

Abstract
Background
Reprogramming of cardiac fibroblasts into induced cardiomyocyte‐like cells represents a promising potential new therapy for treating heart disease, inducing significant improvements in postinfarct ventricular function in rodent models. Because reprogramming factors effective in transdifferentiating rodent cells are not sufficient to reprogram human cells, we sought to identify reprogramming factors potentially applicable to human studies.
Methods and Results
Lentivirus vectors expressing Gata4, Mef2c, and Tbx5 (GMT); Hand2 (H), Myocardin (My), or microRNA (miR)‐590 were administered to rat, porcine, and human cardiac fibroblasts in vitro. induced cardiomyocyte‐like cell production was then evaluated by assessing expression of the cardiomyocyte marker, cardiac troponin T (cTnT), whereas signaling pathway studies were performed to identify reprogramming factor targets. GMT administration induced cTnT expression in ≈6% of rat fibroblasts, but failed to induce cTnT expression in porcine or human cardiac fibroblasts. Addition of H/My and/or miR‐590 to GMT administration resulted in cTNT expression in ≈5% of porcine and human fibroblasts and also upregulated the expression of the cardiac genes, MYH6 and TNNT2. When cocultured with murine cardiomyocytes, cTnT‐expressing porcine cardiac fibroblasts exhibited spontaneous contractions. Administration of GMT plus either H/My or miR‐590 alone also downregulated fibroblast genes COL1A1 and COL3A1. miR‐590 was shown to directly suppress the zinc finger protein, specificity protein 1 (Sp1), which was able to substitute for miR‐590 in inducing cellular reprogramming.
Conclusions
These data support porcine studies as a surrogate for testing human cardiac reprogramming, and suggest that miR‐590‐mediated repression of Sp1 represents an alternative pathway for enhancing human cardiac cellular reprogramming.
Keywords: cardiomyocyte, direct reprogramming, fibroblasts, Mef2c, microRNA, transdifferentiation
Subject Categories: Cellular Reprogramming, Gene Therapy, Myocardial Regeneration, Translational Studies, Stem Cells
Introduction
Cellular reprogramming, wherein one adult somatic cell type can be transdifferentiated into another without passing through a progenitor state, offers the remarkable new possibility of regenerating newly functional cells from damaged or scarred tissue.1 More specifically, cardiac fibroblasts have been shown to be reprogrammable into induced cardiomyocytes (iCMs) through the administration of a variety of reprogramming factors, including the cardiac transcription factors, Gata4, Mef2c, and Tbx5.2, 3 Moreover, iCM generation from scar fibroblasts in situ has been demonstrated to improve postinfarct ventricular function and decrease fibrosis in rodent myocardial infarction MI models.4, 5, 6, 7, 8, 9
In comparison to the relative cellular “plasticity” of lower‐order species, differentiated human cells are relatively resistant to reprogramming, likely attributed to epigenetic stabilization of cell survival and differentiation functions in these cells.10 A variety of factors have been tested and added to “basic” rodent reprogramming cocktails to, at least partially, overcome the resistance of human cells to transdifferentiation,11, 12, 13 but the divergence between human and lower‐order cells highlights the need for an appropriate model to test reprogramming strategies that might ultimately be relevant for human application.
We accordingly compared the reprogramming potential of porcine and human cardiac fibroblasts to rodent cells and analyzed the molecular mechanisms underlying the ability of various reprogramming cocktails to induce cellular transdifferentiation in these species. We found that whereas GMT alone was insufficient to reprogram porcine and human cells, the addition of the cardiotransdifferentiating factors, Hand2 (H) and Myocardin (My), or microRNA (miR)‐590 alone, successfully induced transdifferentiation of porcine and human cardiac fibroblasts into iCMs. We further demonstrated that miR‐590 upregulated a number of genes associated with a cardiomyocyte phenotype, and suppressed other fibroblast‐associated genes, by directly inhibiting expression of the zinc‐finger protein, Sp1 (specificity protein 1). This transcription factor has been shown to play an important regulatory role in expression of several genes relevant to fibrosis, including collagen I (COL1) and transforming growth factor (TGF).14, 15, 16 These findings suggest novel pathways potentially applicable to human cellular reprogramming strategies.
Methods
Isolation of Cardiac Cells
Adult human cardiac fibroblasts were harvested under a protocol approved by the Baylor College of Medicine (Houston, TX) Institutional Review Board (IRB H‐33421) from ventricular tissue obtained from explants of heart failure patients undergoing mechanical assist device placement or cardiac transplantation at Baylor St. Luke's Medical Center. Adult porcine cardiac fibroblasts were harvested from ventricular tissue obtained from adult porcine hearts provided by Center for Comparative Medicine, Baylor College of Medicine, and adult rat cardiac fibroblasts were harvested from 8‐ to 10‐week‐old male Sprague‐Dawley rats (Harlan Laboratories, Inc., Indianapolis, IN) using standard cell isolation protocols.7
Following mincing of these tissues, these explants were cultured in DMEM, 10% FBS, and 1% penicillin/streptomycin, as previously described.12 Fibroblasts were allowed to migrate out from these explants over a period of 2 weeks, after which they were passaged 3 times in M106 medium (Catalog No.: M106500; Thermo Fisher Scientific Inc., Waltham, MA), 10% FBS, and LSGS kit supplements (Catalog No. S‐003‐K; Thermo Fisher Scientific).
Primary mouse neonatal cardiomyocytes were obtained from C57/BL6 black mice, subjected to Percoll gradient centrifugation and differential plating to deplete nonmyocytes, as previously described.17
Vector Preparation
Lentiviral vectors were prepared by the Gene Vector Core at Baylor College of Medicine, as previously described.7 The following open reading frames were used to generate these vectors: GATA4 variant 1 (GenBank: NM_002052); MEF2C variant 2 (NM_001131005); TBX5 variant 1 (NM_000192); HAND2 (NM_021973); and MYOCD variant 1 (NM_001146312). All cDNAs were codon optimized for humans and then synthesized by Life Technologies (Carlsbad, CA). To construct single‐gene lentivirus transfer vector, NheI at the 5′ end and EcoRI sites at the 3′end were added for GATA4 and TBX5 cDNAs and cloned into the NheI/EcoRI sites of pCDH‐CMV‐MSC‐EF1‐copGFP vector (Catalog No.: CD511B‐1; System Biosciences Inc., Palo Alto, CA). For MEF2C, the NheI site was added to the 5′ end and the BamHI site to the 3′ end. To construct HAND2‐MYOCD expression vector, 2 cDNAs were connected by self‐cleaving P2A peptide and cloned into the NheI/NotI sites. All constructs contained a Kozak sequence at the translation initiation sites.
A human pre‐miR‐590 expression vector was purchased from System Biosciences (Catalog No.: PMIRH589PA‐1) and resubcloned into the NheI/NotI sites of a pCDH‐CMV‐MCS‐EF1‐GFP vector (Catalog No.: CD512A‐1). Anti‐mir‐590 was constructed to express antisense RNA against a human mir‐590 precursor (GenBank: LM609567) by an oligo ligation method. After phosphorylation with T4 polynucleotide kinase, oligos were annealed and ligated into the HpaI/XhoI sites of pLL3.7 (Catalog No.: 11795; Addgene, Cambridge, MA). All plasmids clones were verified by sequence analyses.
Final viral titers were determined on HEK293T cells by an endpoint transduction assay.18 In brief, 1×105 HEK293T cells in 0.5 mL of DMEM/5% FBS/polybrene (1 μg/mL)/DNase I (10 μg/mL) were plated in a 24‐well plate and infected with serial dilution of lentivirus vectors. Two days later, cells were collected and DNA was extracted using a Tissue DNA kit (Omega Bio‐Tek Inc., Norcross, GA). Proviral DNA was then quantified by real‐time polymerase chain reaction (PCR) using gag primers: 5′‐ AGCGTCAGTATTAAGCGGGG ‐3′ and 5′‐ AGGCCAGGATTAACTGCGA ‐3′. Transducing units were calculated as lentiviral proviral DNA copy number per diploid cell/μL.
Cell Reprogramming
Cardiac fibroblasts (1×106 cells/dish) were seeded onto 6‐ or 10‐cm culture dishes or onto 6‐well plates that were precoated with SureCoat (Cellutron Life Technologies, Baltimore, MD). Twenty‐four hours after cells were 70% to 80% confluent, fresh lentivirus supernatant (multiplicity of infection=50) was added to these plates in a mixture with polybrene at a final concentration of 5 μg/μL. Two days later, this initial transfer medium (DMEM/199 [4:1], 10% FBS, and 1% penicillin/streptomycin) was replaced with induction medium, containing DMEM/199 (4:1), 10% FBS, 5% horse serum, 1% penicillin/streptomycin, 1% nonessential amino acids, 1% essential amino acids, 1% B‐27, 1% insulin‐selenium‐transferrin, 1% vitamin mixture, and 1% sodium pyruvate, as previously described.12 Induction media was replaced every 2 days until cells were harvested.
For coculture studies, 4 weeks after treatment with reprogramming factors, porcine fibroblasts were harvested and replated onto cultures of neonatal mouse cardiomyocytes at a ratio of 1:4 in DMEM/M199/10% FBS medium.
Flow Cytometry
To perform flow cytometry, adherent cells were first washed with DPBS and trypsinized with 0.25 Trpsin/EDTA. Cells were then fixed with fixation buffer (BD Biosciences, San Jose, CA) for 15 minutes at room temperature. Fixed cells were washed with Perm/Wash buffer (BD Biosciences) and then incubated with mouse monoclonal anti‐cardiac troponin T (cTnT) antibody (Thermo Fisher Scientific) at 1:100 dilution in Perm/Wash buffer for 90 minutes at room temperature. They were then incubated with donkey antimouse Alexa Fluor 647 (Invitrogen, Carlsbad, CA) at 1:200, then washed with Perm/Wash buffer again, and then analyzed for cTnT expression using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ) using Diva software (version 6.0).7 For Thy1/CD31/c‐kit cell sorting, cells were incubated with APC‐conjugated ant‐CD31 (eBiosciences, San Diego, CA), brilliant violet conjugated anti‐Thy1 (BioLegend, San Diego, CA), and APC‐conjugated anti‐c‐kit antibodies (BD Biosciences) for sorting with a FACSCalibur flow cytometer (Becton Dickinson).2
Immunocytochemistry
To perform immunocytochemistry, cells were fixed in 4% paraformaldehyde for 15 minutes at room temperature and then permeabilized with permeabilization buffer (0.5% Triton‐X) for 15 minutes. They were then blocked with 10% goat serum for 30 minutes and incubated with primary antibodies against cTnT (1:400 dilution; Thermo Fisher Scientific) or α‐actinin (1:300 dilution; Sigma‐Aldrich, St. Louis, MO). After washing with DPBS, Alexa fluorogenic secondary antibodies (Invitrogen) were used to detect signal.7
Real‐Time PCR
Real‐time PCR was performed by first extracting total RNA using the TRIzol method (Invitrogen), after which relative quantification was performed using SYBR‐green detection of PCR products in real time with the ABI ViiA 7 (Applied Biosystems Inc., Foster City, CA). The following primers were used: TNNT2, MYH6, TNNC1, NPPA, RYR2, COL1A1, COL3A1, Sp1 (sc‐29487; Santa Cruz Biotechnology, Santa Cruz, CA), and Has‐miR‐590 (478168_mir; Thermo Fisher Scientific). mRNA levels were normalized by comparison to GAPDH. miRNA levels were normalized to expression of U6 snRNA.
Transfection With siRNA or Plasmid
For siRNA administration studies, once cells had reached 60% confluence, siRNA‐control (sc37007) and siRNA‐Sp1 (sc29487) were administered to cells (1, 5, and 10 μmol/L) together with Lipofectamine 2000 transfection reagent (Life Technologies), according to the manufacturer's instructions. Sp1 expression plasmid was constructed by inserting Sp1 cDNA (HG12024‐G, Sino Biological Inc.) with hemagglutinin tag into pGEM‐T expression plasmid under the control of the T7 promoter.19 Transfection for overexpressing Sp1 (1 μg) in fibroblasts was performed with Lipofectamine 2000 (Invitrogen), following the manufacturer's instructions.
Luciferase Reporter Assay
To detect direct binding of miR‐590 to the 3′‐untraslated region (UTR) of Sp1, HEK cells were transfected with plasmid DNA (PGL3‐Sp1‐3′UTR/PGL3‐Sp1‐3′UTR Mutant 200 ng) and miRNA mimics (5 nmol; RibioBio Co., Ltd., Guangzhou, China) using Lipofectamine 2000 in OptiMEM medium, according the manufacturer's methods. The dual‐luciferase reporter assay system (Promega Corp., Madison, WI) was used for analyses, and the data are shown as the ratio of luciferase activity normalized against that of Renilla.20 The miRNA target prediction program, TargetScan, (http://www.targetscan.org) was used to identify putative targets.
Western Analysis
To perform western analyses, cell lysates were prepared by homogenization of cells in cell lysis buffer (Cell Signaling Technology, Danvers, MA) and run on SDS‐PAGE. After transfer to nitrocellulose membrane, immune detection was performed with antibodies to Sp1 (Santa Cruz Biotechnology) and actin (Sigma‐Aldrich), followed by treatment with appropriate HRP‐conjugated secondary antibodies (Millipore, Billerica, MA). Antibody‐bound proteins were visualized by chemiluminescence detection (Thermo Fisher Scientific).
Measurements of Contractility and Calcium Transient
Cell shortening and Ca2+ transients were measured at room temperature (22–23°C) in separate experiments. Myocytes were field stimulated to contract by using a Grass S5 stimulator through platinum electrodes placed alongside the bath (0.5 Hz, bipolar pulses with voltages 50% above myocyte voltage threshold). Contractions of myocytes from random fields were videotaped and digitized on a computer. For Ca2+ signal measurements, cells were loaded with 2 μmol/L of Fura‐2/AM (Life Technologies) and alternately excited at 340 and 380 nm by use of a Delta Scan dual‐beam spectrophotofluorometer (Photon Technology International, Edison, NJ) at baseline conditions. Ca2+ transients were expressed as the 340/380‐nm ratios of the resulting 510‐nm emissions. Data were analyzed by the use of Felix software (Photon Technology International).21
Statistical Analyses
Results are presented as mean±SEM. Three independent experiments with triplicate determinations were performed. The nonparametric Mann–Whitney U test (also called the Wilcoxon rank‐sum test) was used when comparing 2 groups, and the Kruskal–Wallis test was used when comparing >2 groups, to analyze statistical significance for data that were not normally distributed. A P<0.05 was considered to indicate significance.
Results
Porcine and Human Cardiac Fibroblast Reprogramming Equivalence
Human and porcine cardiac fibroblasts were isolated using the explant culture method described in Methods, after which cells were passaged 3 times to exclude the possibility of contamination with rare c‐kit‐positive cardiac progenitors and endothelium cells. Consistent with this expectation, flow cytometric analysis revealed that ≈90% of porcine and human cardiac fibroblasts expressed Thy1+, a cardiac fibroblast‐specific surface marker,2 whereas less than 1% of cells were c‐kit positive (cardiac progenitor cell marker) or CD31 positive, an endothelium cell marker (Figure S1).
In order to determine whether porcine cells could be used as a model system, or “surrogate” for human cell reprogramming, we first tested whether they exhibited similar resistances to cellular reprogramming. Accordingly, we tested whether cardiac reprogramming factors known to induce rodent (but not human) cellular cardiac transdifferentiation would also induce porcine cardiac fibroblast transdifferentiation into iCMs. We showed that whereas lentiviral‐mediated administration of Gata4, Mef2c, and Tbx5 (GMT) to rat cardiac fibroblasts upregulated expression of the cardiomyocyte marker, cTnT, both human and porcine cardiac fibroblasts failed to express cTnT after GMT administration (Figure 1). In comparison, the addition of the transcription factors, Hand2 and Myocardin (H/My), to GMT administration resulted in cTnT expression in ≈5% of porcine as well as human cells (Figure 1). These data demonstrate that porcine cells possess resistances to reprogramming that are similar to human cells.
Figure 1.
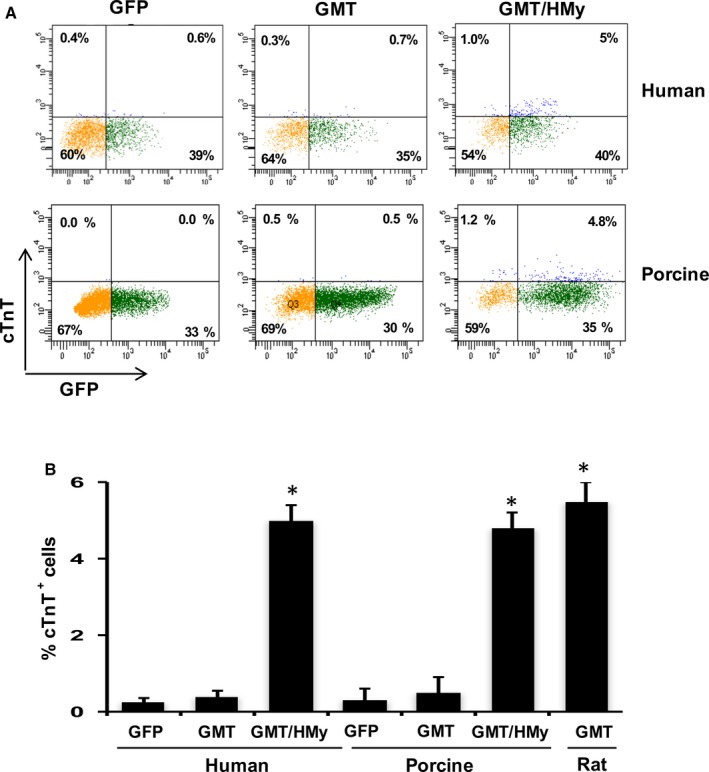
Gata4, Mef2c, Tbx5 (GMT), Hand2 (H), and Myocd (My) induce cardiac gene expression in human cardiac fibroblasts and porcine cardiac fibroblasts. A, Representative flow cytometry plots for analyses of cTnT + cells 2 weeks after infection of human and porcine cardiac fibroblasts with lentiviruses expressing indicated combinations of factors. Cells were infected with: left—GFP vector lentivirus negative control, middle—GMT, and right—GMT and H/My. B, Summary of flow cytometry analyses. Percentage of cTnT + cells following infection of human or porcine or rat cardiac fibroblasts (n=3). Data are presented as mean±SEM. *P<0.05 versus GFP (Kruskal–Wallis test). cTnT indicates cardiac troponin T; GFP, green fluorescent protein.
Influence of MiR‐590 on Cardiac Cellular Reprogramming
Given the challenges of administering complex transcription factor cocktails to induce human cardiac cellular reprogramming, we explored whether an appropriate miRNA could be used to more efficiently induce cardiac reprogramming. Based on the role of miR‐590 in inducing adult cardiac cellular proliferation,22 we accordingly treated human and porcine cardiac fibroblasts with GMT, together with miR‐590, as a substitute for H/My. Whereas neither GMT nor miR‐590 administration alone induced cTnT expression in porcine or human cells, the addition of miR‐590 to GMT induced cTnT expression in ≈5% of cells, as assessed by fluorescence‐activated cell sorting (FACS; Figure 2). The addition of H/My to GMT/miR‐590 yielded somewhat higher levels of cTnT expression.
Figure 2.
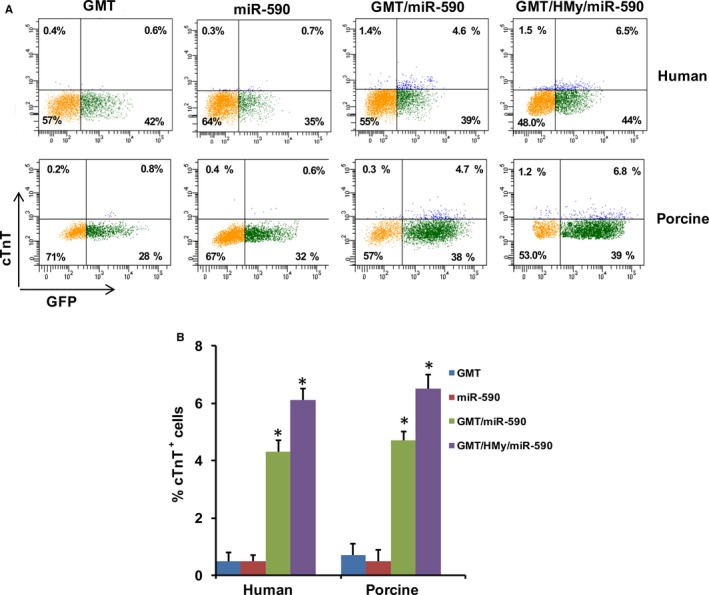
miR‐590 can replace Hand2 and Myocardin in inducing cardiac gene expression in human cardiac fibroblasts and porcine cardiac fibroblasts. A, Representative flow cytometry plots for analyses of cTnT + cells 2 weeks after infection of human or porcine cardiac fibroblasts with lentiviruses expressing indicated combinations of factors. Cells were infected with (left to right): GMT, miR‐590, GMT plus miR‐590, or GMT plus H/My plus miR‐590. B, Summary of flow cytometry analyses. Percentage of cTnT + cells following infection of human or porcine cardiac fibroblasts (n=3). Data are presented as mean±SEM. *P<0.05 versus GMT (Kruskal–Wallis test). cTnT indicates cardiac troponin T; miR, microRNA.
Immunofluorescence studies likewise demonstrated that GMT plus miR‐590 induced expression of the cardiomyocyte marker, α‐sarcomeric actinin, as well as cTnT expression in porcine and human cardiac fibroblasts to an extent similar to that observed after administration of GMT plus H/My (Figure 3).
Figure 3.
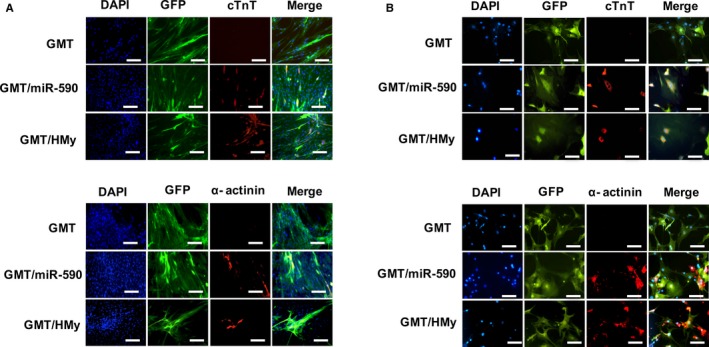
Immunostaining for cardiomyocyte markers in porcine and human cardiac fibroblasts. A, Representative immunofluorescence staining for DAPI ([blue] nuclear marker), GFP ([green] lentivirus infection marker) and the (red) cardiomyocyte markers cTnT (top) and α‐actinin (bottom) in porcine cardiac fibroblasts transduced with GMT, GMT plus miR‐590 or GMT plus H/My. GMT plus miR‐590 and GMT plus H/My induced abundant cTnT and α‐actinin expression 4 weeks after transduction (n=3). B, Immunofluorescence staining for DAPI ([blue] nuclear marker), GFP ([green] lentivirus infection marker) and the (red) cardiomyocyte markers cTnT (top) and α‐actinin (bottom) in human cardiac fibroblasts transduced with GMT, GMT plus miR‐590 or GMT plus H/My. GMT plus miR‐590 and GMT plus H/My induced abundant cTnT and α‐actinin expression 4 weeks after transduction (n=3). Scale bars: 100 μm. cTnT indicates cardiac troponin T; DAPI, 4′,6‐diamidino‐2‐phenylindole; GFP, green fluorescent protein.
Interestingly, although neither GMT plus H/My nor GMT plus miR‐590 induced spontaneous iCM beating or contractility when isolated cultures of cardiac fibroblasts were treated, ≈3% of porcine cardiac fibroblasts treated with GMT plus H/My and GMT plus miR‐590 contracted synchronously with surrounding cardiomyocytes 14 days after being placed in coculture with murine cardiomyocytes. In comparison, green fluorescent protein (GFP)+ fibroblasts generated by treatment of these cells with a lenti‐GFP‐negative control vector failed to demonstrate contractions in coculture experiments, suggesting that the cells identified as beating iCMs did not reflect artifact from the surrounding myocyte coculture (Figure S2A and Videos S1 through S3). In addition, cells also did not develop contractile properties when only media conditioned by cultures of murine cardiomyocytes were added to cells treated with GMT plus H/My or GMT plus miR‐590.
We next measured excitation‐contraction coupling to further characterize the functional properties of induced cardiomyocytes generated through coculture. In this analysis, beating iCMs visually identified by GFP expression revealed contractility and calcium transient under basal conditions (Figure S2B). Consistent with this finding, we also observed expression of the cardiac contractile protein, α‐sarcomeric actinin, in GMT plus H/My and GMT plus miR‐590 transduced porcine cardiac fibroblasts, but not in GFP or GFP/miR‐590 transduced cells (Figure S2C).
Downstream Gene Activation Induced by Reprogramming Factors
GMT plus H/My and GMT plus miR‐590 both initiated the expression of a similarly broad range of cardiac genes in treated porcine cells, including those specific to cardiomyocyte structure in function, including TNNT2, MYH6, TNNC1, natriuretic peptide (NPPA) and ryanodine receptor‐2 (RYR2) (Figure 4A). Each of these reprogramming cocktails also downregulated expression of fibroblast‐specific genes, including COL1A1 and COL3A1 (Figure 4B). These results further substantiate the cardiac‐like phenotype evoked by the GMT plus mir‐590 or GMT plus H/My in porcine and human cardiac fibroblasts.
Figure 4.
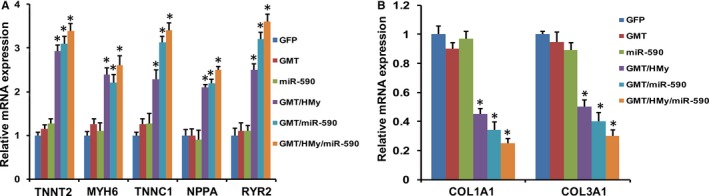
Cardiac and fibroblast marker gene expression profile following reprogramming factor administration. A, mRNA expression of cardiomyocyte marker genes (TNNT2, MYH6, TNNC1, NPPA, and RYR2) in porcine cardiac fibroblasts 2 weeks after transduction with GMT plus other reprogramming factors, as determined by qRT‐PCR (n=3). B, mRNA expression of fibroblast marker genes (COL1A1 and COL3A1) in porcine cardiac fibroblasts 2 weeks after transduction with GMT plus other reprogramming factors, as determined by qRT‐PCR (n=3). Data were normalized against the GFP values. All the data presented as mean±SEM. *P<0.05 versus GFP (Kruskal–Wallis test). COL indicates collagen; cTnT, cardiac troponin T; DAPI, 4′,6‐diamidino‐2‐phenylindole; GFP, green fluorescent protein; miR, microRNA; NPPA, natriuretic peptide; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; RYR2, ryanodine receptor‐2.
In order to better discern the mechanism of action of miR‐590, we used an miRNA target prediction program (Targetscan 7.0), which identified Sp1 as a potential target of miR‐590 (Figure S3A). Confirming these data, luciferase reporter assays showed the direct binding of miR‐590 to the 3′‐UTR of Sp1 (Figure S3B).
We next investigated whether miR590 regulated Sp1 expression levels. Quantitative reverse‐transcription PCR (qRT‐PCR) demonstrated that downregulation of Sp1 in porcine and human cardiac fibroblasts by GMT was significantly reduced further by the addition of miR‐590 (Figure 5A). Western blot analyses likewise demonstrated that Sp1 protein expression was strongly downregulated by miR‐590 or GMT/miR‐590 administration (Figure 5B). Conversely, Sp1 mRNA expression was significantly increased by anti‐miR590 (Figure 5C).
Figure 5.
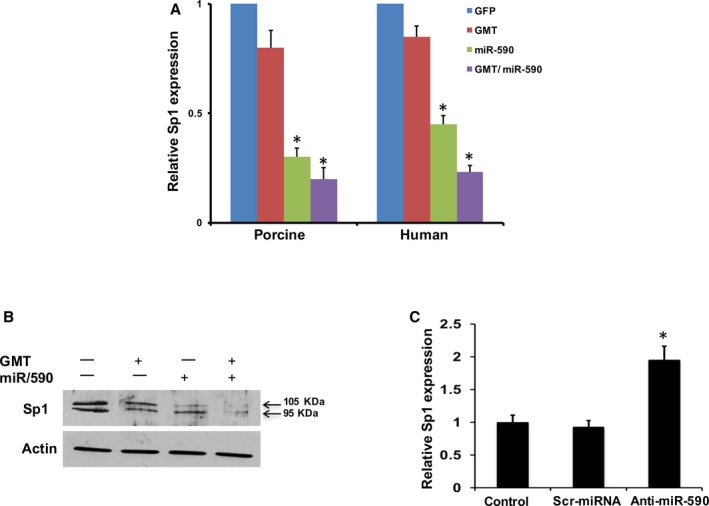
miR‐590 directly represses Sp1 expression. qPCR analysis of relative Sp1 mRNA expression in porcine cardiac fibroblasts and human cardiac fibroblasts (A), treated with GFP, GMT, miR‐590 and GMT plus miR‐590 (n=3). *P<0.05 vs relevant control (Kruskal–Wallis test). B, Western blot analyses for Sp1 expression in porcine cardiac fibroblasts treated with GFP, GMT, miR‐590 and GMT plus miR‐590 (n=3). C, qPCR analysis of relative Sp1 mRNA expression in porcine cardiac fibroblasts transfected with control (scrambled) and anti‐miR590 (n=3; Wilcoxon rank‐sum test). All data are presented as mean±SEM. *P<0.05 vs relevant control. GFP indicates green fluorescent protein; miR, microRNA; qPCR, quantitative polymerase reaction; Sp1, specificity protein 1.
Role of Sp1 in Cardiac Reprogramming
Based on the above findings, we hypothesized that miR‐590 may downregulate Sp1 expression as a potential intermediary in cardiac cellular reprogramming. To test this, we downregulated Sp1 expression in GMT‐transduced porcine cardiac fibroblasts using Sp1 siRNA, and demonstrated that inhibition of Sp1 strongly upregulated the same panel of cardiac genes, including TNNT2 and MYH6, and downregulated expression of fibroblast genes, including COL1A1 and COL3A1, that had been analogously modified by miR‐590 administration (Figure 6A through 6C).
Figure 6.
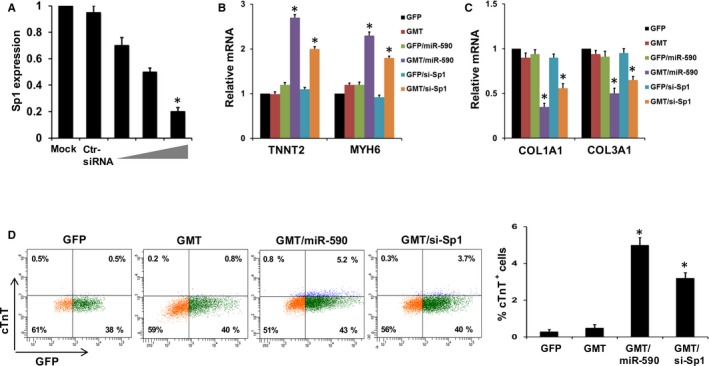
miR‐590‐mediated Sp1 is critical for cardiac reprogramming. A, Relative mRNA expression of Sp1 in untreated (mock) porcine cardiac fibroblasts and porcine cardiac fibroblasts transfected with scrambled siRNA or anti‐Sp1 siRNA (1, 5, and 10 μmol/L; n=3). *P<0.05 versus mock (Wilcoxon rank‐sum test). B, Relative mRNA expression of cardiomyocyte marker gene (TNNT2, MYH6) in and porcine cardiac fibroblasts transduced with GMT and/or miR‐590, and with or without si‐Sp1 (n=3). *P<0.05 versus relevant control (Kruskal–Wallis test). C, Relative mRNA expression of fibroblast marker genes (COL1A1 and COL3A1) in, and porcine cardiac fibroblasts transduced with, GMT and/or miR‐590, and with or without si‐Sp1 (n=3). *P<0.05 versus relevant control (Kruskal–Wallis test). D, Left: Representative flow cytometry plots for analyses of cTnT + cells 2 weeks after transduction of porcine cardiac fibroblasts with GMT and si‐Sp1 or miR‐590. Left to right: GFP, GMT, GMT plus miR‐590, and GMT plus si‐Sp1. Right: Summary of flow cytometry analyses. Percentage of cTnT + cells following infection of porcine cardiac fibroblasts (n=3; Wilcoxon rank‐sum test). Data are presented as mean±SEM. *P<0.05 versus relevant control. COL indicates collagen; cTnT, cardiac troponin T; GFP, green fluorescent protein; miR, microRNA; Sp1, specificity protein 1.
Consistent with the findings of similar transdifferentiating gene expression patterns induced by miR‐590 and Sp‐1, we demonstrated that inhibition of Sp1 with Sp1 siRNA significantly increased the percentage of GMT‐treated porcine cardiac fibroblasts expressing cTnT (Figure 6D). In contrast, Sp1 overexpression induced by Sp1 cDNA inhibited cardiac gene expression, activated fibroblast gene expression (Figure 7A and 7B), and inhibited induction of cTnT expression in GMT/miR‐590‐transduced cells (Figure 7C). These results suggest that miR‐590‐mediated suppression of Sp1 may represent an important cardiac transdifferentiation pathway, albeit one that also requires the presence of GMT.
Figure 7.
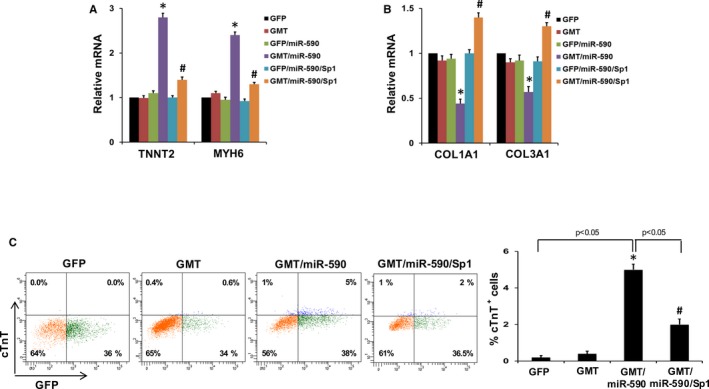
Overexpression of Sp1 disrupts cardiac reprogramming in GMT/miR‐590‐transduced cells. A, Relative mRNA expression of cardiomyocyte marker genes (TNNT2, MYH6) in porcine cardiac fibroblasts transduced with GMT and/or miR‐590, and with or without Sp1 overexpression induced by Sp1 cDNA (n=3). *P<0.05 versus relevant control (Kruskal–Wallis test). B, Relative mRNA expression of fibroblast marker genes (COL1A1 and COL3A1) in porcine cardiac fibroblasts transduced with transduced with GMT and/or miR‐590, and with or without Sp1 overexpression induced by Sp1 cDNA (n=3). *P<0.05 versus relevant control (Kruskal–Wallis test). C, Left: Representative flow cytometry plots for analyses of cTnT + cells 2 weeks after transduction of porcine cardiac fibroblasts with GMT with or without miR‐590 transduction, and with Sp1 overexpression. Right: Summary of flow cytometry analyses. Percentage of cTnT + cells following infection of porcine cardiac fibroblasts (n=3). All data are presented as mean±SEM. *P<0.05 versus GFP; # P<0.05 versus GMT/miR‐590 (Wilcoxon rank‐sum test). COL indicates collagen; cTnT, cardiac troponin T; GFP, green fluorescent protein; miR, microRNA; Sp1, specificity protein 1.
Discussion
We and others have previously demonstrated that GMT administration effectively induces reprogramming of rodent cardiac fibroblasts into iCMs,5, 6, 7 but subsequent studies have shown that administration of these factors alone are insufficient to induce cardiac gene expression in human fibroblasts.11, 12, 13 In the present study, we show that porcine, as well as human cardiac, fibroblasts can be converted into iCMs through administration of GMT plus Hand2 and Myocardin, and that miR‐590 can substitute for the latter 2 factors. Importantly, the validation of porcine cells as an appropriate model system, or “surrogate” for studies relevant to human cells in that both of these cell types demonstrated similar resistances to reprogramming, should allow for large animal model studies that will be needed to advance the potential undertaking of clinical trials.
The capacity of miR‐590 to substitute for Hand2 and Myocardin in inducing cellular reprogramming is not entirely surprising. It is well known that microRNAs have numerous epigenetic and downstream gene promoter targets, including transcription factors and cell signaling networks.23 Recently, Jayawardena et al reported that a combination of muscle‐specific miRNAs (miR‐1, ‐133, ‐208, and ‐499) alone reprogrammed neonatal and adult murine cardiac fibroblasts into cardiomyocyte‐like cells.24, 25 Muraoka et al likewise demonstrated that the addition of miR‐133 to reprogramming regimens including GMT Mesp1, and/or Myocd, improved both murine and human cardiac fibroblast reprogramming into iCMs.26
The miR‐590 family, more specifically, which is comprised of 2 miRNAs—miR‐590‐5p and miR‐590‐3p—is thought to play a key role in cardiovascular disease through its protection of blood vessels, regulation of lipid metabolism, and prevention of myocarditis and atherosclerosis through its target genes.27, 28, 29 miR‐590 is expressed in human induced pluripotent stem‐cell‐derived cardiomyocytes and in the human heart.30, 31 miR‐590 regulates signaling pathways (TGF‐β, protein kinase B) involved in cardiac fibrosis/remodeling, embryonic stem cell proliferation/cardiac differentiation, and metabolism by suppressing TGF‐β receptor II.32, 33, 34, 35 Interestingly, whereas overexpression of miR‐590 has been shown to be associated with cardiac regeneration, improved cardiac function and reduced scarring following MI,22 miR‐590 downregulation was correlated with atrial fibrosis and atrial fibrillation.35 Taken together, these data clearly support a role for miR‐590 in cardiac physiology and disease.
Our demonstration that miR‐590 appears to induce cellular reprogramming, at least in part, by inhibiting Sp1 highlights the apparent importance of creating a “critical mass” of appropriately activated (or deactivated) genes in a portfolio favorable to cellular reprogramming. In this regard, we demonstrated that Sp1 suppression downregulated expression of a panel of genes that favor fibroblast‐centric differentiation, and upregulated another series of genes associated with cardiomyocyte differentiation. Nevertheless, even the relatively diverse panel of genes regulated by miR‐590 overexpression required the addition of Gata4, Mef2c, and Tbx5 (GMT) transcription factors to induce human and porcine fibroblast cardiodifferentiation (and vice versa).
Remarkable as the newly discovered “plasticity” of differentiated adult somatic cells may be, the requirement for complex cocktails of reprogramming factors to transdifferentiate cells of higher‐order species highlights the extensiveness of changes to the normal gene expression profile of these cells required to overcome what is (fortunately) their otherwise fairly stable genotypic and phenotypic signatures. The targeting of a multiplicity of genes required to reprogram human cells in particular may be ideally suited to the use of miRs as potential therapeutic agents, given that they, like transcription factors, can regulate expression of multiple gene targets at once.
Whereas the resistance of higher‐order cells to reprogramming is not well understood, it is thought to be related to the relative stability of the epigenetic signature of these cells.10 In this regard, the capability of miR‐590 to enhance cardiomyocyte transdifferentiation is consistent with its previously demonstrated ability to suppress the profibrotic TGFβ signaling pathway, as well as alpha‐smooth muscle actin and COL1 expression associated with myofibroblast generation.35, 36 These observations are, in turn, reminiscent of the findings of Muraoka et al, who reported that addition of miR‐133 to GMT promotes cardiac reprogramming by directly inhibiting Snail 1 and silencing a fibroblast signature.26 The procardiomyocyte differentiating properties of miR‐590 are likewise consistent with its role in suppressing epithelial‐to mesenchymal transition, which has been identified as a morphogenic pathway leading differentiated cells toward a profibrotic state.
It has likewise been shown that an appropriate extracellular matrix milieu can also enhance somatic cell reprogramming (or vice versa).37, 38 One recent report demonstrated that fibroblast growth factor 2 administration improved reprogramming efficiency by inhibiting expression of certain extracellular collagens.37 We and others have similarly demonstrated enhanced iCM generation in vivo versus in vitro, and evidence of iCM contractility in cardiomyocyte cocultures, but not in isolated fibroblast/iCM culture.11, 12, 13 Taken together, these data suggest an important role in enhancing reprogramming of epigenetic factors that may tip the balance of cell fate down one cellular differentiation pathway versus another.
The critical role played more specifically by the Sp1 gene as a potential direct target of miR‐590 in enhancing reprogramming, as demonstrated through the addition/deletion studies described in this report, is consistent with its identified role in cell differentiation, growth, apoptosis, and chromatin remodeling.39, 40 This transcription factor binds to GC‐rich motifs of a number of promoters, including that for human COL1, that are relevant to the putative differentiation pathways considered above. Given as well the role of Sp1 overexpression in inducing cardiac fibrosis, and our observations of the need to favorably (re)balance profibrotic and procardiomyocyte differentiating forces in supporting cardiac transdifferentiation, it is possible that repressing Sp1 through miR‐590 might be a key pathway in cardiac reprogramming.15, 41, 42, 43 Because miR‐590 is nonintegrating and has several other predicted targets relevant to cardiac reprogramming, and given that it supports reprogramming to a greater degree that Sp1 suppression alone, it may be an ideal additive to reprogramming cocktails for human intervention.
Sources of Funding
This study was funded by the National Heart, Lung and Blood Institute (1R01HL121294‐01A1 [Rosengart]) and supported, in part, by the BCM Cytometry and Cell Sorting Core (National Institutes of Health grants P30AI036211, P30CA125123, and S10RR024574; National Center for Research Resources grant S10RR024574; National Institute of Allergy and Infectious Diseases grant AI036211, and National Cancer Institute grant P30CA125123).
Disclosures
None.
Supporting information
Figure S1. FACS analyses for Thy1, CD31, and CD117/c‐kit in human and porcine cardiac fibroblasts. Representative FACS analyses of human cardiac fibroblasts (top) or porcine cardiac fibroblasts (bottom) showing majority of the cells were positive for the cardiac fibroblast‐specific surface marker, Thy1, and negative for cardiac progenitor (c‐kit/CD117) or endothelial cell (CD31) markers. FACS indicates fluorescence‐activated cell sorting.
Figure S2. Reprogramming of porcine cardiac fibroblasts in coculture with murine cardiomyocytes. Porcine cardiac fibroblasts were transduced with lentivirus expressing a GFP marker alone (negative control group; left); lentivirus vectors also expressing miR‐590 as well as vectors expressing Gata4, Mef2c, or Tbx5 (“GMT plus miR‐590” group; middle); or with lentivirus vectors also expressing Hand2 and Myocardin as well as vectors expressing Gata4, Mef2c, or Tbx5 (“GMT plus H/My” group; right). Four weeks after initial transduction, these porcine cardiac fibroblasts were cocultured with (untreated) neonatal murine cardiomyocytes (negative for GFP). A, Representative immunofluorescence staining demonstrating (green) GFP expression, which indicates lentivirus infection. Only (green fluorescent) cells that had been treated with GMT plus miR‐590 or GMT plus H/My contracted synchronously with surrounding cardiomyocytes. Fibroblasts treated with a GFP‐control vector only did not demonstrate contractions in these coculture experiments (Videos S1 through S3). B, Representative curves reflecting contraction (left) and Ca2+ transient trace (right) under basal conditions of (induced cardiomyocyte‐like; iCM) cells derived from porcine cardiac fibroblasts after coculture with murine neonatal cardiomyocytes. For the calcium transients, cells were loaded with Fura‐2 for 30 minutes, and then calcium transients were recorded (n=3). C, Representative immunofluorescence staining for DAPI ([blue] nuclear marker), GFP ([green] lentivirus infection marker), and the (red) cardiomyocyte marker, α‐actinin, in porcine cardiac fibroblasts treated with lentivirus containing a GFP marker and GMT, GMT plus miR‐590, or GMT plus H/My which were then cocultured with neonatal murine cardiomyocytes. Porcine cardiac fibroblasts transduced with GFP, miR‐590, or GMT alone did not express α‐actinin in coculture neonatal murine cardiomyocytes. Porcine cardiac fibroblasts transduced with GMT plus miR‐590 or GMT plus H/My expressed the cardiomyocyte marker, α‐actinin (indicates by arrows), in coculture with murine cardiomyocytes (scale bars, 100 μm). Black arrow indicates GFP‐positive nonbeating cells, yellow arrow indicates GFP‐positive beating cells. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; GFP, green fluorescent protein; miR, microRNA.
Figure S3. miR‐590 directly represses Sp1. A, Predicted interaction sites of miR‐590 at the 3′‐UTR of Sp1 (TargetScan: www.targetscan.org). B, HEK293 cells were cotransfected with (pGL3‐Sp1‐3′UTRWT) and mutated targeting sequence (pGL3‐Sp1‐3′UTR‐Mutant) and miRNA mimic‐control/590 for 48 hours, then luciferase activity was detected. miR‐590 directly repressed WT Sp1 3′UTR in luciferase assay, and the repression was abolished when binding site was mutated (n=3). Data are presented as mean±SEM. *P<0.05 versus control‐miR. miR indicates microRNA; Sp1, specificity protein 1; UTR, untranslated region; WT, wild type.
Video S1. This video shows porcine fibroblasts treated with a GFP‐control vector only did not demonstrate contractions in the coculture experiments.
Video S2. This video shows porcine cardiac fibroblasts contracted synchronously with surrounding murine cardiomyocytes after transduction with GMT plus miR‐590.
Video S3. This video shows porcine cardiac fibroblasts contracted synchronously with surrounding murine cardiomyocytes after transduction with GMT plus H/My.
Acknowledgments
We thank Dr Kazuhiro Oka from the Gene Vector Core at Baylor College of Medicine for the preparation of viral vectors.
(J Am Heart Assoc. 2016;5:e003922 doi: 10.1161/JAHA.116.003922)
References
- 1. Cohenand DE, Melton D. Turning straw into gold: directing cell fate for regenerative medicine. Nat Rev Genet. 2011;12:243–252. [DOI] [PubMed] [Google Scholar]
- 2. Ieda M, Fu JD, Delgado‐Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Addis RC, Ifkovits JL, Pinto F, Kellam LD, Esteso P, Rentschler S, Christoforou N, Epstein JA, Gearhart JD. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J Mol Cell Cardiol. 2013;60:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel‐Duby R, Olson EN. Heart repair by reprogramming non‐myocytes with cardiac transcription factors. Nature. 2012;485:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D, Qian L. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mathison M, Gersch RP, Nasser A, Lilo S, Korman M, Fourman M, Hackett N, Shroyer K, Yang J, Ma Y, Crystal RG, Rosengart TK. In vivo cardiac cellular reprogramming efficacy is enhanced by angiogenic preconditioning of the infarcted myocardium with vascular endothelial growth factor. J Am Heart Assoc. 2012;1:e005652 doi: 10.1161/JAHA.112.005652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mathison M, Singh VP, Gersch RP, Ramirez MO, Cooney A, Kaminsky SM, Chiuchiolo MJ, Nasser A, Yang J, Crystal RG, Rosengart TK. Triplet' polycistronic vectors encoding Gata4, Mef2c, and Tbx5 enhances postinfarct ventricular functional improvement compared with singlet vectors. J Thorac Cardiovasc Surg. 2014;148:1656–1664. [DOI] [PubMed] [Google Scholar]
- 8. Inagawa K, Miyamoto K, Yamakawa H, Muraoka N, Sadahiro T, Umei T, Wada R, Katsumata Y, Kaneda R, Nakade K, Kurihara C, Obata Y, Miyake K, Fukuda K, Ieda M. Induction of cardiomyocyte‐like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ Res. 2012;111:1147–1156. [DOI] [PubMed] [Google Scholar]
- 9. Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, Bursac N, Liu J, Qian L. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ Res. 2015;116:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang P, Zhang L, Gao Y, He Z, Yao D, Wu Z, Cen J, Chen X, Liu C, Hu Y, Lai D, Hu Z, Chen L, Zhang Y, Cheng X, Ma X, Pan G, Wang X, Hui L. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell. 2014;14:370–384. [DOI] [PubMed] [Google Scholar]
- 11. Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, Tohyama S, Yuasa S, Kokaji K, Aeba R, Yozu R, Yamagishi H, Kitamura T, Fukuda K, Ieda M. Induction of human cardiomyocyte‐like cells from fibroblasts by defined factors. Proc Natl Acad Sci USA. 2013;110:12667–12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel‐Duby R, Olson EN. Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci USA. 2013;110:5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fu JD, Stone NR, Liu L, Spencer CI, Qian L, Hayashi Y, Delgado‐Olguin P, Ding S, Bruneau BG, Srivastava D. Direct reprogramming of human fibroblasts toward a cardiomyocyte‐like state. Stem Cell Reports. 2013;1:235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verrecchia F, Rossert J, Mauviel A. Blocking sp1 transcription factor broadly inhibits extracellular matrix gene expression in vitro and in vivo: implications for the treatment of tissue fibrosis. J Invest Dermatol. 2001;116:755–763. [DOI] [PubMed] [Google Scholar]
- 15. Li R, Xiao J, Qing X, Xing J, Xia Y, Qi J, Liu X, Zhang S, Sheng X, Zhang X, Ji X. Sp1 mediates a therapeutic role of MiR‐7a/b in angiotensin II‐induced cardiac fibrosis via mechanism involving the TGF‐β and MAPKs pathways in cardiac fibroblasts. PLoS One. 2015;10:e0125513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li N, Cui J, Duan X, Chen H, Fan F. Suppression of type I collagen expression by miR‐29b via PI3K, Akt, and Sp1 pathway in human Tenon's fibroblasts. Invest Ophthalmol Vis Sci. 2012;53:1670–1678. [DOI] [PubMed] [Google Scholar]
- 17. Harada M, Saito Y, Kuwahara K, Ogawa E, Ishikawa M, Nakagawa O, Miyamoto Y, Kamitani S, Hamanaka I, Kajiyama N, Takahashi N, Masuda I, Itoh H, Nakao K. Interaction of myocytes and nonmyocytes is necessary for mechanical stretch to induce ANP/BNP production in cardiocyte culture. J Cardiovasc Pharmacol. 1998;31:S357–S359. [DOI] [PubMed] [Google Scholar]
- 18. Caburet S, Arboleda VA, Llano E, Overbeek PA, Barbero JL, Oka K, Harrison W, Vaiman D, Ben‐Neriah Z, García‐Tuñón I, Fellous M, Pendás AM, Veitia RA, Vilain E. Mutant cohesin in premature ovarian failure. N Engl J Med. 2014;370:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parks CL, Shenk T. The serotonin 1a receptor gene contains a TATA‐less promoter that responds to MAZ and Sp1. J Biol Chem. 1996;271:4417–4430. [DOI] [PubMed] [Google Scholar]
- 20. Jin Y, Chen Z, Liu X, Zhou X. Evaluating the microRNA targeting sites by luciferase reporter gene assay. Methods Mol Biol. 2013;936:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Singh VP, Rubinstein J, Arvanitis DA, Ren X, Gao X, Haghighi K, Gilbert M, Iyer VR, Kim do H, Cho C, Jones K, Lorenz JN, Armstrong CF, Wang HS, Gyorke S, Kranias EG. Abnormal calcium cycling and cardiac arrhythmias associated with the human Ser96Ala genetic variant of histidine‐rich calcium‐binding protein. J Am Heart Assoc. 2013;2:e000460 doi: 10.1161/JAHA.113.000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. [DOI] [PubMed] [Google Scholar]
- 23. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, Dzau VJ. MicroRNA‐mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res. 2012;110:1465–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson CP, Pratt RE, Rosenberg PB, Mirotsou M, Dzau VJ. MicroRNA induced cardiac reprogramming in vivo: evidence for mature cardiac myocytes and improved cardiac function. Circ Res. 2015;116:418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, Nakashima H, Akiyama M, Wada R, Inagawa K, Nishiyama T, Kaneda R, Fukuda T, Takeda S, Tohyama S, Hashimoto H, Kawamura Y, Goshima N, Aeba R, Yamagishi H, Fukuda K, Ieda M. MiR‐133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 2014;33:1565–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qin B, Xiao B, Jiang T, Yang H. Effects of miR‐590‐5p on ox‐LDL‐induced endothelial cells apoptosis and LOX‐1 expression. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2012;37:675–681. [DOI] [PubMed] [Google Scholar]
- 28. Zhao S, Yang G, Liu P, Deng Y, Zhao Z, Sun T, Zhuo X, Liu J, Tian Y, Zhou J, Yuan Z, Wu Y. miR‐590‐3p is a novel MicroRNA in myocarditis by targeting nuclear factor kappa‐B in vivo. Cardiology. 2015;132:182–188. [DOI] [PubMed] [Google Scholar]
- 29. He PP, OuYang XP, Li Y, Lv YC, Wang ZB, Yao F, Xie W, Tan YL, Li L, Zhang M, Lan G, Gong D, Cheng HP, Zhong HJ, Liu D, Huang C, Li ZX, Zheng XL, Yin WD, Tang CK. MicroRNA‐590 inhibits lipoprotein lipase expression and prevents atherosclerosis in apoE knockout mice. PLoS One. 2015;10:e0138788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin X, Steinberg S, Kandasamy SK, Afzal J, Mbiyangandu B, Liao SE, Guan Y, Corona‐Villalobos CP, Matkovich SJ, Epstein N, Tripodi D, Huo Z, Cutting G, Abraham TP, Fukunaga R, Abraham MR. Common miR‐590 variant rs6971711 present only in African Americans reduces miR‐590 biogenesis. PLoS One. 2016;11:e0156065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aggarwal P, Turner A, Matter A, Kattman SJ, Stoddard A, Lorier R, Swanson BJ, Arnett DK, Broeckel U. RNA expression profiling of human iPSC‐derived cardiomyocytes in a cardiac hypertrophy model. PLoS One. 2014;9:e108051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang X, Tsitsiou E, Herrick SE, Lindsay MA. MicroRNAs and the regulation of fibrosis. FEBS J. 2010;277:2015–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Q, Wang G, Chen Y, Li G, Yang D, Kang J. A miR‐590/Acvr2a/Rad51b axis regulates DNA damage repair during mESC proliferation. Stem Cell Reports. 2014;3:1103–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ekhteraei‐Tousi S, Mohammad‐Soltani B, Sadeghizadeh M, Mowla SJ, Parsi S, Soleimani M. Inhibitory effect of hsa‐miR‐590‐5p on cardiosphere‐derived stem cells differentiation through downregulation of TGFB signaling. J Cell Biochem. 2015;116:179–191. [DOI] [PubMed] [Google Scholar]
- 35. Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai B, Wang N, Li X, Feng T, Hong Y, Yang B. Downregulation of miR‐133 and miR‐590 contributes to nicotine‐induced atrial remodelling in canines. Cardiovasc Res. 2009;83:465–472. [DOI] [PubMed] [Google Scholar]
- 36. Liu T, Nie F, Yang X, Wang X, Yuan Y, Lv Z, Zhou L, Peng R, Ni D, Gu Y, Zhou Q, Weng Y. MicroRNA‐590 is an EMT‐suppressive microRNA involved in the TGFβ signaling pathway. Mol Med Rep. 2015;12:7403–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiao J, Dang Y, Yang Y, Gao R, Zhang Y, Kou Z, Sun XF, Gao S. Promoting reprogramming by FGF2 reveals that the extracellular matrix is a barrier for reprogramming fibroblasts to pluripotency. Stem Cells. 2013;31:729–740. [DOI] [PubMed] [Google Scholar]
- 38. Zhao Y, Londono P, Cao Y, Sharpe EJ, Proenza C, O'Rourke R, Jones KL, Jeong MY, Walker LA, Buttrick PM, McKinsey TA, Song K. High‐efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro‐fibrotic signalling. Nat Commun. 2015;6:8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Safe S, Abdelrahim M. Sp1 transcription factor family and its role in cancer. Eur J Cancer. 2005;41:2438–2448. [DOI] [PubMed] [Google Scholar]
- 40. Reddy VS, Prabhu SD, Mummidi S, Valente AJ, Venkatesan B, Shanmugam P, Delafontaine P, Chandrasekar B. Interleukin‐18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP‐9 in part via EMMPRIN and through AP‐1 and NF‐kappaB activation. Am J Physiol Heart Circ Physiol. 2010;299:H1242–H1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ihn H, Tamaki K. Competition analysis of the human alpha2(I) collagen promoter using synthetic oligonucleotides. J Invest Dermatol. 2000;114:1011–1016. [DOI] [PubMed] [Google Scholar]
- 42. Hitraya EG, Varga J, Artlett CM, Jimenez SA. Identification of elements in the promoter region of the alpha1(I) procollagen gene involved in its up‐regulated expression in systemic sclerosis. Arthritis Rheum. 1998;41:2048–2058. [DOI] [PubMed] [Google Scholar]
- 43. Jiang L, Zhou Y, Xiong M, Fang L, Wen P, Cao H, Yang J, Dai C, He W. Sp1 mediates microRNA‐29c‐regulated type I collagen production in renal tubular epithelial cells. Exp Cell Res. 2013;319:2254–2265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. FACS analyses for Thy1, CD31, and CD117/c‐kit in human and porcine cardiac fibroblasts. Representative FACS analyses of human cardiac fibroblasts (top) or porcine cardiac fibroblasts (bottom) showing majority of the cells were positive for the cardiac fibroblast‐specific surface marker, Thy1, and negative for cardiac progenitor (c‐kit/CD117) or endothelial cell (CD31) markers. FACS indicates fluorescence‐activated cell sorting.
Figure S2. Reprogramming of porcine cardiac fibroblasts in coculture with murine cardiomyocytes. Porcine cardiac fibroblasts were transduced with lentivirus expressing a GFP marker alone (negative control group; left); lentivirus vectors also expressing miR‐590 as well as vectors expressing Gata4, Mef2c, or Tbx5 (“GMT plus miR‐590” group; middle); or with lentivirus vectors also expressing Hand2 and Myocardin as well as vectors expressing Gata4, Mef2c, or Tbx5 (“GMT plus H/My” group; right). Four weeks after initial transduction, these porcine cardiac fibroblasts were cocultured with (untreated) neonatal murine cardiomyocytes (negative for GFP). A, Representative immunofluorescence staining demonstrating (green) GFP expression, which indicates lentivirus infection. Only (green fluorescent) cells that had been treated with GMT plus miR‐590 or GMT plus H/My contracted synchronously with surrounding cardiomyocytes. Fibroblasts treated with a GFP‐control vector only did not demonstrate contractions in these coculture experiments (Videos S1 through S3). B, Representative curves reflecting contraction (left) and Ca2+ transient trace (right) under basal conditions of (induced cardiomyocyte‐like; iCM) cells derived from porcine cardiac fibroblasts after coculture with murine neonatal cardiomyocytes. For the calcium transients, cells were loaded with Fura‐2 for 30 minutes, and then calcium transients were recorded (n=3). C, Representative immunofluorescence staining for DAPI ([blue] nuclear marker), GFP ([green] lentivirus infection marker), and the (red) cardiomyocyte marker, α‐actinin, in porcine cardiac fibroblasts treated with lentivirus containing a GFP marker and GMT, GMT plus miR‐590, or GMT plus H/My which were then cocultured with neonatal murine cardiomyocytes. Porcine cardiac fibroblasts transduced with GFP, miR‐590, or GMT alone did not express α‐actinin in coculture neonatal murine cardiomyocytes. Porcine cardiac fibroblasts transduced with GMT plus miR‐590 or GMT plus H/My expressed the cardiomyocyte marker, α‐actinin (indicates by arrows), in coculture with murine cardiomyocytes (scale bars, 100 μm). Black arrow indicates GFP‐positive nonbeating cells, yellow arrow indicates GFP‐positive beating cells. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; GFP, green fluorescent protein; miR, microRNA.
Figure S3. miR‐590 directly represses Sp1. A, Predicted interaction sites of miR‐590 at the 3′‐UTR of Sp1 (TargetScan: www.targetscan.org). B, HEK293 cells were cotransfected with (pGL3‐Sp1‐3′UTRWT) and mutated targeting sequence (pGL3‐Sp1‐3′UTR‐Mutant) and miRNA mimic‐control/590 for 48 hours, then luciferase activity was detected. miR‐590 directly repressed WT Sp1 3′UTR in luciferase assay, and the repression was abolished when binding site was mutated (n=3). Data are presented as mean±SEM. *P<0.05 versus control‐miR. miR indicates microRNA; Sp1, specificity protein 1; UTR, untranslated region; WT, wild type.
Video S1. This video shows porcine fibroblasts treated with a GFP‐control vector only did not demonstrate contractions in the coculture experiments.
Video S2. This video shows porcine cardiac fibroblasts contracted synchronously with surrounding murine cardiomyocytes after transduction with GMT plus miR‐590.
Video S3. This video shows porcine cardiac fibroblasts contracted synchronously with surrounding murine cardiomyocytes after transduction with GMT plus H/My.