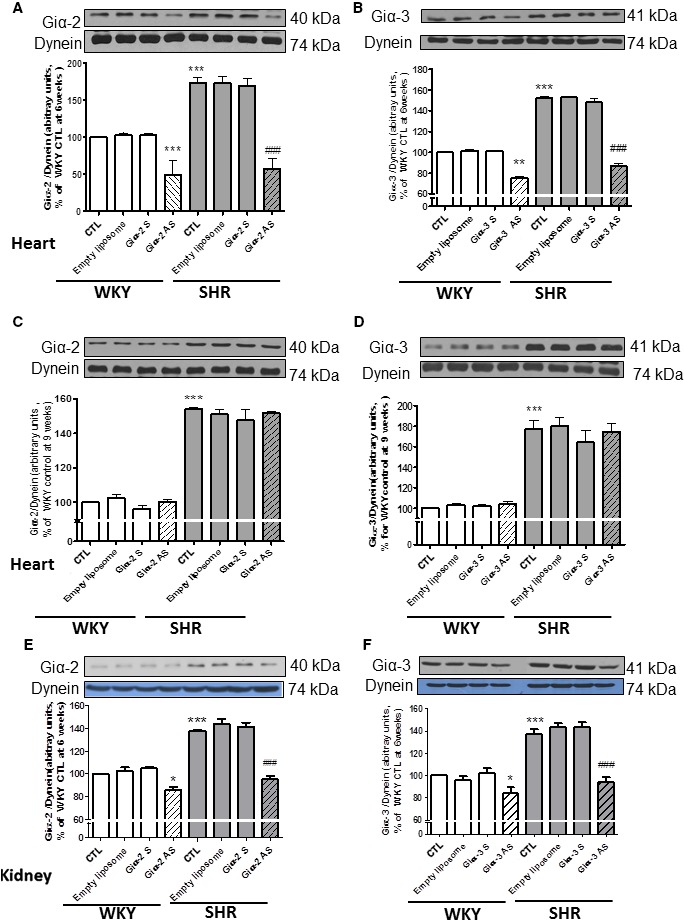
Effect of Giα‐2 and Giα‐3 knockdown on the expression of Giα‐2 or Giα‐3 proteins in hearts from 6‐ or 9 week‐old and in kidney from spontaneously hypertensive rats (SHRs) aged 6 weeks and age‐matched Wistar Kyoto (WKY) rats. Proteins in heart membrane (30 μg) from SHRs aged 6 weeks (A and B) and 9 weeks (C and D) and in kidney (E and F) from 6‐week‐old SHRs and WKY rats with or without treatment were subjected to Western blotting using antibodies against Giα‐2 (A, C, and E) and Giα‐3 (B, D, and F). The protein bands were quantified by densitometric scanning. The results are expressed as the ratio of Gi protein:dynein of WKY rats taken as 100%. Values are mean±SD of 4–6 separate experiments using different rats from each group. *P<0.05, **P<0.01, ***P<0.001vs WKY control (CTL); ### P<0.001 vs SHR CTL.
In kidneys from SHRs and WKY rats, we examined the effect of Giα‐2 and Giα‐3 antisense on the expression of Giα‐2 and Giα‐3 proteins, respectively, which were shown to play an important role in the regulation of renal vascular tone in SHRs.27, 54 Results shown in Figure 4 indicated that the expression of Giα‐2 (Figure 4E) and Giα‐3 (Figure 4F) proteins in kidneys from SHRs was significantly augmented by ≈40% compared with WKY rats, and this increase was completely restored to levels in WKY rats by antisense treatment. In addition, Giα‐2 and Giα‐3 antisense also attenuated the expression of Giα‐2 and Giα‐3, respectively, in kidneys from WKY rats by ≈25%.
Effect of Giα‐2 Knockdown on Giα‐3 Expression and Vice Versa
To examine the specificity of the antisense oligonucleotides, the effect of Giα‐2 antisense treatment on Giα‐3 protein expression and the effect of Giα‐3 antisense treatment on Giα‐2 protein expression was assessed in the hearts (Figure 5A and 5B) and aortas (Figure 5C and 5D) from SHRs aged 6 weeks and age‐matched WKY rats. Enhanced expression of Giα‐2 was not attenuated by the antisense of Giα‐3 (Figure 5A and 5C), and enhanced expression of Giα‐3 was not attenuated by the antisense of Giα‐2 (Figure 5B and 5D). In addition, the antisense of Giα‐3 and Giα‐2 was also ineffective in attenuating the expression of Giα‐2 and Giα‐3 proteins, respectively, in hearts and aortas from WKY rats.
Figure 5.
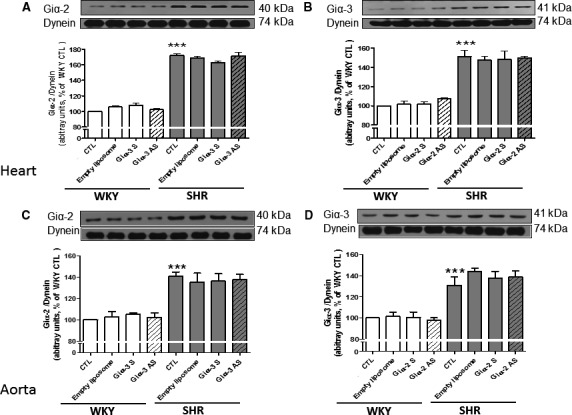
Effect of knockdown of Giα‐2 antisense (AS) on the expression of Giα‐3 protein and vice versa. Heart (A and B) and aorta (C and D) from spontaneously hypertensive rats (SHRs) aged 9 weeks and age‐matched Wistar Kyoto (WKY) rats with or without treatment were subjected to Western blotting using antibodies against Giα‐2 (A and C) and Giα‐3 (B and D). The protein bands were quantified by densitometric scanning. The results are expressed as the ratio of Gi protein:dynein of WKY rats taken as 100%. Values are mean±SEM of 4 separate experiments using different rats from each group. ***P<0.001 vs WKY control (CTL).
Effect of Giα‐2 and Giα‐3 Knockdown on Adenylyl Cyclase Inhibition in Hearts From SHRs and WKY Rats
To investigate whether knockdown of Giα‐2 and Giα‐3 by antisense treatment was reflected in attenuation of Gi‐mediated functions, we examined the effect of antisense treatment on the inhibitory effects of oxotremorine, which interacts with muscarinic receptors coupled to adenylyl cyclase inhibition through Giα proteins.55 Results shown in Figure 6 indicated that oxotremorine inhibited adenylyl cyclase activity in hearts from SHRs and WKY rats aged 6 weeks; however, as reported previously,32 the extent of inhibition was significantly greater in SHRs. Oxotremorine, for example, inhibited adenylyl cyclase activity by ≈30% in WKY rats and ≈45%, in SHRs. Knockdown of Giα‐2 by Giα‐2 antisense attenuated oxotremorine‐mediated inhibition of adenylyl cyclase by ≈55% (13.5+4.9% versus 29+2.0%) in WKY rats and ≈35% (28.0+2.8% versus 43.7%) in SHRs. In contrast, the knockdown of Giα‐3 by Giα‐3 antisense did not have any effect on the percentage of inhibition of adenylyl cyclase by oxotremorine in SHRs and WKY rats (29.0+2.0% versus 28.5+1.0% in WKY rats and 43.7+1.5% versus 41.7+3.8% in SHRs).
Figure 6.
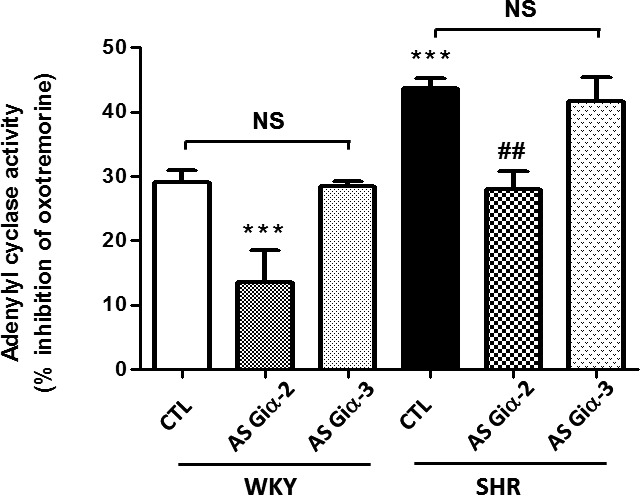
Effect of knockdown of Giα‐2 antisense (AS) on M2 receptor‐dependent Gi functions. Adenylyl cyclase activity was determined in the absence or presence of 5 μmol/L oxotremorine in heart particulate fractions from control (CTL) and Giα‐2 AS‐treated 6‐week‐old spontaneously hypertensive rats (SHRs) and Wistar Kyoto (WKY) rats. Values are mean±SD of 4 separate experiments using different rats from each group. ***P<0.001 vs WKY CTL; ## P<0.01 vs SHR CTL. NS indicates not significant.
Effect of Giα‐2 and Giα‐3 Knockdown on Proliferation of VSMCs From SHRs and WKY Rats
Because enhanced expression of Giα proteins is implicated in hyperproliferation of VSMCs from SHRs,56 it was of interest to examine whether knocking down of Giα proteins by antisense treatment would also result in attenuation of hyperproliferation of VSMCs from SHRs. Results shown in Figure 7 indicated that the proliferation of VSMCs from SHRs was significantly augmented by ≈170% compared with WKY rats, and Giα‐2 and Giα‐3 antisense treatment decreased hyperproliferation by ≈70% and 20%, respectively. In addition, the proliferation of VSMCs from WKY rats was attenuated significantly by both Giα‐2 and Giα‐3 antisense treatments.
Figure 7.
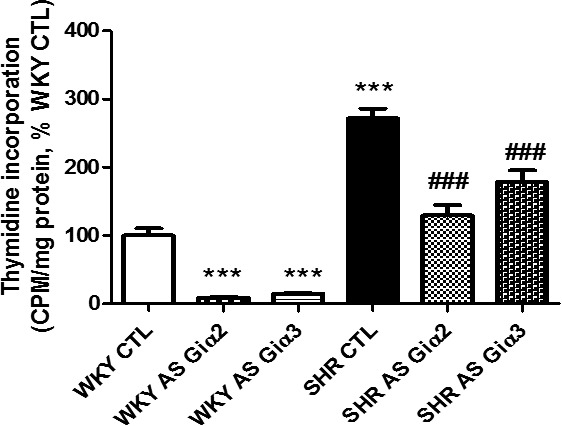
Effect of knockdown Giα‐2 and Giα‐3 antisense (AS) on thymidine incorporation in vascular smooth muscle cells (VSMCs) from spontaneously hypertensive rats (SHRs) aged 6 weeks and age‐matched Wistar Kyoto (WKY) rats. Thymidine incorporation in confluent VSMCs from control (CTL) and AS‐treated SHRs and WKY rats was determined. Results are expressed as percentage of WKY CTL (taken as 100%). Values are mean±SD of 4 separate experiments using different rats from each group. ***P<0.001 vs WKY CTL; ### P<0.001 vs SHR CTL.
Effect of Giα‐2 Knockdown on the Production of Superoxide Anion and NADPH Oxidase Activity in SHRs Aged 6 Weeks and Age‐Matched WKY Rats
Oxidative stress caused by the overproduction of reactive oxygen species contributes to the pathophysiology of cardiovascular diseases including hypertension.57, 58 We previously reported the implication of enhanced expression of Giα proteins in enhanced oxidative stress exhibited by VSMCs from SHRs.51 To investigate whether knockdown of enhanced levels of Giα‐2 and Giα‐3 proteins in SHRs by antisense treatments also resulted in attenuation of oxidative stress, the effect of antisense treatments on superoxide anion and NADPH oxidative activity was examined in hearts, aortas, and kidneys from SHRs aged 6 weeks and age‐matched WKY rats (Figure 8. As reported previously,12, 20, 51 the production of superoxide anion that was increased by about ≈35%, 25%, and 45% in hearts, aortas, and kidneys, respectively, from SHRs compared with WKY rats (Figure 8A through 8C) was significantly attenuated by Giα‐2 and Giα‐3 antisense treatment. In addition, NADPH oxidase activity was also significantly enhanced by ≈70%, 100%, and 45% in hearts, aortas, and kidneys, respectively, from SHRs compared with WKY rats (Figure 8D through 8F), and Giα‐2 and Giα‐3 antisense treatments almost completely restored the enhanced activity to levels in WKY controls. In contrast, Giα‐2 and Giα‐3 antisense decreased the production of superoxide anion and NADPH oxidase activity in these tissues from WKY rats.
Figure 8.
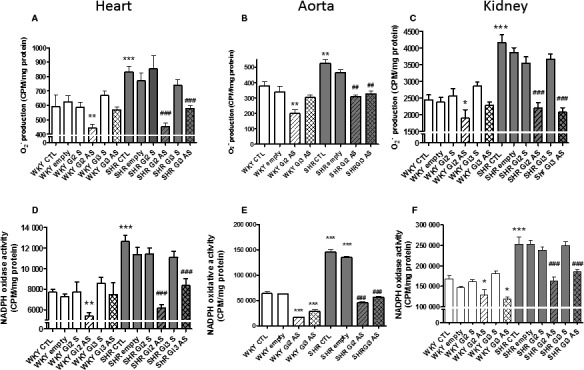
Effect of knockdown of Giα‐2 and Giα‐3 antisense (AS) on the production of superoxide anion () production and NADPH oxidative activity in heart, aorta, and kidney from spontaneously hypertensive rats (SHRs) aged 6 weeks and age‐ matched Wistar Kyoto (WKY) rats. The production and NADPH oxidase activity were determined in heart (A and D), aorta (B and E), and kidney (C and F) from SHRs aged 6 weeks and age‐matched WKY rats with or without AS treatment. Values are mean±SD of 4 separate experiments by using different rats from each group. *P<0.05, **P<0.01, ***P<0.001 vs WKY control (CTL); ## P<0.01, ### P<0.001 vs SHR CTL. CPM indicates count per minute.
Discussion
We previously showed that enhanced expression of Giα proteins may be the contributing factor in the pathogenesis of hypertension in SHRs.15, 16, 17, 18, 19, 20, 21 This finding was supported by our study showing that the intraperitoneal injection of pertussis toxin, which inactivates both Giα‐2 and Giα‐3, in prehypertensive SHRs aged 2 weeks attenuated the development of high BP.32 Furthermore, we showed recently that treatment of SHRs with natriureic peptide receptor C agonist C‐ANP4‐23, which inhibits the enhanced expression of both Giα‐2 and Giα‐3 proteins in VSMCs from SHRs,12, 59 attenuated the development of hypertension.12 Several studies using knockout mice have shown a role of Giα‐2 in physiological functions.60, 61, 62, 63, 64, 65 Nagata et al showed that ablation of Giα‐2 in mice resulted in attenuation of muscarinic M2 receptor–mediated antiadrenergic effect on β‐adrenergic receptor–induced contractility and calcium currents in adult cardiomyocytes.64 The disruption of the Giα‐2 gene in mice was also shown to result in impaired platelet activation and aggregation,61 ulcerative colitis, and adrenocarcinoma of the colon.65 Furthermore, a lack of Giα‐2 proteins has been shown to result in dilated cardiomyopathy and increased mortality in β1‐adrenoceptor–overexpressing mice62 and in increased infarct size in the heart in myocardial ischemia reperfusion injury.63 In addition, transgenic mice overexpressing Gi inhibitor peptide, which causes functional knockout of cardiac Giα‐2 signaling when subjected to ischemia reperfusion, were shown to have increased infarct size.60 In the present study, by using antisense oligodeoxynucleotide encapsulated in PEG‐cationic liposomes to knock down the Giα‐2 and Giα‐3 proteins in SHRs, we reported for the first time that enhanced expression of Giα‐2 and not Giα‐3 plays a major role in the development of hypertension.
We found that treatment of prehypertensive SHRs aged 3 weeks with a single intravenous injection of 1 mg/kg body weight of Giα‐2 antisense prevented the development of hypertension up to age 6 weeks and was associated with attenuation of enhanced expression of Giα‐2 protein, which has been implicated as a contributing factor in the pathogenesis of hypertension.15, 17 In contrast, the increase in BP at age 9 weeks may be due to the elimination of antisense from the system or to the de novo synthesis of Gi proteins. This notion was supported by our results showing that Giα‐2 protein expression that was attenuated by antisense treatment in SHRs aged 6 weeks was enhanced in SHRs aged 9 weeks and was not attenuated by antisense treatment and thus contributed to increased BP. Our results are in accordance with earlier studies showing that the effect of antisense of β1‐adrenoceptor on BP had worn off after 20 days.40 In contrast, antisense treatment decreased the expression of Giα‐2 proteins but not BP in WKY rats, suggesting that enhanced expression of Giα protein is implicated in the development of high BP in SHRs. Our results are consistent with our previous studies showing that pertussis toxin, which attenuated the expression of Giα proteins in WKY rats, did not affect BP.32 We also showed that BP in all SHR groups was slightly higher compared with WKY groups at age 3 weeks. This result may be attributed to enhanced expression of Giα‐2 protein and vascular changes that occur in the prehypertensive state.31, 66, 67 Alternatively, knockdown of Giα‐3 protein in SHRs using Giα‐3 antisense, which decreased the enhanced levels of Giα‐3 proteins, did not significantly decrease BP. It is important to note that the extent of attenuation of enhanced expression of Giα‐2 and Giα‐3 proteins by Giα‐2 and Giα‐3 antisense treatment, respectively, was almost the same, whereas high BP was decreased by knocking down only Giα‐2 and not Giα‐3 proteins. This result suggests the contribution of enhanced expression of Giα‐2 protein to the development of hypertension.
Several studies have shown that knocking out one isoform of Giα protein upregulates the expression of other isoforms of Giα protein in knockout mice.62, 68 In the current study, however, the effect of antisense was very specific because Giα‐2 antisense attenuated the expression of the Giα‐2 protein without affecting the expression of Giα‐3 protein, and Giα‐3 antisense attenuated the expression of Giα‐3 and not Giα‐2 protein. Consequently, the lack of effect of Giα‐3 antisense on attenuation of high BP may not be caused by upregulation of Giα‐2 and further suggests a role for enhanced expression of Giα‐2 in the development of high BP in SHRs.
We also reported for the first time that enhanced expression of Giα‐2 protein in SHRs contributes to increased heart rate in SHRs because the knockdown of Giα‐2 protein attenuated the enhanced heart rate in SHRs aged 6 weeks. Augmented expression of Giα‐3 protein did not appear to play a role in the regulation of heart rate because the knockdown of Giα‐3 protein by antisense treatment was ineffective in attenuating the enhanced heart rate in SHRs. Consequently, it may be suggested that the Giα‐2 protein may be a major contributor to the regulation of heart rate in SHRs. The molecular mechanism responsible for Giα‐2–mediated regulation of heart rate appears to involve cAMP because knockdown of Giα‐2 protein by Giα‐2‐antisense, which attenuated increased heart rate, also attenuated enhanced inhibition of adenylyl cyclase by the Giα‐2–coupled M2 receptor agonist oxotremorine in hearts from SHRs. In this regard, several G protein–coupled receptors have been shown to regulate cardiac contractility via cAMP and the Ca2+ pathway.69, 70, 71 Whether Giα‐2 antisense also attenuates intracellular levels of Ca2+ needs to be investigated. Our results, however, agree with the study by Nagata et al, who showed a role for Giα‐2 but not Giα‐3 in the regulation of cardiac contractility and calcium currents by muscarinic receptor in adult cardiomyocytes.64
In the present study, we demonstrated that Giα‐2 and Giα‐3 antisense treatment also attenuated enhanced expression of Giα‐2 and Giα‐3 proteins in kidney, a target tissue involved in the regulation of BP. Because enhanced expression of Giα proteins in kidneys of SHRs has been shown to contribute to increased renal vascular resistance and decreased renal blood flow that results in augmentation of BP in SHRs,27 it may be suggested that Giα‐2 antisense–induced attenuation of enhanced expression of Giα‐2 proteins in kidney may also participate in attenuation of high BP in SHRs by decreasing renal vascular resistance and increasing renal blood flow.
The attenuation of high BP by knockdown of Giα‐2 is also associated with inhibition of enhanced oxidative stress and hyperproliferation of VSMCs, suggesting that attenuation of oxidative stress and hyperproliferation of VSMCs may be a mechanism contributing to the attenuation of high BP and tachycardia in SHRs. In this regard, increased oxidative stress caused by overproduction of reactive oxygen species and hyperproliferation of VSMCs have been shown to contribute to the pathophysiology of cardiovascular diseases such as hypertension and atherosclerosis.57, 58 Alternatively, knockdown of Giα‐3 in SHRs by Giα‐3 antisense treatment also resulted in attenuation of enhanced oxidative stress and hyperproliferation of VSMCs in SHRs. It appears, however, that inhibition of enhanced oxidative stress and hyperproliferation induced by Giα‐3 antisense may not play a role in decreasing high BP in SHRs.
Perspectives
Hypertension is a multifactorial disease. The present study demonstrated an important role for Giα‐2 protein in the regulation of BP. Moreover, it showed for the first time that a single intravenous injection of Giα‐2 antisense, and not Giα‐3 antisense, encapsulated in PEG‐cationic liposomes prevents the development of high BP and attenuates tachycardia in SHRs. Based on these findings, it may be suggested that the highly specific gene therapeutic agents encapsulated in nanoliposomes targeting Giα‐2 protein may have the potential for use in the treatment and/or prevention of hypertension and tachycardia.
Sources of Funding
This project was supported by fund from the Canadian Institute of Health Research (CIHR) (MOP‐53074).
Disclosures
None.
(J Am Heart Assoc. 2016;5:e004594 doi: 10.1161/JAHA.116.004594)
References
- 1. Gilman AG. G proteins: transducers of receptor‐generated signals. Annu Rev Biochem. 1987;56:615–649. [DOI] [PubMed] [Google Scholar]
- 2. Cockcroft S, Gomperts BD. Role of guanine nucleotide binding protein in the activation of polyphosphoinositide phosphodiesterase. Nature. 1985;314:534–536. [DOI] [PubMed] [Google Scholar]
- 3. Litosch I, Wallis C, Fain JN. 5‐Hydroxytryptamine stimulates inositol phosphate production in a cell‐free system from blowfly salivary glands. Evidence for a role of GTP in coupling receptor activation to phosphoinositide breakdown. J Biol Chem. 1985;260:5464–5471. [PubMed] [Google Scholar]
- 4. Rodbell M, Krans HM, Pohl SL, Birnbaumer L. The glucagon‐sensitive adenyl cyclase system in plasma membranes of rat liver. IV. Effects of guanyl nucleotides on binding of 125I‐glucagon. J Biol Chem. 1971;246:1872–1876. [PubMed] [Google Scholar]
- 5. Pfaffinger PJ, Martin JM, Hunter DD, Nathanson NM, Hille B. GTP‐binding proteins couple cardiac muscarinic receptors to a K channel. Nature. 1985;317:536–538. [DOI] [PubMed] [Google Scholar]
- 6. Neer EJ. Heterotrimeric G proteins: organizers of transmembrane signals. Cell. 1995;80:249–257. [DOI] [PubMed] [Google Scholar]
- 7. Hempel A, Noll T, Bach C, Piper HM, Willenbrock R, Hohnel K, Haller H, Luft FC. Atrial natriuretic peptide clearance receptor participates in modulating endothelial permeability. Am J Physiol. 1998;275:H1818–H1825. [DOI] [PubMed] [Google Scholar]
- 8. Noll T, Hempel A, Piper HM. Neuropeptide Y reduces macromolecule permeability of coronary endothelial monolayers. Am J Physiol. 1996;271:H1878–H1883. [DOI] [PubMed] [Google Scholar]
- 9. Feraille E, Doucet A. Sodium‐potassium‐adenosinetriphosphatase‐dependent sodium transport in the kidney: hormonal control. Physiol Rev. 2001;81:345–418. [DOI] [PubMed] [Google Scholar]
- 10. Kinoshita S, Sidhu A, Felder RA. Defective dopamine‐1 receptor adenylate cyclase coupling in the proximal convoluted tubule from the spontaneously hypertensive rat. J Clin Invest. 1989;84:1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsuda K, Tsuda S, Nishio I, Goldstein M, Masuyama Y. Modulation of [3H]dopamine release by neuropeptide Y in rat striatal slices. Eur J Pharmacol. 1997;321:5–11. [DOI] [PubMed] [Google Scholar]
- 12. Li Y, Sarkar O, Brochu M, Anand‐Srivastava MB. Natriuretic peptide receptor‐C attenuates hypertension in spontaneously hypertensive rats: role of nitroxidative stress and Gi proteins. Hypertension. 2014;63:846–855. [DOI] [PubMed] [Google Scholar]
- 13. Feldman AM, Cates AE, Veazey WB, Hershberger RE, Bristow MR, Baughman KL, Baumgartner WA, Van Dop C. Increase of the 40,000‐mol wt pertussis toxin substrate (G protein) in the failing human heart. J Clin Invest. 1988;82:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bohm M, Gierschik P, Jakobs KH, Pieske B, Schnabel P, Ungerer M, Erdmann E. Increase of Gi alpha in human hearts with dilated but not ischemic cardiomyopathy. Circulation. 1990;82:1249–1265. [DOI] [PubMed] [Google Scholar]
- 15. Anand‐Srivastava MB. Enhanced expression of inhibitory guanine nucleotide regulatory protein in spontaneously hypertensive rats. Relationship to adenylate cyclase inhibition. Biochem J. 1992;288(Pt 1):79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anand‐Srivastava MB, de Champlain J, Thibault C. DOCA‐salt hypertensive rat hearts exhibit altered expression of G‐proteins. Am J Hypertens. 1993;6:72–75. [DOI] [PubMed] [Google Scholar]
- 17. Anand‐Srivastava MB, Picard S, Thibault C. Altered expression of inhibitory guanine nucleotide regulatory proteins (Gi alpha) in spontaneously hypertensive rats. Am J Hypertens. 1991;4:840–843. [DOI] [PubMed] [Google Scholar]
- 18. Di Fusco F, Anand‐Srivastava MB. Enhanced expression of Gi proteins in non‐hypertrophic hearts from rats with hypertension‐induced by L‐NAME treatment. J Hypertens. 2000;18:1081–1090. [DOI] [PubMed] [Google Scholar]
- 19. Ge C, Garcia R, Anand‐Srivastava MB. Altered expression of Gi‐protein and adenylyl cyclase activity in hearts from one kidney one clip hypertensive rats: effect of captopril. J Hypertens. 1999;17:1617–1626. [DOI] [PubMed] [Google Scholar]
- 20. Lappas G, Daou GB, Anand‐Srivastava MB. Oxidative stress contributes to the enhanced expression of Gialpha proteins and adenylyl cyclase signaling in vascular smooth muscle cells from spontaneously hypertensive rats. J Hypertens. 2005;23:2251–2261. [DOI] [PubMed] [Google Scholar]
- 21. Thibault C, Anand‐Srivastava MB. Altered expression of G‐protein mRNA in spontaneously hypertensive rats. FEBS Lett. 1992;313:160–164. [DOI] [PubMed] [Google Scholar]
- 22. Bohm M, Gierschik P, Knorr A, Larisch K, Weismann K, Erdmann E. Role of altered G‐protein expression in the regulation of myocardial adenylate cyclase activity and force of contraction in spontaneous hypertensive cardiomyopathy in rats. J Hypertens. 1992;10:1115–1128. [DOI] [PubMed] [Google Scholar]
- 23. Clark CJ, Milligan G, McLellan AR, Connell JM. Guanine nucleotide regulatory protein levels and function in spontaneously hypertensive rat vascular smooth‐muscle cells. Biochim Biophys Acta. 1992;1136:290–296. [DOI] [PubMed] [Google Scholar]
- 24. McLellan AR, Milligan G, Houslay MD, Connell JM. G‐proteins in experimental hypertension: a study of spontaneously hypertensive rat myocardial and renal cortical plasma membranes. J Hypertens. 1993;11:365–372. [DOI] [PubMed] [Google Scholar]
- 25. Bohm M, Gierschik P, Knorr A, Schmidt U, Weismann K, Erdmann E. Cardiac adenylyl cyclase, beta‐adrenergic receptors, and G proteins in salt‐sensitive hypertension. Hypertension. 1993;22:715–727. [DOI] [PubMed] [Google Scholar]
- 26. Wainford RD, Carmichael CY, Pascale CL, Kuwabara JT. Gαi2‐protein‐mediated signal transduction: central nervous system molecular mechanism countering the development of sodium‐dependent hypertension. Hypertension. 2015;65:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kost CK Jr, Herzer WA, Li PJ, Jackson EK. Pertussis toxin‐sensitive G‐proteins and regulation of blood pressure in the spontaneously hypertensive rat. Clin Exp Pharmacol Physiol. 1999;26:449–455. [DOI] [PubMed] [Google Scholar]
- 28. Tabrizchi R, Triggle CR. Pressor actions of arginine vasopressin in pithed Sprague‐Dawley, Wistar‐Kyoto and spontaneously hypertensive rats before and after treatment with nifedipine or pertussis toxin. J Hypertens. 1991;9:813–818. [DOI] [PubMed] [Google Scholar]
- 29. Jones DT, Reed RR. Molecular cloning of five GTP‐binding protein cDNA species from rat olfactory neuroepithelium. J Biol Chem. 1987;262:14241–14249. [PubMed] [Google Scholar]
- 30. Marcil J, de Champlain J, Anand‐Srivastava MB. Overexpression of Gi‐proteins precedes the development of DOCA‐salt‐induced hypertension: relationship with adenylyl cyclase. Cardiovasc Res. 1998;39:492–505. [DOI] [PubMed] [Google Scholar]
- 31. Marcil J, Thibault C, Anand‐Srivastava MB. Enhanced expression of Gi‐protein precedes the development of blood pressure in spontaneously hypertensive rats. J Mol Cell Cardiol. 1997;29:1009–1022. [DOI] [PubMed] [Google Scholar]
- 32. Li Y, Anand‐Srivastava MB. Inactivation of enhanced expression of Gi proteins by pertussis toxin attenuates the development of high blood pressure in spontaneously hypertensive rats. Circ Res. 2002;91:247–254. [DOI] [PubMed] [Google Scholar]
- 33. Van Oekelen D, Luyten WH, Leysen JE. Ten years of antisense inhibition of brain G‐protein‐coupled receptor function. Brain Res Brain Res Rev. 2003;42:123–142. [DOI] [PubMed] [Google Scholar]
- 34. Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA. 1978;75:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garcia SI, Alvarez AL, Porto PI, Garfunkel VM, Finkielman S, Pirola CJ. Antisense inhibition of thyrotropin‐releasing hormone reduces arterial blood pressure in spontaneously hypertensive rats. Hypertension. 2001;37:365–370. [DOI] [PubMed] [Google Scholar]
- 36. Gyurko R, Wielbo D, Phillips MI. Antisense inhibition of AT1 receptor mRNA and angiotensinogen mRNA in the brain of spontaneously hypertensive rats reduces hypertension of neurogenic origin. Regul Pept. 1993;49:167–174. [DOI] [PubMed] [Google Scholar]
- 37. Kagiyama S, Eguchi S, Frank GD, Inagami T, Zhang YC, Phillips MI. Angiotensin II‐induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation. 2002;106:909–912. [DOI] [PubMed] [Google Scholar]
- 38. Nguyen TT, Cao N, Short JL, White PJ. Intravenous insulin‐like growth factor‐I receptor antisense treatment reduces angiotensin receptor expression and function in spontaneously hypertensive rats. J Pharmacol Exp Ther. 2006;318:1171–1177. [DOI] [PubMed] [Google Scholar]
- 39. Phillips MI, Wielbo D, Gyurko R. Antisense inhibition of hypertension: a new strategy for renin‐angiotensin candidate genes. Kidney Int. 1994;46:1554–1556. [DOI] [PubMed] [Google Scholar]
- 40. Zhang YC, Bui JD, Shen L, Phillips MI. Antisense inhibition of beta(1)‐adrenergic receptor mRNA in a single dose produces a profound and prolonged reduction in high blood pressure in spontaneously hypertensive rats. Circulation. 2000;101:682–688. [DOI] [PubMed] [Google Scholar]
- 41. Crooke RM, Graham MJ, Martin MJ, Lemonidis KM, Wyrzykiewiecz T, Cummins LL. Metabolism of antisense oligonucleotides in rat liver homogenates. J Pharmacol Exp Ther. 2000;292:140–149. [PubMed] [Google Scholar]
- 42. Eder PS, DeVine RJ, Dagle JM, Walder JA. Substrate specificity and kinetics of degradation of antisense oligonucleotides by a 3′ exonuclease in plasma. Antisense Res Dev. 1991;1:141–151. [DOI] [PubMed] [Google Scholar]
- 43. Allen TM, Hansen C, Martin F, Redemann C, Yau‐Young A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half‐lives in vivo. Biochim Biophys Acta. 1991;1066:29–36. [DOI] [PubMed] [Google Scholar]
- 44. Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M. Lipofection: a highly efficient, lipid‐mediated DNA‐transfection procedure. Proc Natl Acad Sci USA. 1987;84:7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kurtz TW, Griffin KA, Bidani AK, Davisson RL, Hall JE; Subcommittee of P, Public Education of the American Heart A . Recommendations for blood pressure measurement in humans and experimental animals. Part 2: blood pressure measurement in experimental animals: a statement for professionals from the subcommittee of professional and public education of the American Heart Association Council on High Blood Pressure Research. Hypertension. 2005;45:299–310. [DOI] [PubMed] [Google Scholar]
- 46. Liau G, Chan LM. Regulation of extracellular matrix RNA levels in cultured smooth muscle cells. Relationship to cellular quiescence. J Biol Chem. 1989;264:10315–10320. [PubMed] [Google Scholar]
- 47. Anand‐Srivastava MB. Downregulation of atrial natriuretic peptide ANP‐C receptor is associated with alterations in G‐protein expression in A10 smooth muscle cells. Biochemistry. 2000;39:6503–6513. [DOI] [PubMed] [Google Scholar]
- 48. Anand‐Srivastava MB, Srivastava AK, Cantin M. Pertussis toxin attenuates atrial natriuretic factor‐mediated inhibition of adenylate cyclase. Involvement of inhibitory guanine nucleotide regulatory protein. J Biol Chem. 1987;262:4931–4934. [PubMed] [Google Scholar]
- 49. Salomon Y. Adenylate cyclase assay. Adv Cyclic Nucleotide Res. 1979;10:35–55. [PubMed] [Google Scholar]
- 50. Hashim S, Li Y, Anand‐Srivastava MB. Small cytoplasmic domain peptides of natriuretic peptide receptor‐C attenuate cell proliferation through Gialpha protein/MAP kinase/PI3‐kinase/AKT pathways. Am J Physiol Heart Circ Physiol. 2006;291:H3144–H3153. [DOI] [PubMed] [Google Scholar]
- 51. Saha S, Li Y, Lappas G, Anand‐Srivastava MB. Activation of natriuretic peptide receptor‐C attenuates the enhanced oxidative stress in vascular smooth muscle cells from spontaneously hypertensive rats: implication of Gialpha protein. J Mol Cell Cardiol. 2008;44:336–344. [DOI] [PubMed] [Google Scholar]
- 52. Dickhout JG, Lee RM. Blood pressure and heart rate development in young spontaneously hypertensive rats. Am J Physiol. 1998;274:H794–H800. [DOI] [PubMed] [Google Scholar]
- 53. Tucker DC, Johnson AK. Development of autonomic control of heart rate in genetically hypertensive and normotensive rats. Am J Physiol. 1984;246:R570–R577. [DOI] [PubMed] [Google Scholar]
- 54. Vyas SJ, Blaschak CM, Chinoy MR, Jackson EK. Angiotensin II‐induced changes in G‐protein expression and resistance of renal microvessels in young genetically hypertensive rats. Mol Cell Biochem. 2000;212:121–129. [PubMed] [Google Scholar]
- 55. Kostenis E, Zeng FY, Wess J. Structure‐function analysis of muscarinic receptors and their associated G proteins. Life Sci. 1999;64:355–362. [DOI] [PubMed] [Google Scholar]
- 56. Bou Daou G, Li Y, Anand‐Srivastava MB. Enhanced expression of Giα proteins contributes to the hyperproliferation of vascular smooth muscle cells from spontaneously hypertensive rats via MAP kinase‐ and PI3 kinase‐independent pathways. Can J Physiol Pharmacol. 2016;94:49–58. [DOI] [PubMed] [Google Scholar]
- 57. Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. [DOI] [PubMed] [Google Scholar]
- 58. Molavi B, Mehta JL. Oxidative stress in cardiovascular disease: molecular basis of its deleterious effects, its detection, and therapeutic considerations. Curr Opin Cardiol. 2004;19:488–493. [DOI] [PubMed] [Google Scholar]
- 59. El Andalousi J, Li Y, Anand‐Srivastava MB. Natriuretic peptide receptor‐C agonist attenuates the expression of cell cycle proteins and proliferation of vascular smooth muscle cells from spontaneously hypertensive rats: role of Gi proteins and MAPkinase/PI3kinase signaling. PLoS One. 2013;8:e76183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. DeGeorge BR Jr, Gao E, Boucher M, Vinge LE, Martini JS, Raake PW, Chuprun JK, Harris DM, Kim GW, Soltys S, Eckhart AD, Koch WJ. Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation. 2008;117:1378–1387. [DOI] [PubMed] [Google Scholar]
- 61. Jantzen HM, Milstone DS, Gousset L, Conley PB, Mortensen RM. Impaired activation of murine platelets lacking G alpha(i2). J Clin Invest. 2001;108:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Keller K, Maass M, Dizayee S, Leiss V, Annala S, Koth J, Seemann WK, Muller‐Ehmsen J, Mohr K, Nurnberg B, Engelhardt S, Herzig S, Birnbaumer L, Matthes J. Lack of Gαi2 leads to dilative cardiomyopathy and increased mortality in β1‐adrenoceptor overexpressing mice. Cardiovasc Res. 2015;108:348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kohler D, Devanathan V, Bernardo de Oliveira Franz C, Eldh T, Novakovic A, Roth JM, Granja T, Birnbaumer L, Rosenberger P, Beer‐Hammer S, Nurnberg B. Gαi2‐ and Gαi3‐deficient mice display opposite severity of myocardial ischemia reperfusion injury. PLoS One. 2014;9:e98325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nagata K, Ye C, Jain M, Milstone DS, Liao R, Mortensen RM. Galpha(i2) but not Galpha(i3) is required for muscarinic inhibition of contractility and calcium currents in adult cardiomyocytes. Circ Res. 2000;87:903–909. [DOI] [PubMed] [Google Scholar]
- 65. Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2‐deficient mice. Nat Genet. 1995;10:143–150. [DOI] [PubMed] [Google Scholar]
- 66. Dickhout JG, Lee RM. Structural and functional analysis of small arteries from young spontaneously hypertensive rats. Hypertension. 1997;29:781–789. [DOI] [PubMed] [Google Scholar]
- 67. Lee RM. Vascular changes at the prehypertensive phase in the mesenteric arteries from spontaneously hypertensive rats. Blood Vessels. 1985;22:105–126. [DOI] [PubMed] [Google Scholar]
- 68. Dizayee S, Kaestner S, Kuck F, Hein P, Klein C, Piekorz RP, Meszaros J, Matthes J, Bjrnbaumer L, Nurnberg B, Herzig S. Gαi2‐ and Gαi3‐specific regulation of voltage‐dependent L‐type calcium channels in cardiomyocytes. PLoS One. 2011;6:e24979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tilley DG. G protein‐dependent and G protein‐independent signaling pathways and their impact on cardiac function. Circ Res. 2011;109:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Salazar NC, Chen J, Rockman HA. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim Biophys Acta. 2007;1768:1006–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kawaguchi H, Sano H, Iizuka K, Okada H, Kudo T, Kageyama K, Muramoto S, Murakami T, Okamoto H, Mochizuki N. Phosphatidylinositol metabolism in hypertrophic rat heart. Circ Res. 1993;72:966–972. [DOI] [PubMed] [Google Scholar]