

Abstract
Background
Aortic stenosis (AS) is a chronic inflammatory disease, and calcification plays an important role in the progression of the disease. Galectin‐3 (Gal‐3) is a proinflammatory molecule involved in vascular osteogenesis in atherosclerosis. Therefore, we hypothesized that Gal‐3 could mediate valve calcification in AS.
Methods and Results
Blood samples and aortic valves (AVs) from 77 patients undergoing AV replacement were analyzed. As controls, noncalcified human AVs were obtained at autopsy (n=11). Gal‐3 was spontaneously expressed in valvular interstitial cells (VICs) from AVs and increased in AS as compared to control AVs. Positive correlations were found between circulating and valvular Gal‐3 levels. Valvular Gal‐3 colocalized with the VICs markers, alpha‐smooth muscle actin and vimentin, and with the osteogenic markers, osteopontin, bone morphogenetic protein 2, runt‐related transcription factor 2, and SRY (sex‐determining region Y)‐box 9. Gal‐3 also colocalized with the inflammatory markers cd68, cd80 and tumor necrosis factor alpha. In vitro, in VICs isolated from AVs, Gal‐3 induced expression of inflammatory, fibrotic, and osteogenic markers through the extracellular signal‐regulated kinase 1 and 2 pathway. Gal‐3 expression was blocked in VICs undergoing osteoblastic differentiation using its pharmacological inhibitor, modified citrus pectin, or the clustered regularly interspaced short palindromic repeats/Cas9 knockout system. Gal‐3 blockade and knockdown decreased the expression of inflammatory, fibrotic, and osteogenic markers in differentiated VICs.
Conclusions
Gal‐3, which is overexpressed in AVs from AS patients, appears to play a central role in calcification in AS. Gal‐3 could be a new therapeutic approach to delay the progression of AV calcification in AS.
Keywords: aortic stenosis, calcification, galectin‐3, inflammation, valve, valvular interstitial cells
Subject Categories: Valvular Heart Disease, Remodeling, Basic Science Research, Fibrosis, Inflammation
Introduction
Aortic stenosis (AS) is the most common heart valve disease (43.1%) and represents a major health care burden.1 With the increase in the aging population, there is a surge in prevalence of calcific aortic valve (AV) disease. A prediction on the number of elderly (≥70) years for the coming decades demonstrated that patients with severe AS will have increased 2.4‐fold by the year 2040 and will be more than triple by the year 2060.2 Patients with AS have an 80% risk of valve replacement, progression to heart failure (HF), or death in the next 5 years after diagnosis.3
The pathophysiology underlying calcific aortic valve disease (CAVD) remains incompletely defined, and there are currently no effective medical treatments capable of altering its course.4 Chronic inflammation, fibrosis, and calcification play an important role in progression of the disease.5 Therefore, it has been shown that CAVD shares features with vascular calcification and atherosclerosis, such as chronic inflammation, increased extracellular matrix (ECM) remodeling, proliferation and differentiation of valvular interstitial cells (VICs), and development of calcific lesions.6, 7 Of note, although retrospective studies had suggested that statins could delay the hemodynamic progression rate of AS,8, 9 randomized controlled studies reported, in contrast, that a lipid‐lowering strategy resulted neither in lower AV‐related events nor in a slower progression of stenosis.10, 11
Galectin‐3 (Gal‐3) is a 29‐ to 35‐kDa protein and member of a β‐galactoside binding lectin family, which interacts with cell‐surface receptors and ECM proteins.12 Gal‐3 levels are increased in patients with HF13, 14 and in myocardial biopsies from AS patients with depressed ejection fraction.15 In addition, it has been demonstrated that patients with high Gal‐3 plasma levels are associated with adverse outcome after transcatheter aortic valve implantation, suggesting that Gal‐3 could play an important role in patients with AS.16 Gal‐3 plays an important role in cardiac and vascular remodeling through its ability to stimulate ECM deposition as well as by the amplification of key proinflammatory molecules.17, 18 Moreover, Gal‐3 modulated vascular smooth muscle cell (VSMC) osteogenic differentiation, playing an important role in the development of atherosclerosis.19
Therefore, our hypothesis is that Gal‐3 could promote AV calcification, modulating VICs osteogenic differentiation. Furthermore, Gal‐3 could be a new biotarget in CAVD, and its blockade could prevent AV calcification. We tested this hypothesis using human AVs samples and human VICs.
Methods
Patient Population
This prospective, observational study included a total of 77 consecutive patients with severe AS (AV area ≤1 cm2 [and/or] transaortic mean pressure gradient >40 mm Hg), referred to our center for AV replacement from June 2013 to February 2015. Exclusion criteria were moderate or severe concomitant valvular disease, malignant tumor, and chronic inflammatory diseases. All patients were evaluated by echocardiography. Venous blood was drawn for measurement of brain natriuretic peptide (BNP) and other routine laboratory parameters on admission for surgery.
Human AVs were obtained after surgery (AS, n=77). As controls, noncalcified human AVs were obtained at autopsy (control, n=11) and were rapidly cut in 2 pieces. One piece was fixed in formol and embedded in paraffin and the other was frozen in liquid nitrogen.
Informed consent was obtained from each patient and control, and the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a previous approval by the institution's human research committee.
Aortic Human VIC Experiments
For the in vitro approach, we obtained aortic VICs from 7 patients operated on for severe AS. For the experiments, we used at least VICs from 5 different patients. Aortic VICs were isolated using sequential collagenase digestion to eliminate endothelial cells, and then interstitial cells were collected by centrifugation. Inmunocytochemistry was routinely performed on Lab‐Tek II Chamber Slides (Nalge Nunc International, Penfield, NY). Cells were positive for expression of alpha‐smooth muscle actin (α‐SMA) and vimentin and negative for CD31. VICs were cultured in DMEM F‐12 medium supplemented with 10% FBS. All assays were done at 37°C, 95% sterile air, and 5% CO2 in a saturation humidified incubator.
Cells were treated with Gal‐3 (10−8 mol/L; R&D Systems, Minneapolis, MN) for 1 to 6 days to assess the effect of Gal‐3 on inflammatory, fibrotic, and calcification markers. For the intracellular pathways study, cells were treated with Gal‐3 for 15, 30, and 60 minutes. The following chemical inhibitors were added at 10−5 mol/L 1 hour preceding Gal‐3 stimulation: Wortmannin (Sigma‐Aldrich, St. Louis, MO), PD98059 (Sigma Aldrich), SB203580 (Sigma Aldrich), and BAY 11‐7082 (Sigma Aldrich).
To induce calcification, VICs were cultured in conditioning medium DMEM F‐12 supplemented with 10 mmol/L of β‐glycerophospate, 50 μg/mL of ascorbic acid, 10 nmol/L of dexamethasone, 10% FBS, and treated in the presence or absence of the pharmacological Gal‐3 activity inhibitor, modified citrus pectin (MCP; 10−6 mol/L; ecoNugenics, Santa Rosa, CA) every 3 days. At 7, 14, and 21 days of treatment, cell extracts and supernatants were collected to evaluate inflammatory, fibrotic, and calcification markers.
Clustered Regularly Interspaced Short Palindrome Repeats/Cas9 Genome Editing‐Mediated Deletion of Gal‐3
The knockdown of Gal‐3 in VICs was performed by clustered regularly interspaced short palindrome repeats (CRISPR)/Cas9‐guided genome editing. Cells were seeded into 6‐well plates at 70% confluence and transfected with a pool of three plasmids, each encoding the Cas9 nuclease and a target‐specific 20‐nucleotide guide RNA designed for maximum knockout efficiency according to the manufacturer's instructions (Santa Cruz Biotechnology, Santa Cruz, CA). Scramble gRNA CRISPR/Cas9 plasmid was used as a control.
Once Gal‐3 knockdown was generated, VICs were cultured in osteogenic medium as described above. At 7, 14, and 21 days, cell extracts and supernatants were collected to evaluate inflammatory, fibrotic, and calcification markers.
Histological Analysis
Tissue staining was performed on transversal sections of human and rat AV leaflets. All grossly calcified valves were decalcified in 10% formic acid solution for 24 hours. Samples were dehydrated, embedded in paraffin, and cut in 5‐μm‐thick sections.
Slides were treated with H2O2 for 10 minutes to block peroxidase activity. All sections were blocked with 5% normal goat serum in PBS for 1 hour and incubated overnight with Gal‐3, α‐SMA, vimentin, osteopontin, bone morphogenetic protein (BMP) 2, BMP‐4, runt‐related transcription factor 2 (Runx2), SRY (sex‐determining region Y)‐box 9 (Sox‐9), cd68, cd80, and tumor necrosis factor alpha (TNF‐α), washed 3 times, and then incubated for 30 minutes with the HRP‐labeled polymer conjugated to secondary antibodies (Dako Cytomation, Carpentaria, CA). The signal was revealed by using a DAB Substrate Kit (BD Pharmingen, San. Diego, CA). Reference samples of all stains before and after decalcification were compared, thereby ensuring that the process of decalcification did not interfere with the staining and hence the result/interpretation of the staining.
ELISA
Gal‐3 was measured in serum samples by ELISA according to the manufacturer's instructions (BG Medicine, Inc., Waltham, MA).
Interleukin (IL) 6 and IL‐1β concentrations were measured in valve extracts and cells supernatants by ELISA according to the manufacturer's instructions (R&D Systems).
Real‐Time Reverse‐Transcription Polymerase Chain Reaction
Total RNA from AVs and VICs was extracted with TRIzol Reagent (Euromedex, Strasbourg, France) and purified using the RNeasy kit, according to the manufacturer's instructions (Qiagen, Hilden, Germany). First‐strand cDNA was synthesized according to the manufacturer's instructions (Roche, Indianapolis, IN). Quantitative polymerase chain reaction (PCR) analysis was then performed with SYBR green PCR technology (ABgene, Portsmouth, NH) (Table S1). Relative quantification was achieved with MyiQ (Bio‐Rad, Hercules, CA) software according to the manufacturer's instructions. Data were normalized by hypoxanthine‐guanine phosphoribosyltransferase (HPRT) and β‐actin levels and expressed as percentage relative to controls. All PCRs were performed at least in triplicate for each experimental condition.
Western Blot
Total proteins were prepared from either human valve homogenates or VIC extracts. Proteins were separated by SDS‐PAGED on 10% polyacrylamide gels and transferred to Hybond‐c Extra nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ). Membranes were probed with primary antibodies for Gal‐3 (dilution, 1:1000; Thermo Fisher Scientific, Waltham, MA), collagen I (dilution, 1:500; Sigma‐Aldrich), collagen III (dilution 1:500; Santa Cruz Biotechnology), α‐SMA (dilution, 1:2000; Sigma‐Aldrich), fibronectin (dilution, 1:1000; Millipore, Billerica, MA), transforming growth factor β (TGF‐β; dilution, 1:500; Sigma‐Aldrich), BMP‐2 (dilution, 1:500; Thermo Fisher Scientific), BMP‐4 (dilution, 1:500; Thermo Fisher Scientific), osteopontin (dilution, 1:500; Abcam, Cambridge, MA), Runx2 (dilution, 1:500; Sigma‐Aldrich), Sox‐9 (dilution, 1:500; Sigma‐Aldrich), TNF‐α (dilution, 1:500; R&D Systems), cd68 (dilution, 1:500; R&D Systems), cd45 (dilution, 1:500; R&D Systems), cd80 (dilution, 1:500; R&D Systems), extracellular signal‐regulated kinase 1 and 2 (ERK1/2) and ERK1/2‐P (Thr202/Tyr204) at 1:1000 (Cell Signaling Technology, Danvers, MA), p38MAPK (mitogen‐activated protein kinase) and p38MAPK‐P (Thr180/Tyr182) at 1:1000 (Cell Signaling Technology), protein kinase B (Akt) and Akt‐P (Ser473) at 1:1500 (Cell Signaling Technology), nuclear factor kappa B (NFκB) and NFκB‐P (Ser536) at 1:1000 (Cell Signaling Technology), and β‐actin (dilution, 1:5000; Sigma‐Aldrich). Western blots were performed with stain‐free gels for loading control. Signals were detected using the ECL system (Amersham Pharmacia Biotech, Piscataway, NJ). Results are expressed as an n‐fold increase over the values of the control group in densitometric arbitrary units.
Gelatin Zymography
Aliquots of protein tissue samples or culture media containing 25‐μg human valve homogenates or 30 μL of supernatant, respectively, were resolved on a 10% SDS polyacrylamide gel containing 0.3% gelatin. The gel was rinsed 3 times for 15 minutes with a solution of 2.5% Triton X‐100 to remove SDS and renature the proteins, followed by incubation for 48 hours at 37°C in 1000 mmol/L of Tris‐HCl (pH 7.5) with 1000 mmol/L of CaCl2 and 5000 mmol/L of NaCl to promote degradation of gelatin. Gels were fixed in 40% methanol and 10% acetic acid and then stained for 30 minutes in 0.25% Coomassie blue R‐250 to identify proteolytic activity of matrix metalloproteinase (MMP) 1, MMP‐2, and MMP‐9.
Statistical Analysis
Continuous variables were expressed as mean±SD or median (25th–75th percentile) and compared using an unpaired Student t test. Pearson correlation coefficients were calculated to determine correlations. Categorical variables were expressed as percentages and compared using the chi‐square test. In the human study, we had more than 80% power with less than 5% error risk to find the significant differences observed (>0.95 SD).
In vitro data are expressed as mean±SEM. Normality of distributions was verified by means of the Kolmogorov–Smirnov test. Data were analyzed using a 1‐way ANOVA, followed by a Newman–Keuls test to assess specific differences among groups or conditions.
All analysis was done with SPSS software (version 20.0; IBM SPSS Statistics, Armonk, NY), and 2‐tailed P‐value of <0.05 was considered statistically significant.
Results
Baseline Characteristics
Concomitant diseases and causes of death from controls are summarized in Table, as well as the baseline characteristics of the entire cohort. In keeping with the typical characteristics of patients presenting with CAVD, mean age was 73±8 years and 58% were male. A significant proportion suffered from concomitant coronary artery disease, hypertension, diabetes mellitus, and hyperlipidemia. Echocardiographic variables were those expected in patients with severe AS. BNP levels indicated compensated disease. Gal‐3 levels, inflammatory markers (C‐reactive protein [CRP], IL‐6, and TNF‐α), leukocyte activation markers (leukocyte number, myeloperoxidase, IL‐8, and L‐selectin) and renal parameters (glomerular filtration rate; GFR) are shown. Interestingly, serum Gal‐3 levels positively correlated with serum TNF‐α (r=0.621; P<0.001) and IL‐6 (r=0.521; P<0.001) levels.
Table 1.
Baseline Characteristics of Patients
| Controls | AS Patients | |
|---|---|---|
| Age, y | 76±10 | 73±8 |
| Male (%) | 6 (55) | 44 (58) |
| Hypertension (%) | 1 (9) | 57 (75) |
| Hyperlipidemia (%) | 2 (18) | 49 (67) |
| Diabetes mellitus (%) | 1 (9) | 23 (30) |
| Coronary artery disease (%) | 1 (9) | 33 (43) |
| Lung disease (%) | 4 (36) | |
| Cause of death (%) | ||
| Bronchopneumonia | 3 (27) | |
| Sepsis | 1 (9) | |
| Cancer | 5 (45) | |
| Trauma | 1 (9) | |
| Old age | 1 (9) | |
| Bicuspid aortic valves (%) | 33 (43) | |
| Treatment (%) | ||
| ACEi | 42 (55) | |
| MR antagonists | 1 (1) | |
| β‐blockers | 21 (28) | |
| Statins | 47 (62) | |
| Diuretics | 50 (66) | |
| NYHA (%) | ||
| I | 8 (10) | |
| II | 41 (54) | |
| III | 24 (31) | |
| IV | 3 (4) | |
| LVMI, g/m2 | 90±25 | |
| LVEDD (LVEDD/BSA) | 49±6 mm (27±4 mm/m2) | |
| LVESD (LVESD/BSA) | 32±7 mm (18±4 mm/m2) | |
| LVEDV (LVEDV/BSA) | 132±49 mL (72±24 mL/m2) | |
| LVESV (LVESV/BSA) | 51±39 mL (28±20 mL/m2) | |
| LV mass (LV mass/BSA) | 167±58 g (90±25 g/m2) | |
| LVEF, % | 64±14 | |
| Transaortic maximum gradient | 78±21 mm Hg | |
| Transaortic mean gradient | 51±15 mm Hg | |
| Aortic valve area (AVA/BSA) | 0.39±0.19 cm2/m2 | |
| BNP, pg/mL | 130 (60–263) | |
| Gal‐3, ng/mL | 17.08±5.2 | |
| Inflammatory markers | ||
| CRP, mg/L | 2.22±2.04 | |
| IL‐6, pg/mL | 9.6±5.6 | |
| TNF‐α, pg/mL | 31.48±8.86 | |
| Leukocyte activation markers | ||
| Leukocyte number (109/L) | 7.2±2.3 | |
| Myeloperoxidase, pg/mL | 265.49±127.4 | |
| IL‐8, pg/mL | 9.51 (5.12–15.25) | |
| L‐selectin, pg/mL | 1130.75±309.7 | |
| Renal parameters | ||
| Creatinine, mg/dL | 0.93±0.25 | |
| GFR, mL/min per m2 | 76.8±27 | |
Values are mean±SD or median (interquartile range). ACEi indicates angiotensin‐converting enzyme inhibitors; AS, aortic stenosis; BNP, brain natriuretic peptide; BSA, body surface area; CRP, C‐reactive protein; Gal‐3, galectin‐3; GFR, glomerular filtration rate; IL, interleukin; LV, left ventricular; LVEDD, left ventricular end‐diastolic diameter; LVEDV, left ventricular end‐diastolic volume; LVEF, left ventricular ejection fraction; LVESD, left ventricular end‐systolic diameter; LVESV, left ventricular end‐systolic volume; LVMI, left ventricular mass index; MR, mineralocorticoid receptor; NYHA, New York Heart Association classification of heart failure; TNF‐α, tumor necrosis factor alpha.
Stenotic Valves Exhibit Higher Inflammation, ECM Remodeling, and Calcification
As shown in Figure 1, an increase in ECM components was observed in AV from AS patients as compared to controls. Collagen type I alpha 1 chain (Col1a1), collagen type III alpha 1 chain (Col3a1), α‐SMA, TGF‐β, and connective tissue growth factor (CTGF) mRNA levels were increased in AVs from AS patients (Figure 1A). AV protein expressions of collagens (types I and III), α‐SMA, fibronectin, and TGF‐β were higher in AS patients than in controls (Figure 1B). Furthermore, AS samples presented an increased in MMP‐1, MMP‐2, and MMP‐9 mRNA levels accompanied by decreased tissue inhibitor of metalloproteinase (TIMP) 1 mRNA levels without modifications in TIMP‐2 levels (Figure 1C). These results were confirmed by zymography showing an increase in MMP‐1, MMP‐2, and MMP‐9 activities in AVs from AS patients (Figure 1D).
Figure 1.
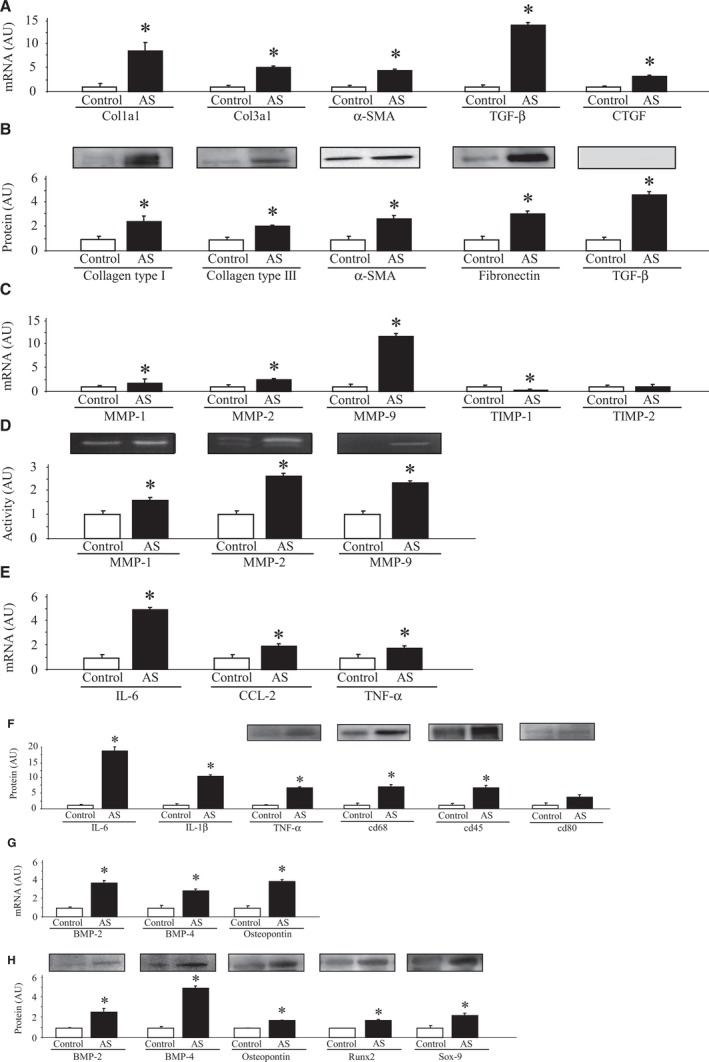
Fibrosis, inflammation, and calcification in AS valves compared to controls. ECM components in AVs at mRNA (A) and protein (B) levels. mRNA levels of MMPs and its inhibitors (TIMPs) in AVs (C). MMPs activities in controls and AS valves (D). Inflammatory markers were measured in stenotic and control AVs at mRNA (E) and protein (F) levels. mRNA levels of calcification markers in AVs (G) and protein expressions of calcification markers in AVs (H). All conditions were performed at least in triplicate. Histogram bars represent the mean±SEM of each group of subjects (control n=11 and patients with severe calcific AS n=77) in arbitrary units (AU) normalized to HPRT and stain‐free gel for cDNA and protein, respectively. *P<0.05 versus control group. AS indicates aortic stenosis; AVs, aortic valves; BMP, bone morphogenetic protein; CCL2, C‐C motif chemokine ligand 2; Col1a1, collagen type I alpha 1 chain; Col3a1, collagen type III alpha 1 chain; CTGF, connective tissue growth factor; ECM, extracellular matrix; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; IL, interleukin; MMPs, matrix metalloproteinases; Runx2, runt‐related transcription factor 2; Sox‐9, SRY (sex‐determining region Y)‐box 9; α‐SMA, alpha‐smooth muscle actin; TGF‐β, transforming growth factor beta; TIMPs, tissue inhibitors of matrix metalloproteinases; TNF‐α, tumor necrosis factor alpha.
Stenotic AVs exhibited higher expression of inflammatory markers at mRNA levels in IL‐6, C‐C motif chemokine ligand 2 (CCL2), and TNF‐α (Figure 1E) and an increase at protein levels of IL‐6, IL‐1β, TNF‐α, cd68, and cd45 as compared to controls without modifications in cd80 levels (Figure 1F).
AVs from AS patients exhibited an increase in calcification markers, such as BMP‐2, BMP‐4, and osteopontin mRNA levels as compared to controls (Figure 1G). At the protein level, BMP‐2, BMP‐4, osteopontin, Runx2, and Sox‐9 were upregulated in AVs from AS patients as compared to controls (Figure 1H).
AVs From AS Patients Presented Higher Gal‐3 Levels Associated With Calcification
Hematoxilin/eosin staining was used to analyze the microstructure of human AVs from controls (left panel) and AS patients (right panel, Figure 2A). As shown in Figure 2B, Gal‐3 was spontaneously expressed in the AVs with a higher expression in AS patients (right panel) as compare to controls (left panel).
Figure 2.
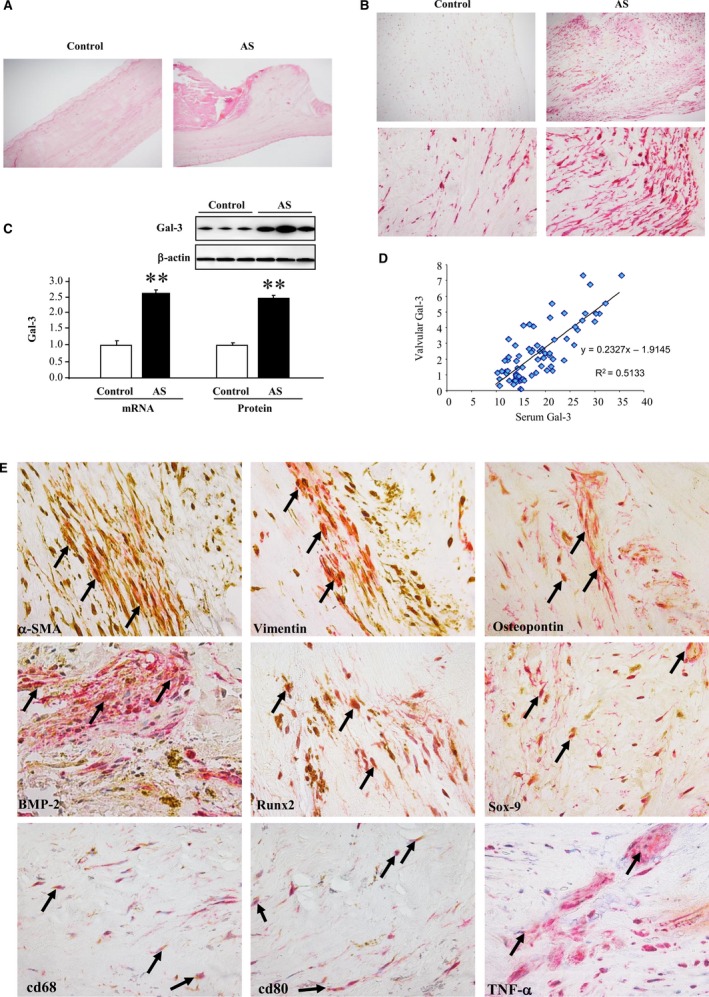
Gal‐3 is associated with osteogenic markers in AS patients. Hematoxilin/eosin staining in AVs from controls and AS patients (A). Representative picture of an AV section from controls and AS patients stained with Gal‐3 at low (×10) and high (×40) magnification (B). Gal‐3 mRNA and protein expressions in AVs from patients (C) and correlation between valvular Gal‐3 protein levels and serum Gal‐3 levels (D). Representative pictures of AV sections stained with Gal‐3 (red) and α‐SMA, vimentin, osteopontin, BMP‐2, Runx2, Sox‐9, cd68, cd80, and TNF‐α (brown). Magnification, ×40 (E). All conditions were performed at least in triplicate. Histogram bars represent the mean±SEM of each group of patients (control n=11 and patients with severe calcific AS n=77) in arbitrary units (AU) normalized to HPRT and β‐actin for cDNA and protein respectively. *P<0.05 versus control group. AS indicates aortic stenosis; AVs, aortic valves; BMP, bone morphogenetic protein; Gal‐3, galectin‐3; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; Runx2, runt‐related transcription factor 2; Sox‐9, SRY (sex‐determining region Y)‐box 9; α‐SMA, alpha‐smooth muscle actin; TGF‐β, transforming growth factor beta; TNF‐α, tumor necrosis factor alpha.
There was a 2.5‐fold significant increase in Gal‐3 mRNA and protein levels in AVs from AS patients as compared to controls (Figure 2C). Strong and positive correlations were found between AV Gal‐3 protein levels and serum Gal‐3 levels (Figure 2D).
Gal‐3 in AVs was colocalized with the VICs markers, α‐SMA and vimentin, with calcification markers osteopontin, BMP‐2, Runx2, and Sox‐9 and with inflammatory markers cd68, cd80, and TNF‐α (Figure 2E).
Gal‐3 Induced Proinflammatory, Profibrotic and Pro‐osteogenic Response in Human VICs
Inflammation, ECM components, and calcification markers were studied in VICs treated with Gal‐3 (10−8 mol/L) for 1, 2, 3, and 6 days. Treatment with Gal‐3 augmented IL‐6 and IL‐1β secretion in VICs (Figure 3A). Moreover, Gal‐3 increased collagen type I, TGF‐β, and fibronectin protein expressions in a time‐dependent manner (Figure 3A). As shown in Figure 3B, Gal‐3 enhanced the expression of the calcification markers, BMP‐2, BMP‐4, and SOX‐9, in a time‐dependent manner, reaching the peak for the factors at 6 days of treatment without modification of Runx2 levels (Figure 3B).
Figure 3.
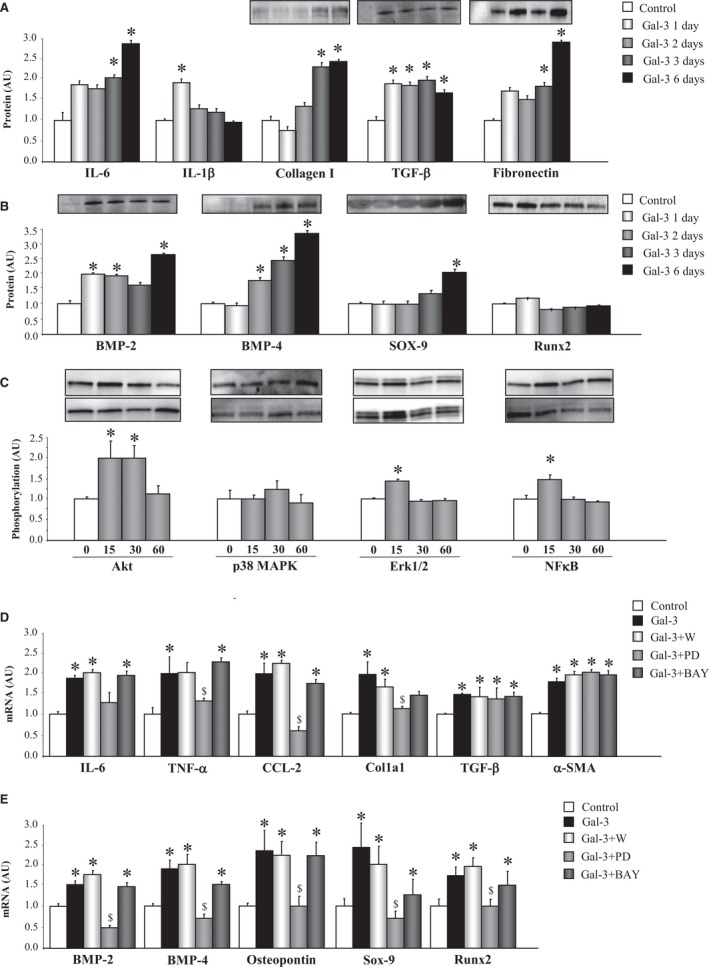
Gal‐3 induces inflammation, ECM components, and calcification markers in VICs. Effects of Gal‐3 on inflammatory markers, ECM components (A), and calcification markers (B) in VICs. Effects of Gal‐3 on phosphorylation of intracellular pathways in VICs (C). Effects of cell signaling chemical inhibitors on the proinflammatory and profibrotic effect of Gal‐3 in VICs (D). Effects of cell signaling chemical inhibitors on the pro‐osteogenic effect of Gal‐3 in VICs (E). All conditions were performed at least in triplicate. Histogram bars represent the mean±SEM of 6 assays in arbitrary units (AU) normalized to stain free for protein and to β‐actin and HPRT for cDNA. *P<0.05 versus control; $ P<0.05 vs Gal‐3. Akt indicates protein kinase B; BMP, bone morphogenetic protein; CCL2, C‐C motif chemokine ligand 2; Col1a1, collagen type I alpha 1 chain; ECM, extracellular matrix; ERK1/2, extracellular signal‐regulated kinase 1 and 2; Gal‐3, galectin‐3; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; IL, interleukin; MAPK, mitogen‐activated protein kinase; NFκB, nuclear factor kappa B; Runx2, runt‐related transcription factor 2; Sox‐9, SRY (sex‐determining region Y)‐box 9; α‐SMA, alpha‐smooth muscle actin; TNF‐α, tumor necrosis factor alpha; VICs, valvular interstitial cells.
The possible intracellular mechanism by which Gal‐3 exerts the proinflammatory, profibrotic, and pro‐osteogenic effects in VICs was analyzed. Gal‐3 induced phosphorylation of Akt (at 15 and 30 minutes of stimulation), ERK1/2 (at 30 minutes of stimulation), and NFκB (at 15 minutes of stimulation; Figure 3C). Phosphorylation of p38 MAPK did not change after Gal‐3 incubation (Figure 3C). Presence of the specific inhibitor of ERK1/2, PD98059, blocked the increases induced by Gal‐3 in the inflammatory molecules, TNF‐α and CCL2, as well as in the fibrotic molecule, collagen type 1 (Figure 3D). However, none of the inhibitors tested decreased TGF‐β or α‐SMA expressions (Figure 3D). Gal‐3 enhancement of the osteogenic markers, BMP‐2, BMP‐4, osteopontin, Sox‐9, and Runx2 was abolished by PD98059 (Figure 3E).
Gal‐3 Inhibition Reduces Inflammation, Fibrosis, and Osteogenesis in Human VICs
We studied the effects of the Gal‐3 inhibitor, MCP, in VICs cultured in an osteogenic medium at different days (7, 14, and 21 days). Presence of MCP in the medium was able to diminish IL‐6 and IL‐1β secretion as well as Gal‐3 protein levels at all of days studied (Figure 4A). This decrease in Gal‐3 was accompanied by a decrease in fibronectin protein levels at 21 days of treatment with MCP (Figure 4A). In addition, VICs treated with MCP presented a decrease in calcification markers in a time‐dependent manner. MCP decreased BMP‐2 protein levels at 14 and 21 days of treatment and BMP‐4, SOX‐9, and Runx2 protein levels at 21 days (Figure 4B).
Figure 4.
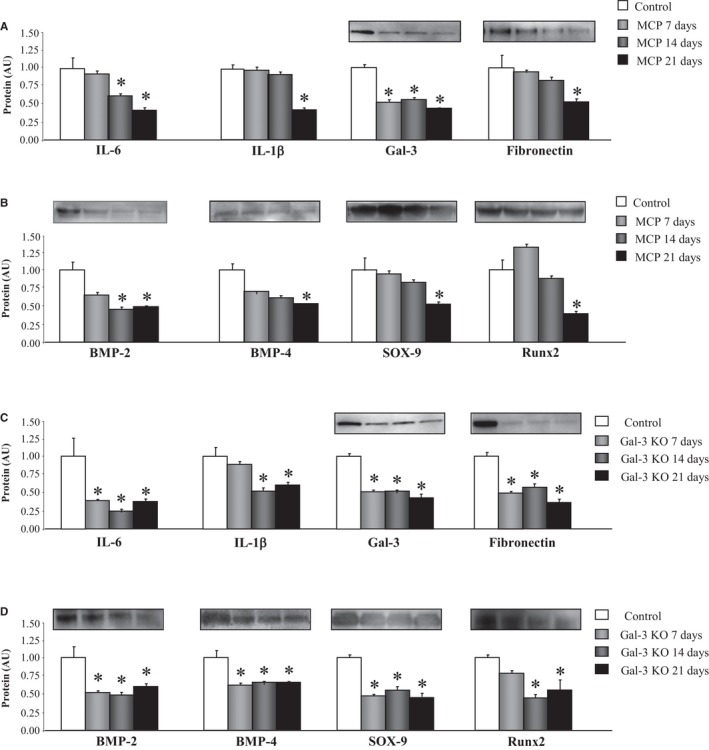
Gal‐3 inhibition reduces inflammation, ECM components, and calcification markers in VICs. Effects of the pharmacological inhibitor of Gal‐3, MCP, on inflammation, Gal‐3, fibronectin protein levels (A), and calcification markers (B) in VICs cultured in osteogenic medium. Effects of the Gal‐3 knockout (KO) on inflammation, Gal‐3, fibronectin protein levels (C), and calcification markers (D) in VICs cultured in osteogenic medium. All conditions were performed at least in triplicate. Histogram bars represent the mean±SEM of 4 assays in arbitrary units (AU) normalized to stain free for protein. *P<0.05 versus control. BMP, bone morphogenetic protein; ECM, extracellular matrix; Gal‐3, galectin‐3; IL, interleukin; MCP, modified citrus pectin; Runx2, runt‐related transcription factor 2; Sox‐9, SRY (sex‐determining region Y)‐box 9; VICs, valvular interstitial cells.
In another set of experiments, to confirm the consequences of Gal‐3 inhibition on VIC osteogenic differentiation, Gal‐3 knockout VICs were generated using CRISPR/Cas9 technology. As compared to scramble, Gal‐3 knockout VICs presented lower levels of IL‐6 and IL‐1β secretion as well as diminished expression of Gal‐3 and fibronectin protein levels at all of days studied (Figure 4C). Moreover, BMP‐2, BMP‐4, SOX‐9, and Runx2 expressions were lower in Gal‐3 knockout VICs at 7, 14, and 21 days of osteogenic differentiation (Figure 4D).
Discussion
The aim of this study was to evaluate the role of Gal‐3 in AV calcification in patients with severe CAVD. Our results demonstrate that Gal‐3 is increased in stenotic AV tissue and correlates with its serum levels. Moreover, Gal‐3 is colocalized with markers of VICs, as well as calcification and inflammation in stenotic AVs, suggesting that it plays a role in these processes. In aortic VICs in vitro, Gal‐3 increases inflammatory, fibrotic, and calcification markers through the ERK1/2 pathway. The Gal‐3 inhibitor, MCP, as well as Gal‐3 silencing delays AV calcification in aortic VICs. Taken together, these findings reveal a role for Gal‐3 in AV calcification associated with progression of AS.
The AV leaflets are a highly specialized structure consisting mostly of VICs and complex ECM structures.6, 20 An inflammatory and fibrotic process in AV in humans and animal models has been previously reported on.21, 22, 23 Aberrant remodeling of the ECM is also brought about by deregulated expression of MMPs, facilitating inflammation.24 These events are associated with activation of VICs toward an osteogenic‐like phenotype associated with upregulation of the BMP pathway.25 In agreement with these data, AS patients presented an increase in inflammation, ECM remodeling, MMP activities, and calcification markers. All these processes are accompanied by enhanced Gal‐3 levels in serum and AVs from AS patients. In addition, AV Gal‐3 protein levels and serum Gal‐3 levels were correlated, suggesting that circulating Gal‐3 levels could indicate AV calcification in AS. Moreover, Gal‐3 colocalized with expression of VICs markers, as well as with osteogenic and inflammatory markers, proposing a role for this lectin in the calcification and inflammatory processes associated with AS progression. Accordingly, Gal‐3 enhanced inflammatory, fibrotic, and osteogenic markers in VICs by activating the ERK1/2 pathway. The ERK1/2 pathway plays an important role in regulating the calcification of VICs.26 Similarly, in human cardiac fibroblasts, Gal‐3 showed proinflammatory and profibrotic properties increasing inflammatory markers, MMP activity, and ECM components.27 Importantly, and in line with our findings, Gal‐3 has been shown to promote vascular calcification associated with atherosclerosis.19 When cultured in an osteogenic medium, Gal‐3‐deficient VSMCs exhibited defective osteogenic differentiation,19 suggesting that Gal‐3 was essential for a complete transdifferentiation of VSMCs into osteoblast‐like cells. In fact, Gal‐3 has also been related with Runx2,28 and it has been suggested to participate in the process of endochondral bone formation.29
CAVD shares features with vascular calcification and atherosclerosis.6 However, lipid‐lowering therapy with statins did not reduce the progression of AS.10, 11 In addition, AS patients present left ventricle (LV) remodeling in response to chronically increased afterload. Because of this fact, angiotensin‐converting enzyme inhibitors (ACEis) could promote beneficial effects in AS patients.30 In the RIAS study, ACE inhibition leads to a modest reduction in LV mass in asymptomatic patients with AS, with trends toward slower progression of valvular stenosis.31 Although the data of the RIAS study are promising, routine use of ACEis in patients with asymptomatic AS cannot be systematically recommended at this time. Importantly, circulating Gal‐3 levels positively correlated with cardiac fibrosis in AS patients.32 Moreover, Gal‐3 may serve as a prognostic biomarker after transcatheter AV implantation by reflecting the degree of myocardial fibrosis.16 Additionally, cardiac Gal‐3 expression is associated with inflammatory markers in myocardial biopsies from AS patients.7 However, a recent study concludes that Gal‐3 did not provide prognostic information on the occurrence of AS‐related events.33
Previous studies in our group have demonstrated that pharmacological Gal‐3 inhibition using MCP prevented cardiovascular remodeling in several hypertensive models17, 18, 27 as well as in a normotensive model of obesity.34 Similar beneficial effects of Gal‐3 inhibition have been reported on cardiac fibrosis, remodeling, and dysfunction in angiotensin II–treated animals.35 Our results have shown that Gal‐3 not only acts on ECM components and inflammation, but also in the calcification process. In vitro, in human VICs, Gal‐3 inhibition with MCP attenuated the proinflammatory, profibrotic, and pro‐osteogenic response. Complementary, Gal‐3‐deficient VICs presented lower inflammation and osteogenic differentiation, indicating that Gal‐3 may play a role as a downstream mediator in the regulation of calcification.
Perspectives
The present study demonstrates that Gal‐3 is elevated in aortic VICs of stenotic valves. Enhanced Gal‐3 activation plays an important role in the augmentation of the proinflammatory, profibrotic, and pro‐osteogenic response. Furthermore, Gal‐3 blockade delays AV calcification. These novel findings obtained in diseased AVs and in human aortic VICs indicate that Gal‐3 may be a new potential biotarget for delaying AV calcification and for prevention of CAVD.
Limitations
Several limitations of our study must be acknowledged. First, this study involves relatively small amounts of tissue obtained from patients, making it difficult to undertake an extensive range of analyses. Second, at the time of valve replacement, patients were under treatment, which may influence the results, although Gal‐3 levels did not seem to be influenced by therapy. Finally, the very small number of controls with either hyperlipidemia, hypertension, or diabetes mellitus in the control precluded us for any adjustment.
Sources of Funding
This work was supported by Miguel Servet contract CP13/00221 from the “Instituto de Salud Carlos III‐FEDER”, Fondo de Investigaciones Sanitarias (PI15/02160), FIBROTARGETS project (grant agreement number FP7 #602904), Fondo de Investigaciones Sanitarias (PI12/01729), and Red de Investigación Cardiovascular (RD12/0042/0033). The study was cofunded by Fondo Europeo de Desarrollo Regional (FEDER), a way to build Europe. P.R. was supported by the RHU Fight‐HF, a public grant overseen by the French National Research Agency (ANR) as part of the second “Investissements d'Avenir” programme (reference: ANR‐15‐RHUS‐0004).
Disclosures
None.
Supporting information
Table S1. Primers Used in Real‐Time PCR Analysis
(J Am Heart Assoc. 2016;5:e004360 doi: 10.1161/JAHA.116.004360)
References
- 1. Iung B, Baron G, Butchart EG, Delahaye F, Gohlke‐Barwolf C, Levang OW, Tornos P, Vanoverschelde JL, Vermeer F, Boersma E, Ravaud P, Vahanian A. A prospective survey of patients with valvular heart disease in Europe: the Euro Heart Survey on Valvular Heart Disease. Eur Heart J. 2003;24:1231–1243. [DOI] [PubMed] [Google Scholar]
- 2. Danielsen R, Aspelund T, Harris TB, Gudnason V. The prevalence of aortic stenosis in the elderly in iceland and predictions for the coming decades: the AGES‐Reykjavik study. Int J Cardiol. 2014;176:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Otto CM, Burwash IG, Legget ME, Munt BI, Fujioka M, Healy NL, Kraft CD, Miyake‐Hull CY, Schwaegler RG. Prospective study of asymptomatic valvular aortic stenosis. Clinical, echocardiographic, and exercise predictors of outcome. Circulation. 1997;95:2262–2270. [DOI] [PubMed] [Google Scholar]
- 4. Bosse Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet. 2009;2:489–498. [DOI] [PubMed] [Google Scholar]
- 5. Mohler ER III, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. [DOI] [PubMed] [Google Scholar]
- 6. Back M, Gasser TC, Michel JB, Caligiuri G. Biomechanical factors in the biology of aortic wall and aortic valve diseases. Cardiovasc Res. 2013;99:232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. 2012;60:1854–1863. [DOI] [PubMed] [Google Scholar]
- 8. Rajamannan NM, Subramaniam M, Springett M, Sebo TC, Niekrasz M, McConnell JP, Singh RJ, Stone NJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia‐induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation. 2002;105:2660–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, McConnell JP, Singh RJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart. 2005;91:806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121:306–314. [DOI] [PubMed] [Google Scholar]
- 11. Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A randomized trial of intensive lipid‐lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. [DOI] [PubMed] [Google Scholar]
- 12. Ochieng J, Furtak V, Lukyanov P. Extracellular functions of galectin‐3. Glycoconj J. 2004;19:527–535. [DOI] [PubMed] [Google Scholar]
- 13. van Kimmenade RR, Januzzi JL Jr, Ellinor PT, Sharma UC, Bakker JA, Low AF, Martinez A, Crijns HJ, MacRae CA, Menheere PP, Pinto YM. Utility of amino‐terminal pro‐brain natriuretic peptide, galectin‐3, and apelin for the evaluation of patients with acute heart failure. J Am Coll Cardiol. 2006;48:1217–1224. [DOI] [PubMed] [Google Scholar]
- 14. Lopez‐Andres N, Rossignol P, Iraqi W, Fay R, Nuee J, Ghio S, Cleland JG, Zannad F, Lacolley P. Association of galectin‐3 and fibrosis markers with long‐term cardiovascular outcomes in patients with heart failure, left ventricular dysfunction, and dyssynchrony: insights from the CARE‐HF (cardiac resynchronization in heart failure) trial. Eur J Heart Fail. 2012;14:74–81. [DOI] [PubMed] [Google Scholar]
- 15. Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JP, Schroen B, Andre S, Crijns HJ, Gabius HJ, Maessen J, Pinto YM. Galectin‐3 marks activated macrophages in failure‐prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. 2004;110:3121–3128. [DOI] [PubMed] [Google Scholar]
- 16. Baldenhofer G, Zhang K, Spethmann S, Laule M, Eilers B, Leonhardt F, Sanad W, Dreger H, Sander M, Grubitzsch H, Baumann G, Stangl K, Stangl V, Knebel F. Galectin‐3 predicts short‐ and long‐term outcome in patients undergoing transcatheter aortic valve implantation (TAVI). Int J Cardiol. 2014;177:912–917. [DOI] [PubMed] [Google Scholar]
- 17. Calvier L, Martinez‐Martinez E, Miana M, Cachofeiro V, Rousseau E, Sadaba JR, Zannad F, Rossignol P, Lopez‐Andres N. The impact of galectin‐3 inhibition on aldosterone‐induced cardiac and renal injuries. JACC Heart Fail. 2015;3:59–67. [DOI] [PubMed] [Google Scholar]
- 18. Calvier L, Miana M, Reboul P, Cachofeiro V, Martinez‐Martinez E, de Boer RA, Poirier F, Lacolley P, Zannad F, Rossignol P, Lopez‐Andres N. Galectin‐3 mediates aldosterone‐induced vascular fibrosis. Arterioscler Thromb Vasc Biol. 2013;33:67–75. [DOI] [PubMed] [Google Scholar]
- 19. Menini S, Iacobini C, Ricci C, Blasetti Fantauzzi C, Salvi L, Pesce CM, Relucenti M, Familiari G, Taurino M, Pugliese G. The galectin‐3/RAGE dyad modulates vascular osteogenesis in atherosclerosis. Cardiovasc Res. 2013;100:472–480. [DOI] [PubMed] [Google Scholar]
- 20. Merryman WD, Youn I, Lukoff HD, Krueger PM, Guilak F, Hopkins RA, Sacks MS. Correlation between heart valve interstitial cell stiffness and transvalvular pressure: implications for collagen biosynthesis. Am J Physiol Heart Circ Physiol. 2006;290:H224–H231. [DOI] [PubMed] [Google Scholar]
- 21. Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013;113:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chakraborty S, Wirrig EE, Hinton RB, Merrill WH, Spicer DB, Yutzey KE. Twist1 promotes heart valve cell proliferation and extracellular matrix gene expression during development in vivo and is expressed in human diseased aortic valves. Dev Biol. 2010;347:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miller JD, Weiss RM, Serrano KM, Brooks RM II, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vanhoutte D, van Almen GC, Van Aelst LN, Van Cleemput J, Droogne W, Jin Y, Van de Werf F, Carmeliet P, Vanhaecke J, Papageorgiou AP, Heymans S. Matricellular proteins and matrix metalloproteinases mark the inflammatory and fibrotic response in human cardiac allograft rejection. Eur Heart J. 2013;34:1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poggio P, Sainger R, Branchetti E, Grau JB, Lai EK, Gorman RC, Sacks MS, Parolari A, Bavaria JE, Ferrari G. Noggin attenuates the osteogenic activation of human valve interstitial cells in aortic valve sclerosis. Cardiovasc Res. 2013;98:402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gu X, Masters KS. Role of the MAPK/ERK pathway in valvular interstitial cell calcification. Am J Physiol Heart Circ Physiol. 2009;296:H1748–H1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martinez‐Martinez E, Calvier L, Fernandez‐Celis A, Rousseau E, Jurado‐Lopez R, Rossoni LV, Jaisser F, Zannad F, Rossignol P, Cachofeiro V, Lopez‐Andres N. Galectin‐3 blockade inhibits cardiac inflammation and fibrosis in experimental hyperaldosteronism and hypertension. Hypertension. 2015;66:767–775. [DOI] [PubMed] [Google Scholar]
- 28. Stock M, Schafer H, Stricker S, Gross G, Mundlos S, Otto F. Expression of galectin‐3 in skeletal tissues is controlled by Runx2. J Biol Chem. 2003;278:17360–17367. [DOI] [PubMed] [Google Scholar]
- 29. Colnot C, Sidhu SS, Poirier F, Balmain N. Cellular and subcellular distribution of galectin‐3 in the epiphyseal cartilage and bone of fetal and neonatal mice. Cell Mol Biol (Noisy‐le‐grand). 1999;45:1191–1202. [PubMed] [Google Scholar]
- 30. Davin L, Dulgheru R, Lancellotti P. ACE inhibitors in aortic stenosis: no fear just hope. Eur Heart J Cardiovasc Imaging. 2015;16:828–830. [DOI] [PubMed] [Google Scholar]
- 31. Bull S, Loudon M, Francis JM, Joseph J, Gerry S, Karamitsos TD, Prendergast BD, Banning AP, Neubauer S, Myerson SG. A prospective, double‐blind, randomized controlled trial of the angiotensin‐converting enzyme inhibitor ramipril in aortic stenosis (RIAS trial). Eur Heart J Cardiovasc Imaging. 2015;16:834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou K, Zhou Y, Zhao Y, Tan C, Yuan Z, Li J, Liao X, Gu L, Zhou X. The relationship between galectin‐3 and different patterns of ventricular geometry remodelling in aortic valve stenosis. Heart Lung Circ. 2016;25:371–377. [DOI] [PubMed] [Google Scholar]
- 33. Arangalage D, Nguyen V, Robert T, Melissopoulou M, Mathieu T, Estellat C, Codogno I, Huart V, Duval X, Cimadevilla C, Vahanian A, Dehoux M, Messika‐Zeitoun D. Determinants and prognostic value of galectin‐3 in patients with aortic valve stenosis. Heart. 2016;102:862–868. [DOI] [PubMed] [Google Scholar]
- 34. Martinez‐Martinez E, Lopez‐Andres N, Jurado‐Lopez R, Rousseau E, Bartolome MV, Fernandez‐Celis A, Rossignol P, Islas F, Antequera A, Prieto S, Luaces M, Cachofeiro V. Galectin‐3 participates in cardiovascular remodeling associated with obesity. Hypertension. 2015;66:961–969. [DOI] [PubMed] [Google Scholar]
- 35. Yu L, Ruifrok WP, Meissner M, Bos EM, van Goor H, Sanjabi B, van der Harst P, Pitt B, Goldstein IJ, Koerts JA, van Veldhuisen DJ, Bank RA, van Gilst WH, Sillje HH, de Boer RA. Genetic and pharmacological inhibition of galectin‐3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ Heart Fail. 2013;6:107–117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers Used in Real‐Time PCR Analysis