

Abstract
Background
We previously identified peritoneal B1a cells that secrete natural IgM as a key atheroprotective B cell subset. However, the molecules that activate atheroprotective B1a cells are unknown. Here, we investigated whether Toll‐like receptors (TLRs) TLR2, TLR4, and TLR9 expressed by B1a cells are required for IgM‐mediated atheroprotection.
Methods and Results
We adoptively transferred B1a cells from wild‐type mice or from mice deficient in TLR2, TLR4, TLR9, or myeloid differentiation primary response 88 (MyD88) into ApoE−/− mice depleted of peritoneal B1a cells by splenectomy and fed a high‐fat diet for 8 weeks. Elevations in plasma total, anti‐oxLDL (oxidized low‐density lipoprotein), anti‐leukocyte, anti‐CD3, anti‐CD8, and anti‐CD4 IgMs in atherosclerotic mice required B1a cells expressing TLR4 and MyD88, indicating a critical role for TLR4‐MyD88 signaling for IgM secretion. Suppression of atherosclerosis was also critically dependent on B1a cells expressing TLR4‐MyD88. Atherosclerosis suppression was associated not only with reductions in lesion apoptotic cells, necrotic cores, and oxLDL, but also with reduced lesion CD4+ and CD8+ T cells. Transforming growth factor beta 1 (TGF‐β1) expression, including macrophages expressing TGF‐β1, was increased, consistent with increased IgM‐mediated phagocytosis of apoptotic cells by macrophages. Reductions in lesion inflammatory cytokines tumor necrosis factor alpha (TNF‐α), interleukin (IL) 1β, and IL‐18 were consistent with augmented TGF‐β1 expression.
Conclusions
TLR4‐MyD88 expression on B1a cells is critical for their IgM‐dependent atheroprotection that not only reduced lesion apoptotic cells and necrotic cores, but also decreased CD4 and CD8 T‐cell infiltrates and augmented TGF‐β1 expression accompanied by reduced lesion inflammatory cytokines TNF‐α, IL‐1β, and IL‐18.
Keywords: atherosclerosis, B1a cells, cytokine, IgM, inflammation, Toll‐like receptor 4/MyD88
Subject Categories: Atherosclerosis, Inflammation, Vascular Biology, Vascular Disease
Introduction
Atherosclerosis is a chronic inflammatory disease of medium and large arteries responsible for heart attacks and strokes that remain leading causes of global mortality.1 The inflammation is driven by immune responses following lipid entry into the arterial wall.2 Among immune cells, conventional B2 cells are atherogenic3, 4, 5 and B1a cells are atheroprotective.6, 7
B1a cells are innate‐like B cells expressing CD5 that arise from fetal liver and are found mainly in peritoneal and pleural cavities.8 The spleen is required for their survival and maintenance.9 B1a cells are long‐lived, self‐replenishing B cells that produce most of the circulating natural low‐affinity IgM.10 B1a‐cell repertoire is selected by recognition of self‐antigen that results in evolutionarily important antibody specificities responding to pathogen‐related signals, crucial for providing immediate, early humoral protection against pathogens.11
We identified B1a cells as an atheroprotective B‐cell subset, demonstrating that adoptive transfer of wild‐type (WT), but not IgM‐deficient, B1a lymphocytes ameliorates atherosclerosis.6 Atheroprotection was accompanied by increases in lesion IgM deposits and reductions in apoptotic cells, oxidized low‐density lipoprotein (oxLDL), and necrotic core size. We suggested that IgM scavenges apoptotic debris and oxLDL, attenuating inflammation and reducing necrotic cores in atherosclerotic lesions, effects consistent with scavenger properties of natural IgM.12, 13, 14, 15 However, the molecules that activate these atheroprotective B1a cells remain unknown.
B1a cells are innate‐like immune cells, responding rapidly and strongly to pathogen‐encoded signals that constitute pathogen‐associated molecular patterns (PAMPs). Strong selective pressure for recognition and responses to PAMPs are largely dependent on Toll‐like receptors (TLRs).16 Fourteen TLRs have been identified,16 with some, such as TLR2 and TLR4, expressed on cell membranes and others, such as TLR3, TLR7, TLR8, and TLR9, expressed endosomally.17, 18 TLRs, with the exception of TLR3, utilize myeloid differentiation primary response 88 (MyD88), an adaptor protein to activate transcription factor nuclear factor kappa B.17, 18, 19, 20
Using genetic knockout or hematopoietic depletion models, global TLR deficiency affects atherosclerosis development. TLR9 activation appears essential for protecting against atherosclerosis,21 TLR2 modulates atherosclerosis,22 and TLR4, depending on conditions, can either protect23 or exacerbate atherosclerosis.24 Given that B1a cells express and respond to activation of multiple TLRs, including by TLR2, TLR4, and TLR9,25 we set out to determine whether these particular TLRs have roles in atheroprotection mediated by B1a cells.
To this end, we examined the effects of adoptively transferring B1a cells selectively deficient in TLR2, TLR4, or TLR9 or deficient in MyD88 into ApoE−/− mice rendered B1a cell deficient by splenectomy. Our findings provide the novel report that B1a cell expression of TLR4 and MyD88, but not TLR2 or TLR9, are critical for activation of these cells to suppress atherosclerosis. Furthermore, we report that in addition to reducing apoptotic cells and necrotic cores in lesions,6 polyclonal natural IgM produced by B1a cells also suppressed atherosclerosis development by increasing anti‐inflammatory cytokines transforming growth factor‐beta (TGF‐β1) and interleukin (IL) 10, reducing proinflammatory tumor necrosis factor alpha (TNF‐α), IL‐1β, and IL‐18 cytokines and reducing CD4+ and CD8+ T‐cell infiltrates in lesions.
Methods
Animals and Ethics
All mice used in experiments were on a C57BL/6 background. ApoE−/−, WT, TLR2−/−, TLR4−/−, and TLR9−/− mice were maintained at the Alfred Medical Research, and Education Precinct (AMREP), Prahran, Melbourne, Australia. MyD88−/− mice were from the Walter and Eliza Hall Institute, Melbourne, Australia. ApoE−/− mice (6‐ to 8‐week‐old male) were subjected to sham operation or splenectomy; splenectomized mice received (3×), over the ensuing 8 weeks, either PBS or purified B1a cells from WT mice or mice deficient in TLR2, TLR4, TLR9, or MyD88. All mice were fed a high‐fat diet (HFD) containing 21% fat and 0.15% cholesterol (Specialty Feeds, Glen Forrest, Western Australia, Australia) and sterile water under specific pathogen‐free conditions. At the end of the experiment, mice were killed by using slow‐fill carbon‐dioxide asphyxiation. All experimental procedures and study protocols complied with national guidelines for care and use of laboratory animals were approved by the AMREP Animal Ethics Committee.
Splenectomy
Splenectomies were performed on 6‐ to 8‐week‐old male ApoE−/− mice under aseptic conditions.6 Briefly, under anesthesia induced by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (16 mg/kg), the spleen identified through a 10‐mm left‐flank incision was removed using diathermy. After confirming no intra‐abdominal bleeding, the peritoneum and skin were closed using a 2‐0 monofilament suture, atipamezole HCl (anti‐sedan, 100 mg/kg) was administered subcutaneously as recovery agent and mice were placed in 37°C recovery chambers before returned to their cages.
Cell Isolation and Adoptive Cell Transfers
Peritoneal fluid collected from donor mice as described previously6; cells were stained with fluorochrome‐labeled anti‐CD3, anti‐CD19, and anti‐CD5 antibodies. CD3– CD19+ CD5+ B1a cells were then isolated using a FACS Aria Cell Sorter (BD Biosciences, San Jose, CA), and after assessing cell viability (>95%), 105 B1a cells were adoptively transferred by tail vein injection into splenectomized ApoE−/− mice at weeks 1, 4, and 7 after splenectomy,6 while feeding them an HFD. In some experiments, ApoE−/− mice were subjected to splenectomy first, 3 weeks later adoptively transferred withTLR4−/− B1a cells, and challenged for 3 weeks either TLR4 against (LPS; 10 μg, intraperitoneally weekly) or TLR9 against (type B CpG oligodeoxynucleotide [ODN‐1668]; 200 ng, intravenously, weekly).
Generation of Apoptotic Cells and IgM‐Complement Binding Assay
Apoptotic binding assay was modified from IgM‐dependent complement activation assay.26 Briefly, thymocytes prepared from WT mice (2×106 cells/mL) were incubated in serum‐free DMEM containing etoposide (Sigma‐Aldrich, St. Louis, MO) at the concentration of 40 μmol/L for 16 hours to generate early apoptotic cells. These early apoptotic cells were incubated in DMEM containing 10% plasma isolated from different experimental mice for 30 minutes, followed by staining with Annexin V, 7‐aminoactinomycin D (7AAD), IgM, and late complement proteins (C5b‐9) antibodies (BD Pharmingen, San Diego, CA). Early apoptotic cell‐binding capacity of IgM and complement proteins was assessed using a FACS Canto‐II (BD Biosciences).
Plasma Anti‐Leukocyte IgM Detection Using Splenic Leukocytes
In a modified protocol to detect anti‐leukocyte IgM antibody,27, 28 splenocytes from C57Bl/6 mice (2×106 cells/0.2 mL of complete 10% FBS‐RPMI per well) were initially activated with 10 μg/mL of LPS (Sigma‐Aldrich) for 24 hours at 37°C in 5% CO2. After Fc blocking for 30 minutes, splenocytes were incubated with plasma samples (diluted at 1:300) at 37°C for 2 hours using U‐bottomed plates previously blocked with 1% BSA. After washing for 3 times, cells were incubated with HRP‐conjugated goat anti‐mouse IgM antibody (ICL, Portland, OR) again for 1 hour, followed by washing cells again. Color development was done using TMB substrate for color development. The OD at 450 nm was read by ELISA reader.
Plasma Anti‐Lymphocyte IgM Detection Using Recombinant Proteins (CD3, CD4, and CD8)
In a modified protocol to detect anti‐lymphocyte IgM antibodies,27, 28 recombinant CD3, CD4, and CD8 extracellular domain proteins (Life Technologies, Carlsbad, CA) were coated in flat‐bottomed 96‐well ELISA plates (50 μL at 5 μg/mL in 1× PBS/well) at 4°C for 18 hours. Wells were blocked with 1% BSA for 2 hours and plasma diluted at 1:300 in 1% BSA were added and incubated for 2 hours at room temperature. HRP‐conjugated goat anti‐mouse IgM antibody (ICL) was added into the wells, followed by addition of TMB substrate for colour development. The optical density (OD) at 450 nm was read by ELISA reader.
Tissue Collection
At the end of the experiments, peritoneal fluids were collected for lymphocyte analysis. Plasma and thoracic aortas were kept at −80°C in a freezer for further analysis and aortic sinuses embedded in optimal cutting temperature compound for histological and immunohistochemical staining.
Flow Cytometry
B lymphocytes and non‐B‐lymphocyte populations in the peritoneal cavity and peripheral lymph nodes (inguinal and axillary) were analyzed using fluorochrome‐conjugated antibodies (BD Pharmingen) on a FACS Canto‐II (BD Biosciences) as previously described by us.3, 6, 29 For B cells, PE‐conjugated anti‐CD19, APC‐conjugated anti‐CD5, and APC‐Cy7–conjugated anti‐CD11b antibodies were used. For non‐B‐lymphocyte populations, Pacific Blue–conjugated anti‐CD4, PerCP‐conjugated anti‐CD8a, FITC‐conjugated anti‐TCR‐β, and PE‐Cy7–conjugated anti‐NK1.1 antibodies were used.
Atherosclerosis Assessment and Histological Staining
Frozen aortic sinus sections of 6 μm in thickness were fixed in 10% buffered formalin for 5 minutes at room temperature, air‐dried for 10 minutes, and then stained with hematoxylin and eosin (H&E) and Oil Red O (ORO). ORO‐stained sections were used to measure total intimal lesion size and lesion lipid accumulation as described previously.3 To determine necrotic core areas in atherosclerotic lesions, acellular areas from H&E‐stained atherosclerotic lesions were measured as previously described.6, 30
Immunohistochemical and Immunofluorescence Staining
Frozen aortic sinus sections were stained using different antibodies. Macrophages were stained with anti‐CD68 antibody (Serotec, Raleigh, NC); CD4 and CD8 T cells with anti‐CD4 and CD8 antibodies, respectively (BD Biosciences), IgM with anti‐IgM antibody (BD Pharmigen), oxLDL with malondealdehyde (MDA)‐oxLDL antibody (Abcam, UK) as described before.3, 30 Apoptotic cells identified by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) under light microscopy were expressed per lesion areas as described previously.3, 30 Apoptotic macrophages in lesions were identified by dual‐label (TUNEL/CD68) immune fluorescence as were macrophages expressing TGF‐β (Novus Biologicals LLC, Littleton, CO).30
Total and OxLDL‐Specific Immunoglobulins
An ELISA was used to determine plasma levels of total immunoglobulins (total Ig, IgG, and IgM) and oxLDL‐specific immunoglobulins (total Ig, IgG, and IgM) levels at endpoint as described previously.6
Lipid Profiles
Cholesterol profiles (total cholesterol, high‐density lipoprotein cholesterol [HDL‐C], very‐low‐density lipoprotein [VLDL]/low‐density lipoprotein [LDL] cholesterol, and triglycerides) in plasma were measured as described previously.6
Real‐Time Polymerase Chain Reaction Analysis
Total RNAs were extracted from thoracic aortas using RNeasy fibrous tissue mini kit (Qiagen, Hilden, Germany) as described.30 Expression of targeted gene in 20 μg of total RNA was carried out using a one‐step QuantiFast SYBR Green RT‐PCR kit (Qiagen) on a 7500 Fast Real‐Time PCR system (Applied Biosystems, Foster City, CA) to determine the expression of each gene. The comparative cycle threshold (Ct) method was used to determine target‐gene expression with housekeeping gene 18S (Applied Biosystems) as an endogenous control. Relative amounts of each mRNA for each of the genes in lesions from control and test mice were calculated using comparative Ct values.31 The sequences of oligonucleotides used are shown in Table 1.
Table 1.
Primer Sequences Used for Quantitative RT‐PCR
| TNF‐α: | Sense (S), 5′‐TATGGCCCAGACCCTCACA‐3′ |
| Anti‐sense (AS), 5′‐TCCTCCACTTGGTGGTTTGC‐3′ | |
| IFN‐γ: | S, 5′‐TCCTCAGACTCATAACCTCAGGAA‐3′ |
| AS, 5′‐GGGAGAGTCTCCTCATTTGTACCA‐3′ | |
| IL‐1β: | S, 5′‐CCACCTCAATGGACAGAATATCAA‐3′ |
| AS, 5′‐GTCGTTGCTTGGTTCTCCTTGT‐3′ | |
| IL‐18: | S, 5′‐GATCAAAGTGCAGTGAACC‐3′ |
| AS, 5′‐AACTCCATCTTGTTGTGTCC‐3′ | |
| MCP‐1: | S, 5′‐CTCAGCCAGATGCAGTTAACG‐3′ |
| AS, 5′‐GGGTCAACTTCACATTCAAAGG‐3′ | |
| VCAM‐1: | S, 5′‐AGAACCCAGACAGACAGTCC‐3′ |
| AS, 5′‐GGATCTTCAGGGAATGAGTAGAC‐3′ | |
| TGF‐β: | S, 5′‐AGCCCTGGATACCAACTATTGC‐3′ |
| AS, 5′‐TCCAACCCAGGTCCTTCCTAA‐3′ | |
| IL‐10: | S, 5′‐GAAGACAATAACTGCACCCA‐3′ |
| AS, 5′‐CAACCCAAGTAACCCTTAAAGTC‐3′ |
Statistical Analysis
Results are expressed as mean±SEM. Comparisons between groups were carried out using the Student t test or Mann–Whitney U test, depending on whether the data were normally distributed, as assessed using the Kolmogorov–Smirnov test. For multiple comparisons, results were analyzed using 1‐way ANOVA (after confirming normality of distribution) followed by Bonferroni post‐test. A value of P<0.05 was considered statistically significant.
Results
TLR4 and MyD88 Are Required by B1a Cells to Suppress Atherosclerosis Development
To investigate the role of TLRs in atheroprotection conferred by B1a cells, ApoE−/− mice were subjected to splenectomy to deplete peritoneal B1a cells,6, 9 without affecting peritoneal B1b cells9 or sham operation. Then, 1 week later, the splenectomized mice received vehicle or B1a cells isolated from WT, TLR2−/−, TLR4−/−, or TLR9−/− donor mice and fed an HFD for 8 weeks. After the different B1a cell transfer and 8 weeks of HFD, lymphocyte populations in the peritoneal cavity and peripheral lymph nodes were similar (P>0.05; Table 2); body weights and plasma cholesterols did not differ among the mouse groups (P>0.05; Table 2). Transfer of WT B1a cells attenuated atherosclerosis to levels observed in sham‐operated mice, measured as total lesion area; lipid accumulation in lesions was also reduced (both P<0.05; Figure 1A and 1B). Transfer of B1a cells deficient in TLR2 and TLR9 also attenuated lesions, to a similar extent as WT B1a cells with reductions in total lesion size averaging 35% and reductions in lesion lipid accumulation averaging 45% (P<0.05; Figure 1A and 1B) without affecting lipid percent area (P>0.05; Figure 1C). Macrophage accumulation in lesions was also reduced after transfer of WT, TLR2‐, or TLR9‐deficient B1a cells (P<0.05; Figure 1D). In contrast, B1a cells deficient in TLR4 did not affect atherosclerotic lesion size, lesion lipid accumulation, or macrophage accumulation within lesions. Lesion size as well as lipid and macrophage accumulation in lesions of mice that received TLR4‐deficient B1a cells were similar to those that received PBS (P>0.05; Figure 1A, 1B, and 1D). Similar to lipid percent area, macrophage percent area was unaffected (P>0.05; Figure 1E), suggesting that plaque quality was unchanged. Differential survival of B1a cells deficient in TLR4 could not account for these effects given that their numbers in the peritoneal cavity level after adoptive transfer were similar to transfer of WT B1a cells or B1a cells deficient in TLR2 or TLR9 (P>0.05; Table 2). Plasma cholesterol levels and body weights were also similar (P>0.05; Table 2).
Table 2.
Lymphocyte Population, Body Weight, and Lipid Profile of Splenectomized ApoE−/− Mice Received TLR‐Deficient B1a Cells
| SO | SX‐PBS | SX‐WTB1a | SX‐TLR2−/− B1a | SX‐TLR4−/− B1a | SX‐TLR9−/− B1a | |
|---|---|---|---|---|---|---|
| Peritoneal cavity, ×104 | ||||||
| B2 | 28.1±1.57 | 27.4±1.2 | 30.2±3.0 | 28.6±2.2 | 26.1±1.5 | 27.8±2.1 |
| B1a | 11.9±0.6 | 2.5±0.2a | 9.99±1.1 | 9.8±1.2 | 8.6±0.9 | 9.95±1.1 |
| CD4 | 10.1±0.67 | 8.6±0.37 | 10.5±0.47 | 8.1±0.83 | 10.2±1.0 | 9.7±0.82 |
| CD8 | 7.2±0.38 | 6.4±0.34 | 7.2±0.57 | 6.4±0.46 | 7.2±0.53 | 6.8±0.4 |
| NKT | 0.53±0.08 | 0.59±0.09 | 0.42±0.07 | 0.32±0.05 | 0.42±0.09 | 0.31±0.04 |
| NK | 2.5±0.3 | 3.3±0.3 | 2.72±0.27 | 2.73±0.28 | 3.1±0.32 | 2.7±0.32 |
| Lymph nodes, ×104 | ||||||
| B2 | 444.7±68.1 | 434.2±57.1 | 419.1±35.2 | 484.7±57.2 | 412.6±23.1 | 402.5±47.9 |
| B1a | 14.8±3.1 | 2.6±0.3a | 25.2±1.1 | 24.9±1.6 | 21.1±1.03 | 21.9±2.3 |
| CD4 | 244.5±19.5 | 264.2±43.4 | 260±22.3 | 214.9±23.4 | 270.8±21.9 | 212.9±25 |
| CD8 | 163.7±19.7 | 170.7±34.5 | 148.1±11.9 | 159.6±18.7 | 179.2±19.9 | 151.5±18.6 |
| NKT | 3.64±0.9 | 4.31±0.9 | 3.82±0.2 | 3.54±0.5 | 4.13±0.5 | 3.52±0.5 |
| NK | 11.1±1.3 | 12.1±1.3 | 11.3±0.7 | 11.6±1.8 | 10.1±0.8 | 10.5±1.2 |
| Body weight, g | 24.8±1 | 25±1.3 | 26±0.8 | 25±0.9 | 25±0.6 | 26±1.0 |
| Plasma lipids, mmol/L | ||||||
| Total‐C | 20.4±0.9 | 20.8±3.2 | 24.3±2.2 | 21.13±2 | 24.2±0.9 | 24.5±1.7 |
| VLDL/LDL‐C | 15.1±0.5 | 15.8±2.4 | 18±1.6 | 15.9±1.4 | 18.1±0.70 | 18.7±1.3 |
| HDL‐C | 3.2±0.2 | 3.4±0.5 | 4±0.4 | 3.4±0.3 | 4.1±0.2 | 3.9±0.2 |
| Triglycerides | 3.4±0.2 | 3.5±0.6 | 4.8±0.6 | 4±0.7 | 4.5±0.4 | 4.4±0.4 |
Data represented as mean±SEM of 10 to 12 mice in each group. HDL‐C, high‐density lipoprotein cholesterol; NK, natural killer; NKT, natural killer T; SO, sham‐operated group; SX, splenectomized group; TLR, Toll‐like receptor; VLDL, very‐low‐density lipoprotein; WT, wild type.
P<0.05 compared to SO group.
Figure 1.
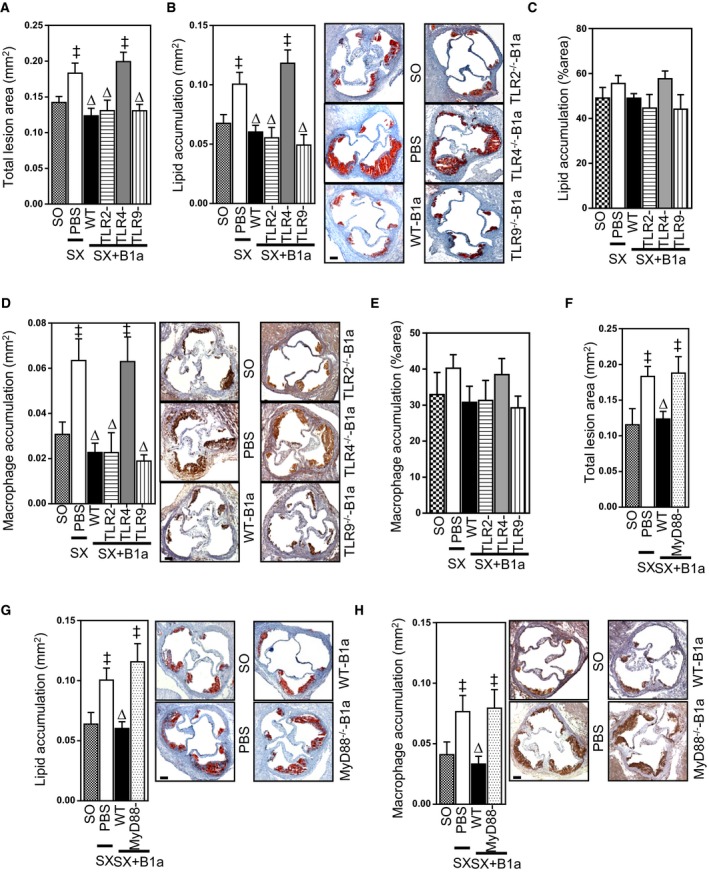
Suppression of atherosclerosis by B1a cells is dependent on expression of TLR4 and MyD88. Splenectomized (SX) ApoE−/− mice received PBS or peritoneal B1a cells isolated from WT, TLR2−/−, TLR4−/−, TLR9−/−, and MyD88−/− donor mice, given an HFD for 8 weeks, and effects on aortic sinus atherosclerotic lesions compared to sham‐operated (SO) ApoE−/− mice given an HFD. A, Total intimal lesion areas in SO mice and splenectomized mice receiving vehicle, WT B1a cells, or B1a cells deficient in TLR2, TLR4, or TLR9. B, ORO‐stained lipid area, (C) lipid percent area in lesions, (D) CD68+‐stained macrophage area, and (E) macrophage percent area in lesions. F, Total intimal lesion areas in SO mice and splenectomized mice receiving PBS, WT B1a cells, or B1a cells deficient in MyD88. G, ORO‐stained lipid accumulation in lesions and (H) CD68+ macrophage accumulation in lesions. Representative photomicrographs accompany bar graphs. Data in bar graphs represented as mean±SEM. Bars present 100 μm. TLR study: 9 to 12 mice/group; MyD88 study: 7 to 10 mice/group. ‡ P<0.05 compared with SO; ∆ P<0.05 compared with PBS. HFD indicates high‐fat diet; MyD88, myeloid differentiation primary response 88; ORO, Oil Red O; TLR, Toll‐like receptor; WT, wild type.
TLR4 can signal through MyD88‐dependent and MyD88‐independent, TIR‐domain‐containing adapter‐inducing interferon (IFN)‐β–dependent pathways.32 To determine the importance of MyD88 for TLR4‐mediated activation of B1a cells, we next compared the effects of adoptively transferring B1a cells from WT mice and mice deficient in MyD88. Similar to effects observed with TLR4‐deficient B1a cells, MyD88‐deficient B1a cells did not attenuate atherosclerosis in splenectomized ApoE−/− mice, measured as total intimal lesion size; lesion lipid content was also unaffected as was macrophage accumulation in lesions (all P>0.05; Figure 1F through 1H). However, both lipid and macrophage accumulation areas corrected as per total lesion area were unaffected (Figure 2A and 2B), similar to the findings in the TLR experiment. The extent of peritoneal B1a cell reconstitution by MyD88‐deficient B1a cells was similar to WT B1a cells (not shown). Other lymphocyte populations in the peritoneal cavity and peripheral lymph nodes, body weights, and plasma cholesterols did not differ among the MyD88−/− and WT mouse groups (not shown). Taken together, our findings indicate that TLR4‐MyD88–dependent signaling is critical for B1a‐cell–mediated atheroprotection.
Figure 2.
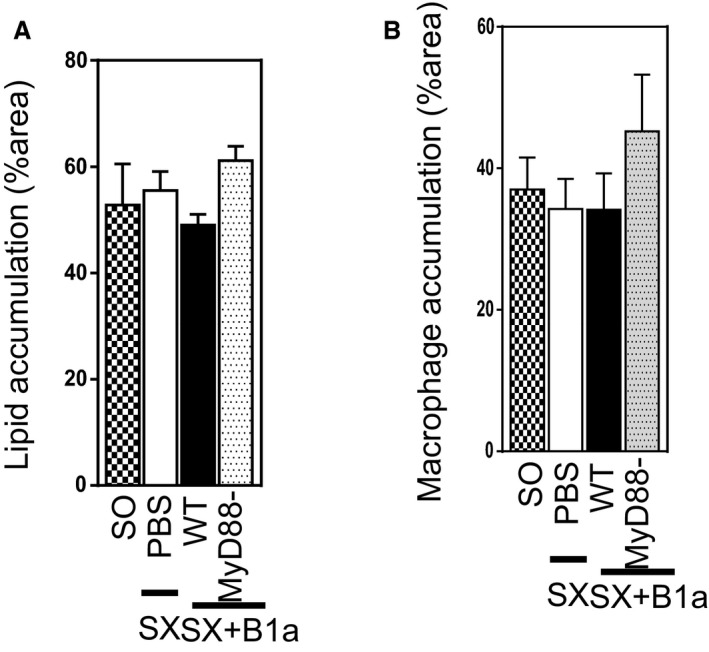
Peritoneal B1a cells are not critical in lesion lipid and macrophage percent area. Splenectomixed (SX) mice received PBS or adoptively transferred with peritoneal B1a cells isolated from WT or MyD88‐deficient donor while feeding a high diet food for 8 weeks. All splenectomixed mice with or without B1a cell transfer showed similar (A) lipid percent area and (B) macrophage percent area that were comparable to sham‐operated (SO) mice. Total of 7 to 10 mice/group. MyD88 indicates myeloid differentiation primary response 88; WT, wild type.
B1a Cell TLR4/MyD88 Expression Is Essential for IgM Secretion During Lesion Development
We next determined whether failure of B1a cells deficient in TLR4 or MyD88 to reduce atherosclerosis is related to their production of natural IgMs; previously, we have shown that B1a cells mediate atheroprotective effects through IgM.6 We compared plasma IgM levels in splenectomized mice that received vehicle, WT‐B1a cells, and B1a cells deficient in TLR4, MyD88, TLR2, and TLR9 during development of atherosclerosis. After 8 weeks of HFD, total plasma IgM levels were reduced by 68% and 75%, respectively, in splenectomized mice that received TLR4−/− or MyD88−/− B1a cells compared to WT B1a cells (P<0.05; Figure 3A), whereas IgM levels in mice that received TLR2‐ or TLR9‐deficient B1a cells were similar to WT B1a cells (P>0.05; Figure 3A). Plasma levels of MDA‐oxLDL IgM were also reduced by ≈45% and 55% in splenectomized mice that received TLR4−/− B1a cells or MyD88−/− B1a cells, respectively, compared with mice receiving WT‐B1a cells or B1a cells deficient in TLR2 or TLR9 (P<0.05; Figure 3B). Given that natural IgMs can include anti‐leukocyte autoantibodies that influence inflammatory disorders,33 we also examined whether such antibodies are also affected by TLR4‐MyD88 expression in B1a cells. Anti‐leukocyte IgM antibodies increased ≈4 times following transfer of WT B1a cells, but were unaffected after transfer of TLR4‐ or MyD88‐deficient B1a cells (P<0.05; Figure 3C). IgMs binding to CD3, CD4, and CD8 were also highly dependent on B1a cells expressing TLR4‐MyD88 (P<0.05; Figure 3D through 3F).
Figure 3.
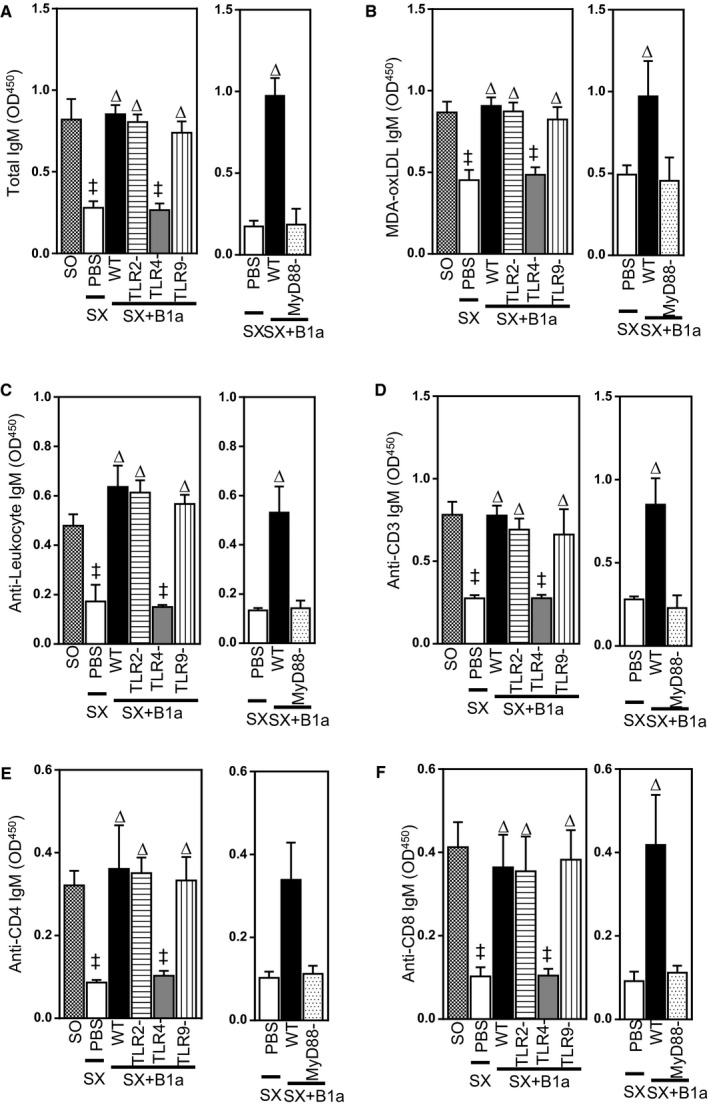
IgM secretion by B1a cells is dependent on TLR4/MyD88 signaling. Plasma IgMs from sham‐operated (SO) mice and splenectomized (SX) mice received PBS or different B1a cells, collected after 8 weeks of HFD, were measured using specific ELISA methodologies (see text for details). A, Plasma total, (B) MDA‐oxLDL, (C) antileukocyte, (D) anti‐CD3, (E) anti‐CD4, and (F) anti‐CD8 IgM antibodies. Data in bar graphs represented as mean±SEM. TLR study, 10 to 12 mice/group; MyD88 study, 8 to 10 mice/group. ‡ P<0.05 compared with SO; ∆ P<0.05 compared with PBS. HFD indicates high‐fat diet; MDA, malondialdehyde; MyD88, myeloid differentiation primary response 88; OD, optical density; oxLDL, oxidized low‐density lipoprotein; TLR, Toll‐like receptor; WT, wild type.
To confirm the importance of TLR4 in B1a‐derived IgM production, ApoE−/− mice were splenectomized to deplete peritoneal B1a cells. Peritoneal B1a cells started to decrease 1 week following splenectomy and remained decreased.9 Three weeks after splenectomy, splenectomized ApoE−/− mice were adoptively transferred with peritoneal B1a cells (105 cells, intravenously, tail vein) isolated from TLR4‐defieicnt donors and challenged with either TLR4 or TLR9 agonist for 3 weeks. Plasma IgM levels were increased in mice that received TLR4 agonist, and this increase was almost similar to the plasma IgM level of untreated C57Bl6 mice at the end of the 3‐week challenge. However, TLR9‐agonist–treated mice failed to increase plasma IgM level (Figure 4).
Figure 4.
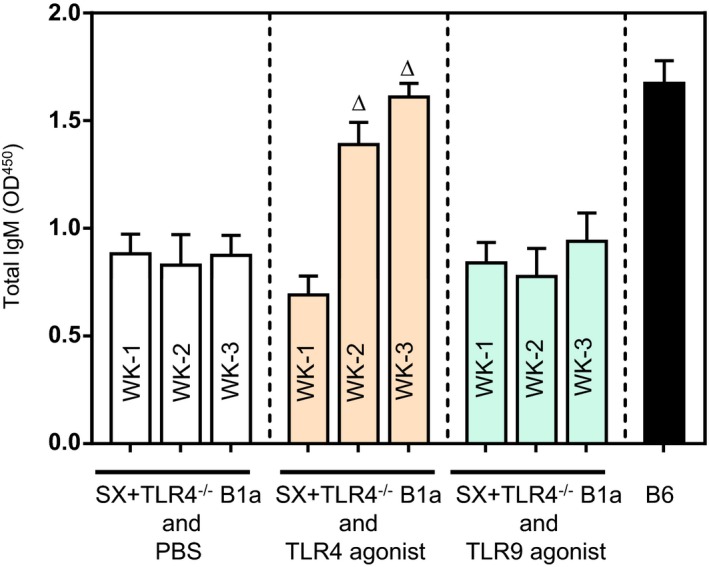
TLR4−/− B1a cells fail to respond TLR9 agonist. ApoE−/− mice were subjected to splenectomy, allowed 3 weeks to deplete spleen‐dependent peritoneal B1a cells, and adoptively transferred with TLR4−/− B1a cells followed by either TLR4 against (LPS) or TLR9 agonist (type B CpG oligodeoxynucleotide) injection for 3 weeks (see text for details). ELISA determination showed that TLR4−/− B1a cells failed to respond TLR9 stimulation. B6 indicates plasma IgM levels in WT mice. Total of 7 to 10 mice/group. ∆ P<0.05 compared with week 1. OD indicates optical density; SX, splenectomy; TLR, Toll‐like receptor; WT, wild type.
Plasma IgG and Ig including anti‐MDA‐oxLDL levels were unaffected by deletion of B1a cells deficient in TLRs or MyD88 (P>0.05; Figure 5A through 5D). Thus, during development of atherosclerosis, only TLR4‐MyD88 expression is critical for B1a cells to secrete multiple IgM antibodies and attenuate atherosclerosis.
Figure 5.
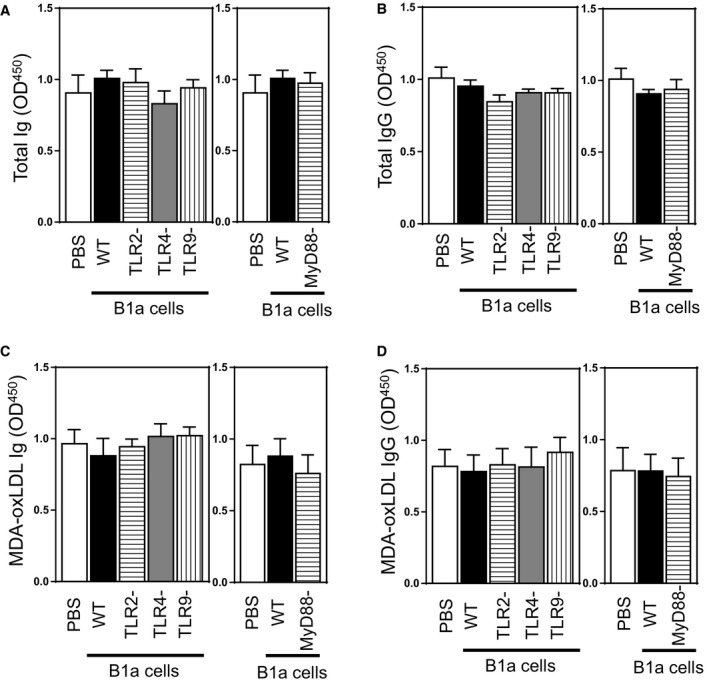
Plasma Ig, MDA‐oxLDL Ig, IgG, and MDA‐oxLDL IgG levels in splenectomized mice given an HFD for 8 weeks after receiving vehicle or B1a cells. Plasma levels of total and MDA‐oxLDL‐specific antibodies at the end of experiments were also determined (see text for details). Plasma (A) total Ig, (B) total IgG, (C) MDA‐oxLDL Ig, and (D) MDA‐oxLDL IgG. Data in bar graphs represented as mean±SEM. TLR study: 10 to 12 mice/group; MyD88 study: 8 to 10 mice/group. HFD indicates high‐fat diet; MDA, malondialdehyde; MyD88, myeloid differentiation primary response 88; oxLDL, oxidized low‐density lipoprotein; TLR, Toll‐like receptor; WT, wild type.
TLR4/MyD88 Expression by B1a Cells Affects IgM Deposition in Lesions
Because circulating levels of IgM were markedly reduced by deletion of TLR4/MyD88 in B1a cells, we next examined whether this also affected IgM accumulation in developing atherosclerotic lesions. Peritoneal B1a cells are known to generate large amounts of circulating antibodies outside their residential site,34 which may include perivascular sites where they have been shown to accumulate.7 Thus, we compared IgM accumulation in lesions following adoptive transfer of WT B1a cells with TLR4‐ or MyD88‐deficient B1a cells. Following transfer of WT B1a cells into splenectomized mice, lesion IgM levels increased 4‐ to 5‐fold (P<0.05; Figure 6A and 6B). In contrast, transfer of B1a cells deficient in either TLR4 or MyD88 only marginally increased lesion IgM levels; compared to transfer of WT B1a cells, the increase in lesion IgM was only 20% of that achieved with WT B1a cells (P<0.05; Figure 6A and 6B). The increase in lesion IgM following transfer of WT B1a cells was associated with a 50% reduction in lesion oxidatively modified MDA‐LDL levels (P<0.05; Figure 6C), contrasting with the lack of effect of transferred TLR4, or MyD88‐deficient B1a cells, whose effects were similar to transferred PBS (P>0.05; Figure 6C and 6D).
Figure 6.
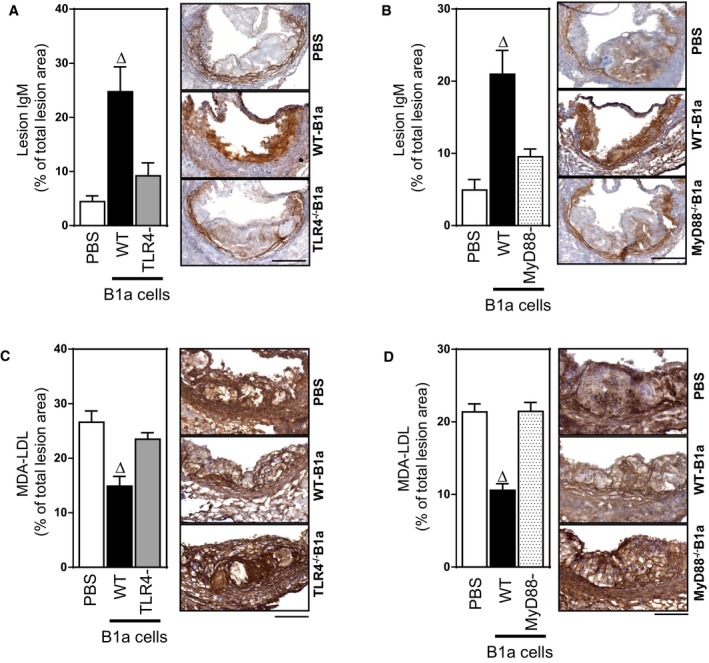
TLR4 and MyD88 activation of B1a cells is required for lesion IgM accumulation and reductions in lesion oxLDL. Photomicrographs and bar graphs showing IgM and MDA‐LDL deposits in aortic sinus atherosclerotic lesions of splenectomized ApoE−/− mice that received either PBS, WT B1a cells, or B1a cells deficient in either TLR4 or MyD88. A, TLR4−/− B1a cells and (B) MyD88−/− B1a cells failed to increase IgM deposits in atherosclerotic lesions compared to WT B1a cells. Transfer of B1a cells deficient in (C) TLR4 or (D) MyD88 does not affect MDA‐LDL in atherosclerotic lesions, compared to reductions following transfer of WT B1a cells. Data in bar graphs represented as mean±SEM. Bars present 100 μm. TLR4 study: 10 to 11 mice/group; MyD88 study: 8 to 10 mice/group. ∆ P<0.05 compared with PBS. MDA indicates malondialdehyde; MyD88, myeloid differentiation primary response 88; LDL, low‐density lipoprotein; oxLDL, oxidized low‐density lipoprotein; TLR, Toll‐like receptor; WT, wild type.
TLR4 and MyD88 Are Essential for B1a‐Cell–Mediated Reductions in Lesion Apoptotic Cells and Increases in Lesion IL‐10 and TGF‐β
We have previously shown that B1a cells promote clearance of lesion apoptotic cells through mechanisms dependent on IgM.6 To confirm these findings and determine their dependence on B1a cells expressing TLR4/MyD88, we examined the extent to which TLR4 expressed by B1a cells influenced lesion apoptotic cell numbers and associated necrotic core development. Adoptive transfer of WT B1a cells into splenectomized mice reduced lesion apoptotic cell numbers by nearly 60% (P<0.05; Figures 7A and 8A). In marked contrast, lesion apoptotic cell numbers were unaffected following transfer of TLR4‐ or MyD88‐deficient B1a cells (P>0.05; Figures 7A and 8A). This B1a‐cell–mediated reduction in lesion apoptotic cell number is accompanied by significant reductions in lesion necrotic core size compared with WT B1a cells (P<0.05; Figures 7B and 8B), but not TLR4‐ or MyD88‐deficient B1a cells (P>0.05; Figures 7B and 8B). Low‐affinity IgM can bind to lesion‐modified LDL or apoptotic cells,35, 36 and one of the predominant functions of IgM is to activate a complement system for subsequent clearance.26 To investigate the role of IgM in complement activation, we conducted early apoptotic cell IgM/complement binding assays using plasma isolates from splenectomized mice that received transfer of WT, TLR4‐, or MyD88‐deficient B1a cells. In contrast to plasma from PBS recipients, WT B1a cell recipient plasma showed an increased IgM/complement binding capacity to early apoptotic 7AAD– Annexin V+ cells. However, plasma from TLR4‐ or MyD88‐deficient B1a cell recipients fail to do so (Figure 9A and 9B), suggesting that low‐affinity IgM is critically important for antibody‐mediated immune complex clearance by complement proteins in accord with literature.26
Figure 7.
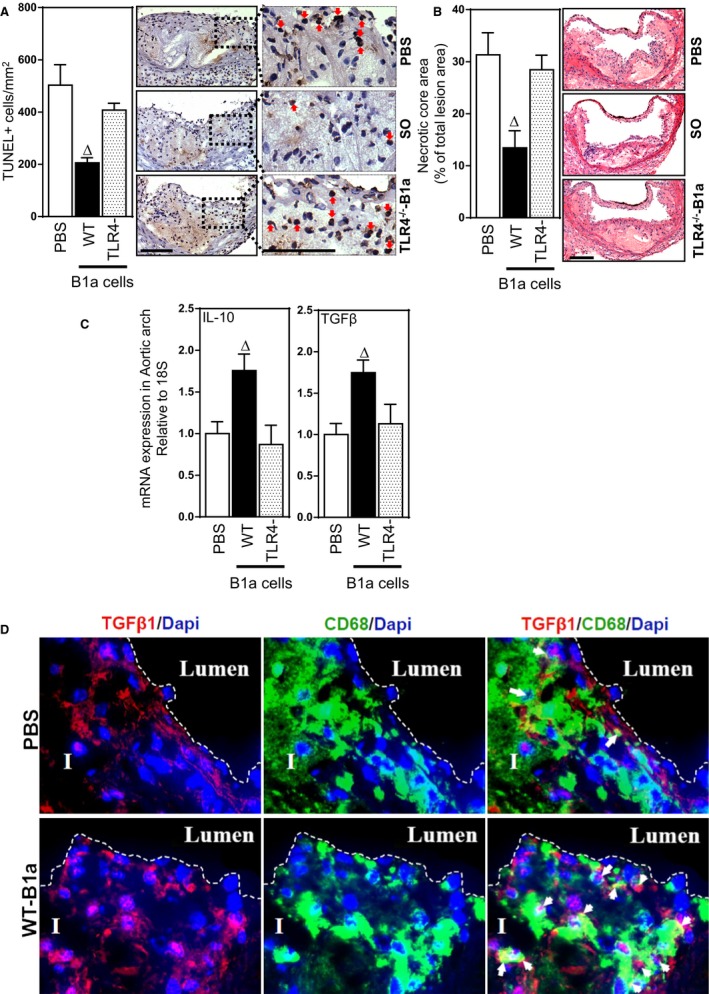
TLR4 expression by B1a cells is required for reductions in lesion apoptotic cells and necrotic cores and increases in lesion IL10 and TGF‐β. Representative photomicrographs and bar graphs showing effects of B1a cells transferred to splenectomized ApoE−/− mice on lesion apoptotic cells, necrotic cores, and IL‐10 and TGF‐β1 expression. A, TUNEL +ve apoptotic cells, (B) lesion necrotic core size, measured as % lesion area, (C) lesion mRNAs encoding IL‐10 and TGF‐β1 measured by RT‐PCR and (D) TGF‐β1 (red), DAPI (blue), and CD68/macrophage (green) immunofluorescence in lesions of splenectomized mice administered either PBS or WT B1a cells. I, Tunica intima; lumen, arterial lumen; arrows indicate macrophages colocalized with TGFβ‐1. Data in bar graphs represented as mean±SEM. Bars present 100 μm. Total of 10 to 11 mice/group. ∆ P<0.05 compared with PBS. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; IL, interleukin; RT‐PCR, reverse‐transcription polymerase chain reaction; SO, sham‐operated; TGF‐β, transforming growth factor beta; TLR, Toll‐like receptor; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; WT, wild type.
Figure 8.
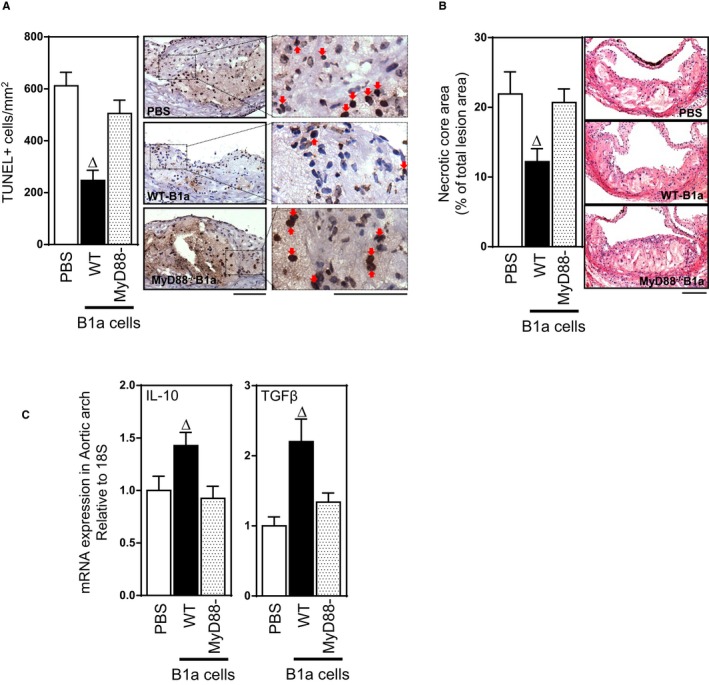
MyD88 expression by B1a cells is required for reductions in lesion apoptotic cells and necrotic cores and increases in lesion IL‐10 and TGF‐β. Representative photomicrographs and bar graphs showing effects of MyD88‐dependent B1a cells transferred to splenectomized ApoE−/− mice on lesion apoptotic cells, necrotic cores, and IL‐10 and TGF‐β1 expression. A, TUNEL +ve apoptotic cells. B, Lesion necrotic core size, measured as % lesion area. C, Lesion expression of mRNAs encoding IL‐10 and TGF‐β1 measured by RT‐PCR. Data in bar graphs represented as mean±SEM. Bars present 100 μm. Total of 8 to 10 mice/group. ∆ P<0.05 compared with PBS. IL indicates interleukin; MyD88, myeloid differentiation primary response 88; RT‐PCR, reverse‐transcription polymerase chain reaction; TGF‐β, transforming growth factor beta; TLR, Toll‐like receptor; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; WT, wild type.
Figure 9.
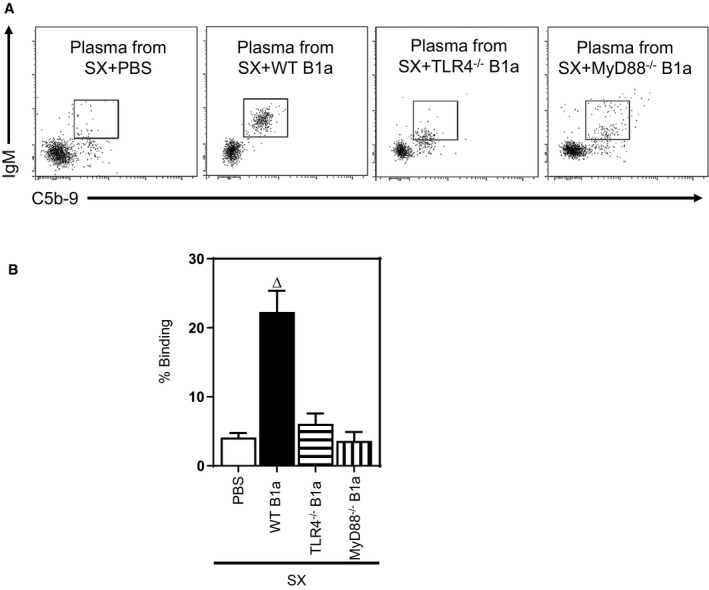
Plasma from TLR4−/− and MyD88−/− B1a recipients initiate less binding capacity of IgM and complement binding to apoptotic cells. Apoptotic cells were incubated for 30 minutes in DMEM containing 10% plasma from different B1a cell recipients after splenectomy (SX) followed by staining with anti–Annexin V, anti‐IgM, anti‐C5b‐9 antibodies and 7‐aminoactinomycin D (7AAD). A‐B Plasma from WT B1a recipients showed increased IgM and complement binding compared to PBS, TLR4−/−, and MyD88−/− B1a recipients. FACS dot plots done on early apoptotic 7AAD – Annexin V+ cells were representative of 4 experiments done at different times. ∆ P<0.05 compared with PBS. MyD88 indicates myeloid differentiation primary response 88; TLR, Toll‐like receptor; WT, wild type.
Given that removal of apoptotic cells involves phagocytosis of IgM‐bound apoptotic cells by macrophages, which then increase their expression of the anti‐inflammatory cytokines TGF‐β and IL‐10,15 we also assessed effects on lesion TGF‐β1 and IL‐10 expression. Compared with splenectomized mice that received PBS, lesions of mice that received WT B1a cells exhibited increased levels of mRNAs encoding IL‐10 and TGF‐ β (P<0.05; Figures 7C and 8C), whereas transfer of either B1a cells deficient in TLR4 or MyD88 were without effect (P>0.05; Figures 7C and 8C). To confirm that macrophages contributed to the increase in TGF‐ β1, we also examined, by immunofluorescence, lesion TGF‐β1 distribution and its collocation with lesion macrophages. As with lesion mRNA encoding TGF‐β1, lesion TGF‐β immunostaining was also increased following transfer of WT‐B1a cells (Figure 7D, top and lower panels), and in these lesions, the TGF‐β:CD68 area staining ratio was increased nearly 3‐fold compared to lesions of splenectomized mice that received PBS, indicating greater macrophage expression of TGF‐β; a small population of unidentified cells also contributed to the increased in lesion TGF‐β expression, suggesting additional yet‐to‐be‐defined mechanisms. Together, these studies indicate that increases in lesion TGF‐β and IL‐10 expression contribute to TLR4‐dependent B1a‐cell–mediated atheroprotection.
Reductions in Lesion CD4‐ and CD8+ T Cells and Proinflammatory Molecules Are Dependent on B1a Cells Expressing TLR4
Because our study indicates that plasma levels of anti‐CD3, anti‐CD4, and anti‐CD8 IgM are dependent on B1a cells expressing TLR4 and anti‐CD4 IgM antibodies inhibit CD4+ T‐cell activation and chemotaxis,37 we also compared the effects of WT and TLR4‐deficient B1a cells on lesion CD4+ and CD8+ T‐cell accumulation during development of atherosclerosis. Transfer of WT, but not TLR4‐deficient, B1a cells, reduced lesion CD4+ and CD8+ T‐cell accumulation (P<0.05; Figure 10A and 10B). Given that our data indicate that TLR4‐expressing B1a cells elevate both lesion IL‐10 and TGF‐β1 expression and these 2 anti‐inflammatory cytokines are known to regulate expression of proinflammatory cytokines,38, 39 we also investigated effects on expression of lesion inflammatory molecules. Transfer of WT, but not TLR4‐deficient, B1a cells into splenectomized ApoE−/− mice reduced expression of proinflammatory cytokines IFN‐γ, TNF‐α, IL‐1β, and IL‐18 in lesions by ≈42%, 60%, 33%, and 52%, respectively (P<0.05; Figure 10C); monocyte chemoattractant protein 1 (MCP‐1) and vascular cell adhesion molecule 1 (VCAM‐1) expression was also reduced, by 50% and 60%, respectively (P<0.05; Figure 10C).
Figure 10.
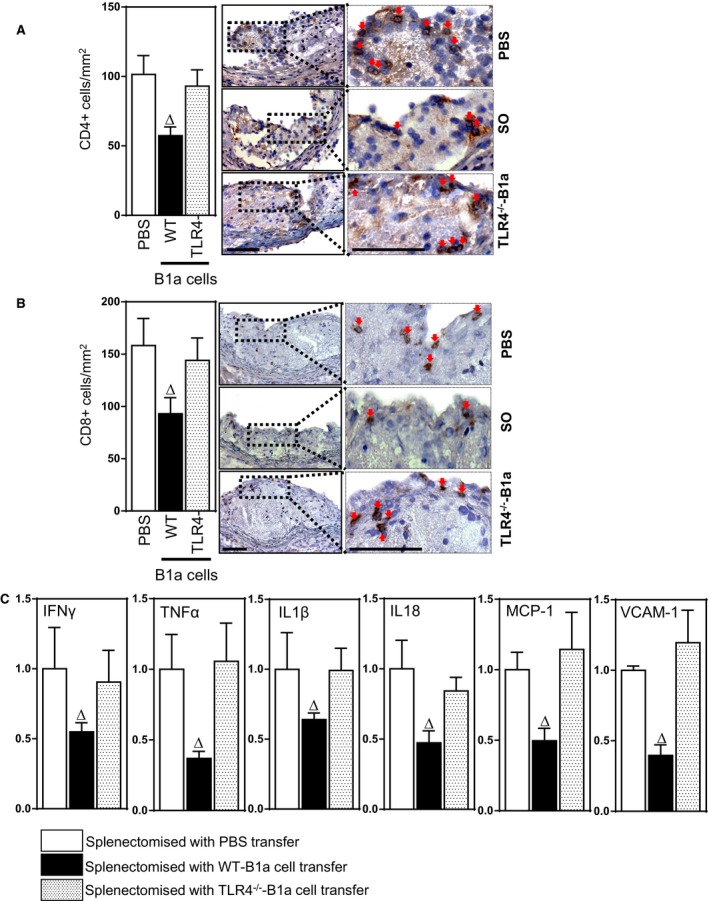
B1a‐cell–mediated reductions in T‐cell numbers and mRNAs encoding inflammatory molecules in splenectomy‐aggravated atherosclerotic lesions are dependent on TLR4 expression. Bar graphs and photomicrographs depicting lesion (A) CD4+ T cells and (B) CD8+ T cells following transfer of PBS, WT B1a cells, and TLR4−/− B1a cells into splenectomized mice. C, mRNA expression of inflammatory cytokines IFN‐γ, TNF‐α, IL‐1β, IL‐18, and MCP‐1 and cell adhesion molecule VCAM‐1 following transfer of PBS or B1a cells. Data in bar graphs represented as mean±SEM; 8 to 10 mice/group. Bars present 100 μm. ∆ P<0.05 compared with PBS. IFN‐γ indicates interferon‐gamma; IL, interleukin; MCP‐1, monocyte chemoattractant protein 1; SO, sham‐operated; TLR, Toll‐like receptor; TNF‐α, tumor necrosis factor alpha; VCAM1, vascular cell adhesion molecule 1; WT, wild type.
Discussion
Previous studies indicate that pathogen‐derived TLR agonists activate B1a cells to produce natural IgM.25 Here, we demonstrate that, during development of atherosclerosis, the ability of B1a cells to secrete atheroprotective IgMs is critically dependent on TLR4 despite the lower expression of this receptor compared with TLR2 or TLR925; the TLR4 signaling pathway used by B1a cells is dependent on MyD88. We found that TLR4‐deficient B1a cells do not attenuate atherosclerosis or produce significant IgMs, contrasting with suppressive effects of WT B1a cells and B1a cells deficient in TLR2 or TLR9. IgMs secreted by TLR4‐expressing B1a cells not only bound to oxLDL, as previously shown by us,6 but also bound to leukocytes as well as CD3 and CD4 T cells. Lesion apoptotic cell numbers, necrotic core development, and lesion anti‐inflammatory and inflammatory cytokine levels were highly dependent on TLR4‐MyD88 expression by B1a cells. Together, our findings indicate that the atheroprotective effects of B1a cells are critically dependent on TLR4‐MyD88 cell signaling during development of atherosclerosis.
We have shown that B1a‐derived IgM antibodies act as a scavenger role to clear apoptotic cells and modified LDL by depositing IgM in lesions and decreasing necrotic core, oxLDL, and apoptotic cells in lesions.6, 40 In accord with our previous findings, the plaques of TLR4−/− and MyD88−/− B1a cell recipients showed a large amount of MDA‐LDL and increased necrotic core area whereas lesion IgM deposits were reduced, suggesting the importance of TLR4‐MyD88 expression by B1a cells for IgM‐mediated atheroprotection. Although lesion IgM promotes clearance of small particles and apoptotic cells by macrophages in the absence of complement activation,41 IgM predominantly activates a complement system for subsequent clearance.26 Our finding of reduced complement binding to early apoptotic cells in IgM‐reduced plasma suggests that IgM is critically important in IgM‐mediated immune complex clearance and complement activation reduced in TLR4−/− and MyD88−/− B1a cell recipients, in agreement with literature.26
Currently, there are no genetic models available to specifically assess the role of TLRs in regulating B1a cell function in atherosclerosis; mixed bone marrow chimeric mouse models involving bone marrow transplantation of B‐cell‐deficient (μMT) mice and TLR knockout mice would result in TLR depletion in all B cells. Thus, we assessed the role of different TLRs in regulating B1a cell function in atherosclerosis by adoptively transferring B1a cells deficient in TLRs into splenectomized mice; similar to congenital asplenia, splenectomy causes depletion of peritoneal B1a cells while leaving both B1b and B2 cells intact.6, 9 Adoptive transfer of B1a cells resulted in ≈80% reconstitution of peritoneal B1a cells and restored plasma IgM to normal levels. Restoration of normal plasma IgM levels most probably explains why atherosclerotic lesions were only reduced to levels observed in sham‐operated mice. It is highly likely that transfer of greater numbers of B1a cells would further reduce atherosclerotic lesions not only in splenectomized mice, but also in mice with intact B‐cell compartments. Indeed, we have recently reported that administration of phosphatidylserine liposomes to atherosclerotic mice, mimicking apoptotic cells, doubles peritoneal B1a cells and reduces lesion size by ≈50%.40
Innate TLR signaling is important for IgM secretion as CD5 expressed by B1a cells prevents signaling through B‐cell receptors. B1a cells express higher levels of TLR2 and TLR9 compared with TLR4.25 TLR2 mediates in vivo B1a cell expansion in spleen and bone marrow,42 and its activation in vitro elevates secretion of IgM to levels observed following activation of TLR4 or TLR9.25 Despite these similar effects on IgM production in vitro, in vivo IgM production by B1a cells during development of atherosclerosis was only dependent on B1a cells expressing TLR4 as were effects on atherosclerotic lesions. Adoptive transfer of TLR2‐deficient (TLR4‐sufficient) B1a cells into splenectomized ApoE−/− mice augmented IgM production and attenuated atherosclerosis similar to WT B1a cells. Also, in vitro stimulation of TLR9 on B1a cells elevates IgM secretion to levels observed after TLR4 activation,25 but during development of atherosclerosis, TLR9 expressed by B1a cells is not required for B1a cells to secrete IgM and its deletion from B1a cells does not influence atherosclerosis development in splenectomized mice. The further finding that TLR9 agonist failed to augment IgM production in splenectomized mice that received TLR9‐sufficient (TLR4‐deficient) B1a cells supported that B1a cells do not utilize TLR9 signaling in their IgM production. We also found that TLR4 agonist challenge increased plasma IgM levels and it might be possibly attributed to (1) residual amount of peritoneal B1a cells in splenectomized mice and (2) CD5– CD19+ B1b cells that are capable of producing atheroprotective IgM.43 Given that B1 cells are exclusively responsible for circulating IgM levels44 and LPS, a TLR agonist, is a potent agent in B1a cell activation,45 collectively B1a cells utilize TLR4 signaling to promote their IgM production.
Also, whereas DNA complexes on the surface of apoptotic cells have been shown to induce tolerogenic IL‐10‐secreting B1a cells through activation of TLR9,46 such a mechanism seems, at best, very minor in B1a‐cell–mediated suppression of atherosclerosis. Studies utilizing global knockout TLR9 ApoE−/− mice indicate that this TLR can protect against atherosclerosis, but the mechanism is yet to be defined.21 Rather, our study indicates that TLR4 expressed on B1a cells is the major TLR responsible for IgM‐mediated atheroprotection by B1a cells. Whereas peritoneal B1a cells require C‐X‐C motif chemokine ligand 13 for homing to the peritoneal and other pleural cavities,47 they require TLR4 to emigrate from the peritoneal cavity and generate large amounts of IgM antibodies outside their normal resident site.34 Whereas sites at which they produce antibodies and the extent to which they differentiate into plasma cells requires further study, our findings indicate that TLR4 is critical for B1a cells to produce IgM, MDA‐oxLDL IgM, anti‐leukocyte, and anti‐T‐cell IgMs in atherosclerotic mice. Although TLR4‐activated peritoneal B1 B cells can also increase their expression of IL‐10,48 it is unclear whether this involves B1a or B1b cells or regulatory B cells, which exhibit some of the characteristics of B1a cells. Although our findings indicate a critical role for IgM in B1a‐cell–mediated atheroprotection, a contribution of IL‐10 by these cells cannot be excluded; however, recently B‐cell‐derived IL‐10 was reported to be dispensable for atherosclerosis development in mice.49
Our study extends previous reports of mechanisms by which B1a cells are atheroprotective.6, 35 In addition to promoting removal of apoptotic cells and oxLDL in atherosclerotic lesions, the present study suggests that natural IgMs also contribute to atheroprotection by acting as anti‐leukocyte antibodies to CD4 and CD8 T cells that impair their recruitment into lesions. Our findings of reduced lesion CD4 and CD8 T cells are consistent with the report that natural IgM immunoprecipitated CD3, CD4, and chemokine receptors C‐C chemokine receptor type 5 and C‐X‐C motif chemokine receptor 4 from cell lysates, inhibited T‐cell proliferation, and inhibited leukocyte chemotaxis.27 Natural IgM anti‐leukocyte antibody also inhibited CD4+ T‐cell differentiation into T‐helper (Th) 1 and Th17 cells,28 effects consistent with our observed reduction in lesion CD4+ T cells. Natural IgM also inhibited cytokine production by activated splenocytes from prediabetic mice.50 Also, TLR4‐activated peritoneal B1 cells possess regulatory B‐cell properties that inhibit the capacity of CD4 T cells to produce inflammatory cytokines.48
Our present findings also suggest that another mechanism by which natural IgM contributes to atheroprotection is by increased expression of anti‐inflammatory TGF‐β1 accompanied by suppression of anti‐inflammatory cytokines TNF‐α, IL‐1β, and IL‐18. These findings are consistent with the report that human macrophages that have ingested apoptotic cells inhibited production of inflammatory cytokines by autocrine/paracrine mechanisms involving TGF‐β1 that was abrogated by neutralizing antibody to TGF‐β1 and restored by exogenous TGF‐β1.51 Similar observations were also reported with mouse macrophages, in that ingestion of apoptotic cells augmented TGF‐β1 production and inhibited the synthesis and secretion of inflammatory cytokines that were also abrogated by neutralizing antibody to TGF‐β1 and restored by exogenous TGF‐β1.52
B1a cells have been found in mouse aorta and represent around 5% of aortic B cells in ApoE‐deficient mice.7 It is not known whether they are localized to the adventitia in association with atherosclerotic lesions, which might be expected, particularly in tertiary lymphoid organs that are found with advanced atherosclerotic lesions.53 Our study indicates that adoptively transferred B1a cells home to the peritoneal cavity, and also lymph nodes, and there are indications that they also home to gut.54 Recently, Gjurich et al7 have shown that B1a‐cell homing to the aorta is L‐selectin dependent. Therefore, it is possible that IgM in lesions may derive through the circulation from extravascular sites as well as from B1a cells in atherosclerotic lesions.
Although our study has identified an important role for TLR4 in mediating B1a‐cell atheroprotection, the natural TLR4 ligands that influence atherosclerosis remain to be identified. Heat shock protein 60 is considered an endogenous TLR4 ligand.55 It has been implicated in both human56 and mouse atherosclerosis57 and has been shown to activate B cells through the TLR4‐MyD88 pathway.58 oxLDL can also be considered a natural TLR4 ligand in atherosclerosis.59 It activates TLR460 and has been associated with enhanced antibody production by lymphocytes in vitro.61 The extent to which these or other yet‐to‐be‐identified natural TLR4 ligands influence B1a‐cell activity in atherosclerosis remains to be elucidated.
Taken together, our findings significantly extend earlier reports of the atheroprotective actions of B1a cells through IgM,62 by demonstrating, for the first time, that IgM production and atheroprotection by these cells is critically dependent on TLR4‐MyD88 expression by B1a cells. Activation of this pathway greatly increases secretion of polyclonal natural IgMs that not only reduce lesion apoptotic and necrotic cells and oxLDL, but also reduce lesion proatherogenic CD4+ and CD8+ T cells as well as promoting increases in lesion anti‐inflammatory cytokines TGF‐β1 and IL‐10 and reducing lesion proinflammatory cytokines TNF‐α, IL‐1β, and IL‐18 to attenuate atherosclerosis. Understanding the factors responsible for activating TLR4/MyD88–expressing atheroprotective B1a cells may lead to novel strategies to expand B1a cells to ameliorate atherosclerosis.
Sources of Funding
This work was supported by a postgraduate scholarship from the National Heart Foundation of Australia to Hosseini; by a project grant from the National Health and Medical Research Council of Australia to Kyaw; and, in part, by the Victorian Government's Operational Infrastructure Support Program.
Disclosures
None.
(J Am Heart Assoc. 2016;5:e002947 doi: 10.1161/JAHA.115.002947)
References
- 1. Mathers CD, Boerma T, Ma Fat D. Global and regional causes of death. Br Med Bull. 2009;92:7–32. [DOI] [PubMed] [Google Scholar]
- 2. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. [DOI] [PubMed] [Google Scholar]
- 3. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, Kehry M, Dunn R, Agrotis A, Tipping P, Bobik A, Toh BH. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. 2010;185:4410–4419. [DOI] [PubMed] [Google Scholar]
- 4. Ait‐Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, Taleb S, Van Vre E, Esposito B, Vilar J, Sirvent J, Van Snick J, Tedgui A, Tedder TF, Mallat Z. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207:1579–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sage AP, Tsiantoulas D, Baker L, Harrison J, Masters L, Murphy D, Loinard C, Binder CJ, Mallat Z. BAFF receptor deficiency reduces the development of atherosclerosis in mice—brief report. Arterioscler Thromb Vasc Biol. 2012;32:1573–1576. [DOI] [PubMed] [Google Scholar]
- 6. Kyaw T, Tay C, Krishnamurthi S, Kanellakis P, Agrotis A, Tipping P, Bobik A, Toh BH. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res. 2011;109:830–840. [DOI] [PubMed] [Google Scholar]
- 7. Gjurich BN, Taghavie‐Moghadam PL, Ley K, Galkina EV. L‐selectin deficiency decreases aortic B1a and Breg subsets and promotes atherosclerosis. Thromb Haemost. 2014;112:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hayakawa K, Hardy RR, Herzenberg LA, Herzenberg LA. Progenitors for Ly‐1 B cells are distinct from progenitors for other B cells. J Exp Med. 1985;161:1554–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wardemann H, Boehm T, Dear N, Carsetti R. B‐1a B cells that link the innate and adaptive immune responses are lacking in the absence of the spleen. J Exp Med. 2002;195:771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baumgarth N. The double life of a B‐1 cell: self‐reactivity selects for protective effector functions. Nat Rev Immunol. 2011;11:34–46. [DOI] [PubMed] [Google Scholar]
- 11. Baumgarth N, Tung JW, Herzenberg LA. Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin Immunopathol. 2005;26:347–362. [DOI] [PubMed] [Google Scholar]
- 12. Kaveri SV, Silverman GJ, Bayry J. Natural IgM in immune equilibrium and harnessing their therapeutic potential. J Immunol. 2012;188:939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, Shaw PX, Palinski W, Witztum JL, Silverman GJ. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. 2003;9:736–743. [DOI] [PubMed] [Google Scholar]
- 14. Binder CJ, Shaw PX, Chang MK, Boullier A, Hartvigsen K, Horkko S, Miller YI, Woelkers DA, Corr M, Witztum JL. The role of natural antibodies in atherogenesis. J Lipid Res. 2005;46:1353–1363. [DOI] [PubMed] [Google Scholar]
- 15. Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. 2010;10:778–786. [DOI] [PubMed] [Google Scholar]
- 16. Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A. The evolution of vertebrate Toll‐like receptors. Proc Natl Acad Sci USA. 2005;102:9577–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barton GM, Kagan JC. A cell biological view of Toll‐like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bekeredjian‐Ding I, Jego G. Toll‐like receptors—sentries in the B‐cell response. Immunology. 2009;128:311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arpaia N, Barton GM. Toll‐like receptors: key players in antiviral immunity. Curr Opin Virol. 2011;1:447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rawlings DJ, Schwartz MA, Jackson SW, Meyer‐Bahlburg A. Integration of B cell responses through Toll‐like receptors and antigen receptors. Nat Rev Immunol. 2012;12:282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koulis C, Chen YC, Hausding C, Ahrens I, Kyaw TS, Tay C, Allen T, Jandeleit‐Dahm K, Sweet MJ, Akira S, Bobik A, Peter K, Agrotis A. Protective role for Toll‐like receptor‐9 in the development of atherosclerosis in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2014;34:516–525. [DOI] [PubMed] [Google Scholar]
- 22. Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll‐like receptor 2. J Clin Invest. 2005;115:3149–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayashi C, Papadopoulos G, Gudino CV, Weinberg EO, Barth KR, Madrigal AG, Chen Y, Ning H, LaValley M, Gibson FC III, Hamilton JA, Genco CA. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J Immunol. 2012;189:3681–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll‐like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. 2004;101:10679–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Genestier L, Taillardet M, Mondiere P, Gheit H, Bella C, Defrance T. TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus‐independent responses. J Immunol. 2007;178:7779–7786. [DOI] [PubMed] [Google Scholar]
- 26. Zwart B, Ciurana C, Rensink I, Manoe R, Hack CE, Aarden LA. Complement activation by apoptotic cells occurs predominantly via IgM and is limited to late apoptotic (secondary necrotic) cells. Autoimmunity. 2004;37:95–102. [DOI] [PubMed] [Google Scholar]
- 27. Lobo PI, Schlegel KH, Spencer CE, Okusa MD, Chisholm C, McHedlishvili N, Park A, Christ C, Burtner C. Naturally occurring IgM anti‐leukocyte autoantibodies (IgM‐ALA) inhibit T cell activation and chemotaxis. J Immunol. 2008;180:1780–1791. [DOI] [PubMed] [Google Scholar]
- 28. Lobo PI, Bajwa A, Schlegel KH, Vengal J, Lee SJ, Huang L, Ye H, Deshmukh U, Wang T, Pei H, Okusa MD. Natural IgM anti‐leukocyte autoantibodies attenuate excess inflammation mediated by innate and adaptive immune mechanisms involving Th‐17. J Immunol. 2012;188:1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kyaw T, Cui P, Tay C, Kanellakis P, Hosseini H, Liu E, Rolink AG, Tipping P, Bobik A, Toh BH. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(‐/‐) mice. PLoS One. 2013;8:e60430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kyaw T, Winship A, Tay C, Kanellakis P, Hosseini H, Cao A, Li P, Tipping P, Bobik A, Toh BH. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE‐deficient mice. Circulation. 2013;127:1028–1039. [DOI] [PubMed] [Google Scholar]
- 31. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 32. Akira S, Takeda K. Toll‐like receptor signalling. Nat Rev Immunol. 2004;4:499–511. [DOI] [PubMed] [Google Scholar]
- 33. Yoshihara R, Aoyama E, Kadota Y, Kawai S, Goto T, Zhong M, Gohda E. Differentiation of murine B cells induced by chondroitin sulfate B. Cell Immunol. 2007;250:14–23. [DOI] [PubMed] [Google Scholar]
- 34. Ha SA, Tsuji M, Suzuki K, Meek B, Yasuda N, Kaisho T, Fagarasan S. Regulation of B1 cell migration by signals through Toll‐like receptors. J Exp Med. 2006;203:2541–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, Choi J, Perkmann T, Backhed F, Miller YI, Horkko S, Corr M, Witztum JL, Binder CJ. Oxidation‐specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009;119:1335–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. I‐PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med. 2002;196:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lobo PI, Schlegal KH, Vengal J, Okusa MD, Pei H. Naturally occurring IgM anti‐leukocyte autoantibodies inhibit T‐cell activation and chemotaxis. J Clin Immunol. 2010;30(suppl 1):S31–S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T. Targeted disruption of the mouse transforming growth factor‐beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Asadullah K, Sterry W, Volk HD. Interleukin‐10 therapy–review of a new approach. Pharmacol Rev. 2003;55:241–269. [DOI] [PubMed] [Google Scholar]
- 40. Hosseini H, Li Y, Kanellakis P, Tay C, Cao A, Tipping P, Bobik A, Toh BH, Kyaw T. Phosphatidylserine liposomes mimic apoptotic cells to attenuate atherosclerosis by expanding polyreactive IgM producing B1a lymphocytes. Cardiovasc Res. 2015;106:443–452. [DOI] [PubMed] [Google Scholar]
- 41. Litvack ML, Post M, Palaniyar N. IgM promotes the clearance of small particles and apoptotic microparticles by macrophages. PLoS One. 2011;6:e17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Komegae EN, Grund LZ, Lopes‐Ferreira M, Lima C. TLR2, TLR4 and the MyD88 signaling are crucial for the in vivo generation and the longevity of long‐lived antibody‐secreting cells. PLoS One. 2013;8:e71185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rosenfeld SM, Perry HM, Gonen A, Prohaska TA, Srikakulapu P, Grewal S, Das D, McSkimming C, Taylor AM, Tsimikas S, Bender TP, Witztum JL, McNamara CA. B‐1b cells secrete atheroprotective IgM and attenuate atherosclerosis. Circ Res. 2015;117:e28–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thurnheer MC, Zuercher AW, Cebra JJ, Bos NA. B1 cells contribute to serum IgM, but not to intestinal IgA, production in gnotobiotic Ig allotype chimeric mice. J Immunol. 2003;170:4564–4571. [DOI] [PubMed] [Google Scholar]
- 45. Koide N, Sugiyama T, Kato Y, Chakravortty D, Mu MM, Yoshida T, Hamano T, Yokochi T. Mouse B1 cell line responds to lipopolysaccharide via membrane‐bound CD14. J Endotoxin Res. 2001;7:39–43. [PubMed] [Google Scholar]
- 46. Miles K, Heaney J, Sibinska Z, Salter D, Savill J, Gray D, Gray M. A tolerogenic role for Toll‐like receptor 9 is revealed by B‐cell interaction with DNA complexes expressed on apoptotic cells. Proc Natl Acad Sci USA. 2012;109:887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ansel KM, Harris RB, Cyster JG. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity. 2002;16:67–76. [DOI] [PubMed] [Google Scholar]
- 48. Margry B, Kersemakers SCW, Hoek A, Arkesteijn GJA, Wieland WH, van Eden W, Broere F. Activated peritoneal cavity B‐1a cells possess regulatory B cell properties. PLoS One. 2014;9:e88869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sage AP, Nus M, Baker LL, Finigan AJ, Masters LM, Mallat Z. Regulatory B cell‐specific interleukin‐10 is dispensable for atherosclerosis development in mice. Arterioscler Thromb Vasc Biol. 2015;35:1770–1773. [DOI] [PubMed] [Google Scholar]
- 50. Chhabra P, Schlegel K, Okusa MD, Lobo PI, Brayman KL. Naturally occurring immunoglobulin M (nIgM) autoantibodies prevent autoimmune diabetes and mitigate inflammation after transplantation. Ann Surg. 2012;256:634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF‐beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McDonald PP, Fadok VA, Bratton D, Henson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF‐beta in macrophages that have ingested apoptotic cells. J Immunol. 1999;163:6164–6172. [PubMed] [Google Scholar]
- 53. Mohanta SK, Yin C, Peng L, Srikakulapu P, Bontha V, Hu D, Weih F, Weber C, Gerdes N, Habenicht AJ. Artery tertiary lymphoid organs contribute to innate and adaptive immune responses in advanced mouse atherosclerosis. Circ Res. 2014;114:1772–1787. [DOI] [PubMed] [Google Scholar]
- 54. Kunisawa J, Kurashima Y, Gohda M, Higuchi M, Ishikawa I, Miura F, Ogahara I, Kiyono H. Sphingosine 1‐phosphate regulates peritoneal B‐cell trafficking for subsequent intestinal IgA production. Blood. 2007;109:3749–3756. [DOI] [PubMed] [Google Scholar]
- 55. Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the Toll‐like receptor‐4 complex. J Immunol. 2000;164:558–561. [DOI] [PubMed] [Google Scholar]
- 56. Kol A, Sukhova GK, Lichtman AH, Libby P. Chlamydial heat shock protein 60 localizes in human atheroma and regulates macrophage tumor necrosis factor‐alpha and matrix metalloproteinase expression. Circulation. 1998;98:300–307. [DOI] [PubMed] [Google Scholar]
- 57. Kanwar RK, Kanwar JR, Wang D, Ormrod DJ, Krissansen GW. Temporal expression of heat shock proteins 60 and 70 at lesion‐prone sites during atherogenesis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1991–1997. [DOI] [PubMed] [Google Scholar]
- 58. Cohen‐Sfady M, Nussbaum G, Pevsner‐Fischer M, Mor F, Carmi P, Zanin‐Zhorov A, Lider O, Cohen IR. Heat shock protein 60 activates B cells via the TLR4‐MyD88 pathway. J Immunol. 2005;175:3594–3602. [DOI] [PubMed] [Google Scholar]
- 59. Li D, Mehta JL. Oxidized LDL, a critical factor in atherogenesis. Cardiovasc Res. 2005;68:353–354. [DOI] [PubMed] [Google Scholar]
- 60. Yang K, He YS, Wang XQ, Lu L, Chen QJ, Liu J, Sun Z, Shen WF. MiR‐146a inhibits oxidized low‐density lipoprotein‐induced lipid accumulation and inflammatory response via targeting Toll‐like receptor 4. FEBS Lett. 2011;585:854–860. [DOI] [PubMed] [Google Scholar]
- 61. Huang YH, Ronnelid J, Frostegard J. Oxidized LDL induces enhanced antibody formation and MHC class II‐dependent IFN‐gamma production in lymphocytes from healthy individuals. Arterioscler Thromb Vasc Biol. 1995;15:1577–1583. [DOI] [PubMed] [Google Scholar]
- 62. Kyaw T, Tipping P, Toh BH, Bobik A. Current understanding of the role of B cell subsets and intimal and adventitial B cells in atherosclerosis. Curr Opin Lipidol. 2011;22:373–379. [DOI] [PubMed] [Google Scholar]