

Abstract
Background
Atherosclerosis is a chronic disease that is closely related to inflammation and macrophage apoptosis, which leads to secondary necrosis and proinflammatory responses in advanced lesions. Caspase‐activated DNase (CAD) is a double‐strand specific endonuclease that leads to the subsequent degradation of chromosome DNA during apoptosis. However, whether CAD is involved in the progression of atherosclerosis remains elusive.
Methods and Results
CAD −/−ApoE−/− and ApoE−/− littermates were fed a high‐fat diet for 28 weeks to develop atherosclerosis. Human specimens were collected from coronary heart disease (CHD) patients who were not suitable for transplantation. CAD expression was increased in the atheromatous lesions of CHD patients and high‐fat diet‐treated ApoE‐deficient mice. Further investigation demonstrated that CAD deficiency inhibited high‐fat diet‐induced atherosclerosis, as evidenced by decreased atherosclerotic plaques, inhibited inflammatory response, and macrophage apoptosis, as well as enhanced stability of plaques in CAD −/−ApoE−/− mice compared to the ApoE−/− controls. Bone marrow transplantation verified the effect of CAD on atherosclerosis from macrophages. Mechanically, the decrease in the phosphorylated levels of mitogen‐activated protein kinase (MAPK) kinase/extracellular signal‐regulated kinase 1 and 2 (MEK‐ERK1/2) that resulted from CAD knockout and the activation of nuclear factor kappa B signaling mediated by CAD stimulation that was suppressed by inhibiting ERK1/2 phosphorylation revealed the potential association between the role of CAD in atherosclerosis and the MAPK signaling pathway.
Conclusions
In conclusion, CAD deficiency protects against atherosclerosis through inhibiting inflammation and macrophage apoptosis, which is partially through inactivation of the MEK‐ERK1/2 signaling pathway. This finding provides a promising therapeutic target for treating atherosclerosis.
Keywords: apoptosis, atherosclerosis, caspase‐activated DNase, inflammation, macrophage
Subject Categories: Atherosclerosis
Introduction
Atherosclerosis is a progressive disease characterized by the accumulation of lipids, recruitment of monocytes, and formation of foam cells.1 It plays a vital role in the development of cardiovascular disease, and it is the leading cause of morbidity and mortality worldwide.2, 3 Monocyte/macrophage‐derived foam cells contribute to the initiation of atherogenesis through uptake of modified forms of low‐density lipoprotein (LDL).4 Previous studies have revealed that atherosclerosis is highly related to inflammation and apoptosis, which can result in the rupture of plaques and occurrence of thrombosis.5, 6 Therefore, identifying the molecular mechanism that contributes to atherogenesis alleviation will provide a novel treatment strategy.
Caspase‐activated DNase (CAD), ICAD also known as DNA fragmentation factor‐40 (DFF40), is a magnesium‐dependent endonuclease that is specific to double‐strand DNA and is responsible for apoptotic degradation and chromatin condensation.7 CAD is expressed in various tissues and cell types, including the heart, kidney, pancreas, spleen, prostate, ovary, and peripheral blood leukocytes.8 CAD usually presents as a complex with its inhibitor, ICAD (also known as DFF45), which can be cleaved by the transactivation of caspases from apoptotic signals, and then this results in the release of CAD, which causes DNA fragmentation in nuclei.7 Moreover, CAD is not only involved in the apoptotic program, but is also regarded as a promoter of cell differentiation,9 which is involved in the initiation of atherosclerotic lesions. A previous study from our group demonstrated that upregulation of CAD aggravates pressure overload‐induced cardiac hypertrophy and heart failure.10 However, it still remains unclear regarding the effect of CAD on the progression of atherosclerosis.
In the current study, we found that CAD expression was dramatically increased in the atherosclerotic lesions of both CHD patients and high‐fat diet (HFD)‐treated ApoE‐deficient mice. We hypothesized that CAD may be involved in the development of atherosclerosis. To investigate this hypothesis, CAD−/−ApoE−/− mice were used to assess the extent of atherosclerotic lesions and explore the related mechanism.
Materials and Methods
Mice and Diets
The 8‐week‐old ApoE knockout mice were obtained from The Jackson Laboratory (Stock No. 002052; The Jackson Laboratory, Bar Harbor, ME), and the CAD knockout mice were purchased from RIKEN BioResource Center (Stock No. 01723; RIKEN BioResource Center, Tsukuba, Japan). The double‐knockout mice were generated by crossing the ApoE−/− mice with the CAD−/− mice. The 30 mice from each group were fed an HFD (15.8% fat and 1.25% cholesterol) for 28 weeks. The animals’ body weight and serum lipid levels were measured at the beginning of the experiment and when they were sacrificed. All of the animal protocols were approved by the Animal Care and Use Committee of the Renmin Hospital of Wuhan University (Wuhan, China).
Human Specimens
Atherosclerotic plaques were collected from the right coronary artery of CHD patients (n=6), whereas the artery was from normal donors (n=4) without cardiac reasons who were inadaptable for transplantation as the control samples. Written informed consent was obtained from relevant families. All the procedures involving human samples were carried out according to the principles outlined in the Declaration of Helsinki and were approved by the Renmin Hospital of Wuhan University Institutional Review Board, Wuhan, and the Institution Review Board for Human Studies of Nanjing Medical University, Nanjing, China.
En Face Analysis of Atherosclerosis and Plaque Histology
All mice were euthanized after 28 weeks on the HFD, and the aortas were collected from the base ascending aorta to the iliac bifurcation. The analysis of lesion areas on the entire aorta, aortic roots, and aortic arches were performed as described11 and quantified with Image‐Pro Plus 6.0 (Image Metrology, Copenhagen, Denmark).
Quantitative Real‐Time Polymerase Chain Reaction and Western Blotting
Isolation of total RNA from the whole aortas was conducted using TRIzol reagent (Roche, Indianapolis, IN). Then, DNase‐treated RNA was reverse‐transcribed with a Transcriptor First Strand cDNA Synthesis Kit (Roche).12 Quantitative real‐time polymerase chain reaction (PCR) reactions were conducted with the LightCycler 480 Real‐time PCR system (Roche) in accord with the manufacturer's guide. The levels of expression of target genes were normalized to GAPDH and then computed and expressed as relative mRNA levels compared to the internal control. Proteins extracted from the aortas were homogenized in lysis buffer and processed as described.13 The protein expression levels were quantified and normalized to the GAPDH control. All antibodies used in this study are listed in Table S1.
Immunofluorescence and TUNEL Staining
The slices from the aortic sinus were incubated with primary antibodies followed by corresponding secondary antibody incubation as described.14 For costaining for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and CD68, tissue sections were first stained with CD68 antibody, followed by TUNEL staining with an ApopTagPlus In Situ Apoptosis Fluorescein Detection Kit (S7111; Millipore, Billerica, MA), according to the manufacturer's protocol. Images were captured using a fluorescence microscope (OLYMPUS DX51; Olympus, Tokyo, Japan) and were analyzed with Image Pro Plus 6.0. All the antibodies used are listed in Table S2.
Irradiation and Bone Marrow Transplantation
In our experiment, ApoE−/− mice (8 weeks old) were used as bone marrow recipients. Bone marrow cells were collected from the femurs and tibias of CAD−/−ApoE−/− mice and ApoE−/− mice, which were used as the donors. The chimeras were obtained as described15, 16 and fed normal chow for 4 to 5 weeks after bone marrow transplantation (BMT) to allow for bone marrow reconstitution and then switched to the HFD for an additional 16 weeks to induce rapid lesion formation.17 Then, all mice were sacrificed, and the entire aortas and hearts were harvested for study.
Foam Cell Formation
Bone marrow cells were isolated from femurs and tibias of CAD−/−ApoE−/− mice and ApoE−/− mice and then cultured in complete RPMI‐1640 medium. After 8 days’ cultivation, bone marrow‐derived macrophages (BMDMs) were generated and incubated in humidified 5% CO2 at 37°C. Then, BMDMs were stimulated with 15 μg/mL of oxidized LDL (Ox‐LDL) for 24 hours. Cells were stained with Oil Red O. Images were then captured and foam cell optical density was quantified.
Statistical Analysis
All data are represented as the mean±SEM. The differences between 2 groups were compared using the 2‐tailed Student t test or the Wilcoxon rank sum, whereas the differences among more than 2 groups were evaluated through 1‐way ANOVA. A value of P<0.05 was considered statistically significant. All of the statistical analyses were performed with SPSS software (version 16.0; SPSS, Inc., Chicago, IL).
Results
CAD Expression Is Upregulated in Both Human and Mouse Atherosclerotic Plaques
To investigate the potential role of CAD in the progression of atherosclerosis, we examined whether CAD expression levels were altered in atheromatous plaques. Interestingly, CAD expression in the right coronary artery of CHD patients was significantly increased compared with that of normal donors according to western blot analysis (Figure 1A). Aortas from the HFD‐fed ApoE−/− mice showed a sharp upregulation of CAD expression in the indicated time that followed the atherogenesis (Figure 1B). Double‐immunofluorescence staining of human coronary artery wall samples indicated that the expression of CAD was increased in atherosclerotic plaques, in contrast to that in healthy donors, and CAD was expressed predominantly in macrophages (Figure 1C). Similarly, CAD expression was enhanced in plaques of HFD‐fed mice compared to that in normal chow‐fed mice, where it associated mainly with plaque macrophages (Figure 1D). However, in both human and mouse plaques, there was little change in CAD expression in smooth muscle cells and endothelial cells (Figure 1E and 1F). Taken together, these results indicate that CAD expression is upregulated in human and mouse atherosclerotic plaques, mainly in macrophages, suggesting that CAD might be involved in the development of atherosclerosis.
Figure 1.
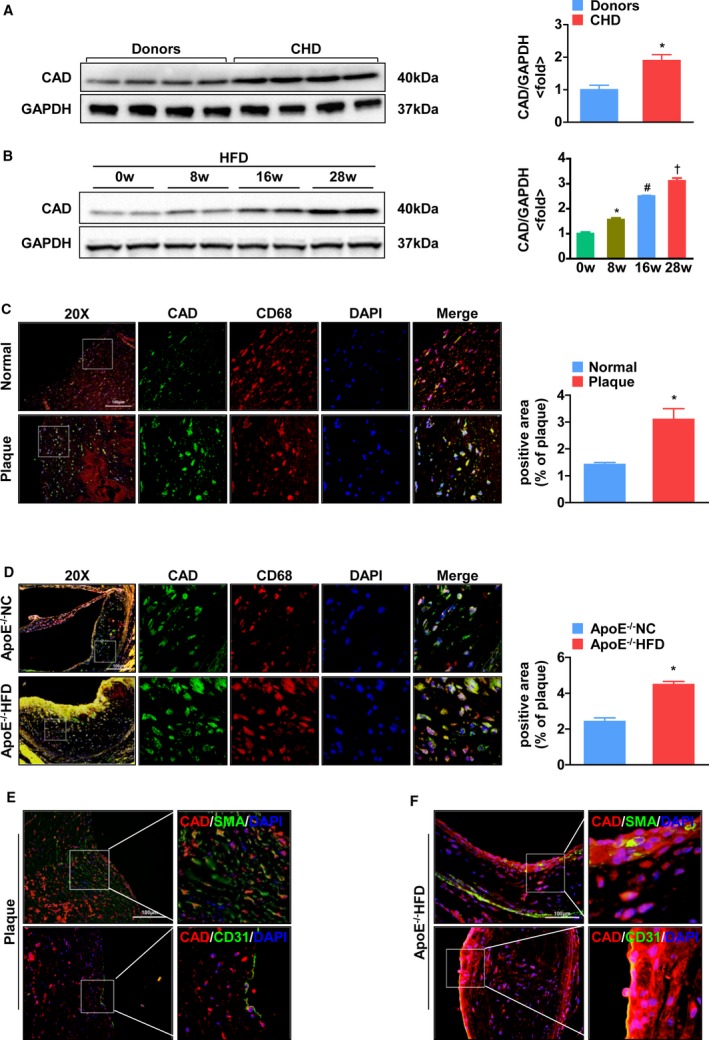
CAD expression is upregulated in both human and mouse atherosclerotic plaques. A, Western blot analysis of CAD in atheromatous plaques from normal donors and patients with coronary heart disease (CHD). Protein levels of CAD were normalized to the loading control. *P<0.05 versus donors. B, Western blot analysis of CAD from ApoE−/− mice fed a high‐fat diet in the indicated time (0, 8, 16, and 28 weeks). Protein levels of CAD were normalized to the loading control. *P<0.05 versus 0 week; # P<0.05 versus 8 weeks; † P<0.05 versus 16 weeks. C and D, (Left panel) Representative images of double‐immunofluorescence staining of human coronary arteries and the aortic sinus from ApoE−/− mice for CAD (green) and CD68 (macrophages, red), respectively. (Right panel) Quantitative analyses of immunofluorescence staining. *P<0.05 versus normal and ApoE−/− mice, respectively. E, Coimmunofluorescence staining of human coronary arteries from patients with CHD for CAD (red) and SMA (smooth muscle cells, green), and CD31 (endothelium, green), respectively. F, Representative images of double‐immunofluorescence staining of cross sections of the aortic sinus from ApoE−/− mice for CAD (red) and SMA (smooth muscle cells, green), and CD31 (endothelium, green), respectively. The original magnification is ×20 or ×40. Scale bar=100 μm in (C) through (F). CAD indicates caspase‐activated DNase; CHD, coronary heart disease; DAPI, 4’,6‐diamidino‐2‐phenylindole; HFD, high‐fat diet; NC, normal controls; SMA, smooth muscle actin.
CAD Deficiency Alleviates the Development of Atherosclerosis
To verify whether CAD expression plays a role in atherosclerosis, a global knockout of the CAD mouse model with an ApoE deficiency background (CAD−/−ApoE−/−) was used. Deficiency of CAD was confirmed by western blot (Figure 2A). After atherogenesis induction, the aortas were collected and stained with Oil Red O to analyze the plaque occupation on entire aortas. The results revealed that smaller atheromatous plaques developed in CAD−/−ApoE−/− mice compared to those in the controls (Figure 2B). In addition, a distinct reduction in atherosclerotic lesions was observed in the aortic sinuses, brachiocephalic arteries, and aortic arches of CAD−/−ApoE−/− mice after hematoxylin and eosin staining (Figure 2C through 2E). Even though an HFD can result in hyperlipidemia, no significant differences in the body weight and serum lipid levels (including triglycerides, total cholesterol, and lipoprotein concentration) were observed between the 2 genotypes (Table S3). Thus, these data indicate that CAD deficiency is protective against atherosclerosis in mice.
Figure 2.
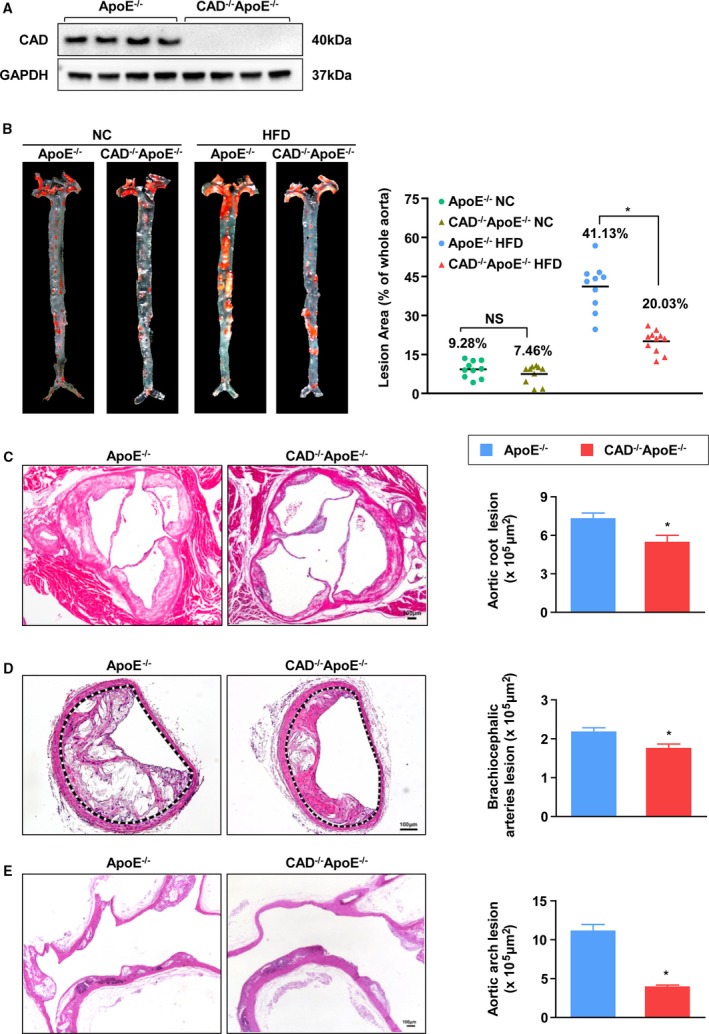
CAD knockout alleviates development of atherosclerosis. A, Loss of CAD expression was confirmed by immunoblotting. B, Representative images of en face with Oil Red O staining of aortas of NC or HFD‐fed CAD −/−ApoE−/− and ApoE−/− littermates are shown in the left panel. Lesion occupation on the whole aorta was quantified (Right panel). n=10. *P<0.05 versus ApoE−/− HF group; NS, no significance. C through E, (Left) Representative images of hematoxylin and eosin–stained cross‐sections of aortic roots (n=10) (C), brachiocephalic arteries (n=4) (D), and aortic arches (n=3) (E). Scale bar=100 μm. (Right) Quantification of areas of atheromatous plagues. *P<0.05 versus ApoE−/− mice. CAD indicates caspase‐activated DNase; HF, high‐fat; HFD, high‐fat diet; NC, normal controls.
CAD Knockout Improves Stability of Atherosclerotic Plaques
The necrotic core is a major determinant of plaque vulnerability, and these cores initially result from the accumulation of apoptotic macrophages.6 Thus, we measured the features of necrotic cores. Representative images of brachiocephalic arteries stained with picrosirius red (PSR) evinced that necrotic areas were obviously decreased in the lesions of CAD−/−ApoE−/− mice compared to ApoE−/− mice (Figure 3A). Additionally, CAD−/−ApoE−/− mice showed thickened fibrous caps (indicated by black markers). These results were reconfirmed in the lesions of aortic roots, which showed smaller necrotic areas, but thicker fibrous caps in CAD‐knockout mice (Figure 3B). The major components of fibrous cap, collagen, and smooth muscle cells, which were stained with PSR and immunofluorescence, respectively, revealed a distinct increase in the lesions of CAD−/−ApoE−/− mice in comparison to those of the controls (Figure 3C and 3D). Meanwhile, the content of macrophages was reduced in the plaques of CAD−/−ApoE−/− mice, and the lipid areas stained with Oil Red O also exhibited a significant decrease in CAD−/−ApoE−/− mice versus ApoE−/− littermates (Figure 3E and 3F). These results indicated that CAD deficiency enhances the stability of atherosclerotic plaques, in addition to protecting ApoE−/− against atherogenesis.
Figure 3.
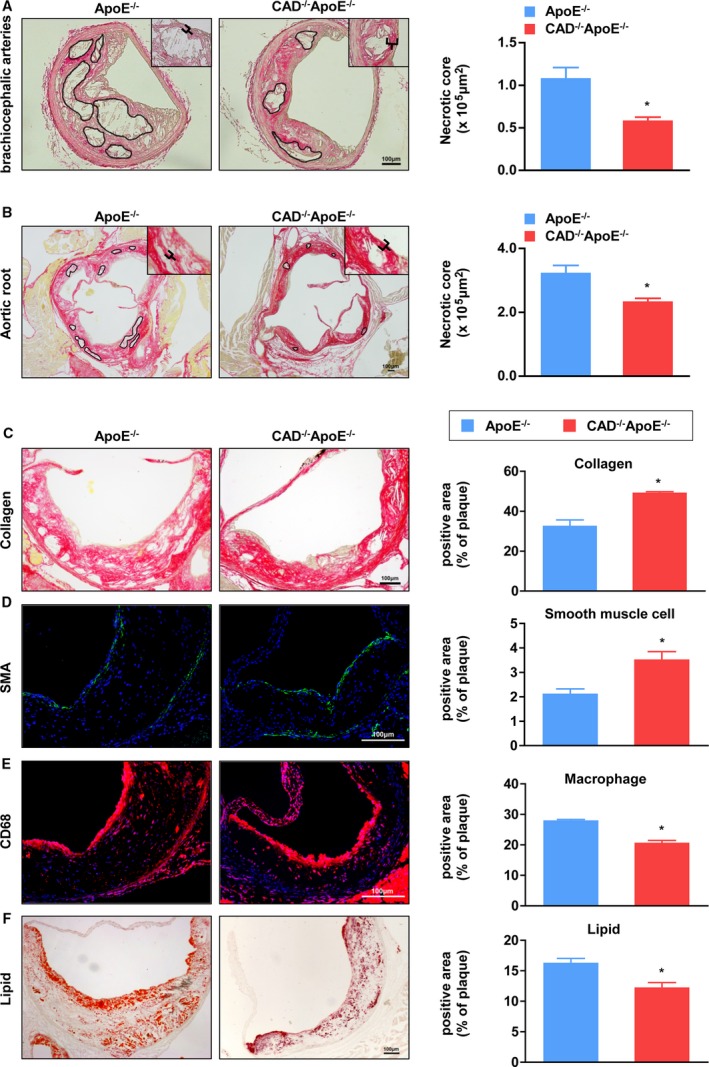
Absence of CAD decreases necrotic areas and enhances stability of atheromatous plaques. A, (Left) PSR staining of histological sections of brachiocephalic arteries from CAD −/−ApoE−/− and ApoE−/− mice. (Right) Quantification of the areas of necrotic cores of brachiocephalic arteries (n=5). *P<0.05 versus ApoE−/− mice. B, (Left) Representative images of PSR staining of aortic roots from CAD −/−ApoE−/− and ApoE−/− mice. (Right) Quantification of the areas of necrotic cores of aortic roots (n=5). *P<0.05 versus ApoE−/− mice. C through F, (Left panel) Representative images of histological sections of aortic sinuses stained with PSR for collagen (C), SMA immunofluorescence for smooth muscle cells (D), CD68 for macrophages (E), and Oil Red O for lipids (F). Scale bar=100 μm. (Right panel) Atherosclerotic lesions from CAD −/−ApoE−/− mice displayed an increase in the percentage of collagen and smooth muscle cells, but a decrease in the percentage of macrophages and lipids compared to ApoE−/− mice (n=5). *P<0.05 versus ApoE−/− mice. CAD indicates caspase‐activated DNase; PSR, picrosirius red; SMA, smooth muscle actin.
CAD Deficiency in Marrow‐Derived Macrophages Attenuates Foam Cell Formation
Considering that the expression of CAD was dramatically and specifically upregulated in the macrophages of the plaques, we performed a BMT experiment to further validate whether CAD in marrow‐derived macrophages exhibits a prominent regulatory function in the development of atherosclerosis. ApoE−/− mice were used as the host, and the bone marrow cells from CAD−/−ApoE−/− mice or ApoE−/− mice were injected into the recipients, which were preconditioned by irradiation. The results of BMT were identified by the PCR assay (Figure 4A). As shown in Figure 4B, the CAD−/−ApoE−/− transplantation mice exhibited fewer atheromatous plaques in their aortas compared with the aortas of the ApoE−/− transplantation mice. Furthermore, the lesions in the aortic sinuses were also decreased in the CAD−/−ApoE−/− chimeras (Figure 4C). These results indicate that the loss of CAD in macrophages derived from the bone marrow plays an important role in protecting against atherogenesis. Moreover, lipoprotein uptake by macrophages facilitates the formation of foam cells, which is critical for the production of inflammatory cytokines and apoptosis.18 Thus, we examined the effect of CAD on foam cells induced by PBS and Ox‐LDL by Oil Red O staining. The representative images revealed a decrease in foam cell formation in macrophages derived from CAD‐deficient mice than those from control littermates (Figure 4D).
Figure 4.
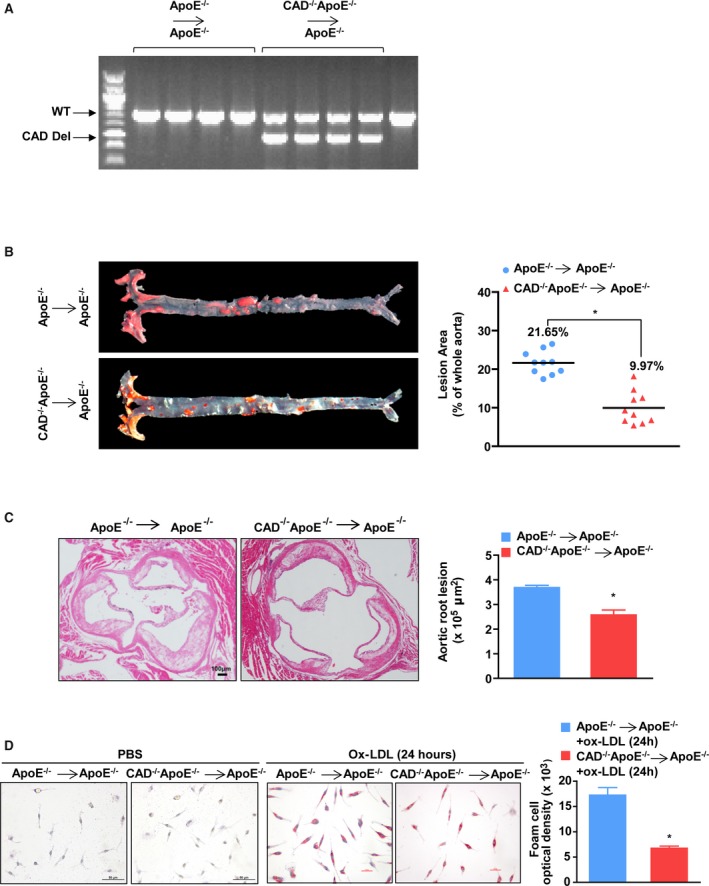
CAD deficiency in marrow‐derived macrophages attenuates foam cell formation. A, PCR assay was performed to identify bone marrow transplantation of CAD −/−ApoE−/− and ApoE−/− mice. B, (Left) Representative images showing Oil Red O staining of the entire aortas from CAD −/−ApoE−/− and ApoE−/− BMT mice. (Right) Quantification of the percentage of lesion areas (n=10). *P<0.05 versus ApoE−/− BMT mice. C, (Left) Representative images of cross‐sections of aortic roots stained with hematoxylin and eosin. Scale bar=100 μm. (Right) Quantification of the areas of plaques of aortic roots (n=5). *P<0.05 versus ApoE−/− BMT mice. D, (Left) Representative images of Oil Red O–stained foam cells treated with PBS and Ox‐LDL for 24 hours from CAD −/−ApoE−/− and ApoE−/− mice. Scale bar=25 μm. (Right) Optical density of foam cells stimulated with Ox‐LDL for 24 hours from CAD −/−ApoE−/− and ApoE−/− mice (n=11). *P<0.05 versus ApoE−/− mice. BMT indicates bone marrow transplantation; CAD, caspase‐activated DNase; CAD Del, CAD deletion; Ox‐LDL, oxidized low‐density lipoprotein; PCR, polymerase chain reaction; WT, wild type.
Loss of CAD Decreases Apoptosis of Macrophages
Macrophage apoptosis occurs throughout all phases of atherosclerosis and exerts distinct effects on the development of atherosclerosis.19 To test whether CAD knockout has an influence on apoptosis of macrophages, TUNEL staining of the aortic roots was performed. We found that the ratio of positive cells displayed a sharp decrease in CAD−/−ApoE−/− mice, in contrast with that of the ApoE−/− littermates (Figure 5A and 5B). In addition, the reverse‐transcriptase (RT)‐PCR assay revealed that the relative mRNA levels of antiapoptotic proteins (eg, B‐cell lymphoma 2 [Bcl2], cellular FLIP long isoform protein [cFLIPL], dolichyl‐diphosphooligosaccharide–protein glycosyltransferase subunit DAD1 [DAD1], and survivin) were increased in CAD−/−ApoE−/− mice, whereas the levels of the proapoptotic proteins Bax and Fas ligand (FasL) were declined (Figure 5C). Furthermore, we treated peritoneal macrophages with PBS and Ox‐LDL for 24 hours to validate the results in vitro. The mRNA levels of antiapoptotic proteins showed an increase, whereas the levels of proapoptotic proteins (eg, cleaved‐caspase 3, Bcl2‐associated X protein [Bax], and FasL) showed a decline in macrophages from CAD−/−ApoE−/− mice treated with Ox‐LDL (Figure 5D), which were in accord with those obtained by immunoblotting (Figure 5E). Taken together, the data suggest that the loss of CAD decreases macrophage apoptosis both in vivo and vitro.
Figure 5.
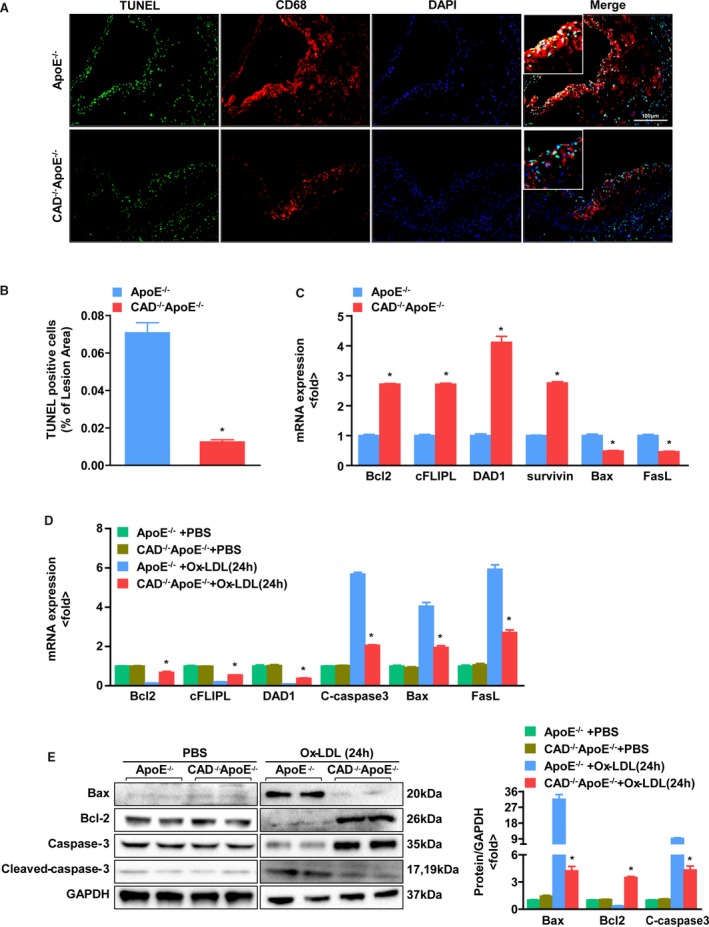
Loss of CAD decreases apoptosis of macrophages. A, Representative images of TUNEL staining of the aortic sinus with CD68 in the lesions from CAD −/−ApoE−/− and ApoE−/− mice. Original magnification, ×20. Scale bar=100 μm. B, Quantification of the percentage of apoptotic cells in lesion areas (n=5). *P<0.05 vs ApoE−/− mice. C, The mRNA expression of apoptotic proteins in vivo, as determined by real‐time PCR. *P<0.05 vs ApoE−/− mice. D, The expression of apoptotic proteins in peritoneal macrophages treated with PBS and Ox‐LDL for 24 hours from CAD −/−ApoE−/− and ApoE−/− mice, as determined by real‐time PCR. *P<0.05 vs ApoE−/− mice. E, (Left) Western blot analysis of apoptotic proteins in the peritoneal macrophages treated with PBS and Ox‐LDL for 24 hours from CAD −/−ApoE−/− and ApoE−/− mice. (Right) The target protein expression levels were normalized to the levels of GAPDH. *P<0.05 vs ApoE−/− mice. Bax indicates Bcl2‐associated X protein; Bcl2, B‐cell lymphoma 2; CAD, caspase‐activated DNase; cFLIPL, cellular FLIP long isoform protein; DAD1, dolichyl‐diphosphooligosaccharide–protein glycosyltransferase subunit DAD1; DAPI, 4’,6‐diamidino‐2‐phenylindole; FasL, Fas ligand; Ox‐LDL, oxidized low‐density lipoprotein; PCR, polymerase chain reaction; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
CAD Ablation Inhibits the Inflammatory Response
Given that atherosclerosis is a chronic inflammatory disease,20, 21 we examined whether CAD deficiency makes a difference in the expression of inflammatory mediators, including cytokines and adhesion molecules, which contribute to the formation of atherosclerotic lesions.11 Immunofluorescence staining of aortic roots revealed that the expression levels of the adhesion molecule, intercellular adhesion molecule 1 (ICAM‐1), and the proinflammatory mediators, interleukin (IL)‐6 and nuclear factor kappa B (NF‐κB) P65 subunit were markedly reduced in CAD−/−ApoE−/− mice versus ApoE−/− mice (Figure 6A). Meanwhile, we measured the serum concentrations of inflammatory cytokines. The results exhibited that the serum level of proinflammatory markers (eg, tumor necrosis factor alpha [TNF‐α], monocyte chemoattractant protein 1 [MCP‐1], IL‐6, and IL‐1β) were significantly reduced in the CAD‐deficient mice compared to that in ApoE−/− controls (Figure 6B), which is consistent with the mRNA expression data obtained by RT‐PCR (Figure 6C). Further analysis of mRNA levels of the chemokine, inducible nitric oxide synthase (iNOS), and adhesion molecule vascular cell adhesion molecule 1 (VCAM‐1) displayed a remarkable reduction in CAD−/−ApoE−/− mice compared to that in the ApoE−/− mice (Figure 6C). At the same time, the levels of phosphorylation of IκB kinase beta (IKKβ), nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor, alpha (IκBα), and P65 were much lower in CAD‐knockout mice than in ApoE ablation littermates (Figure 6D and 6E). Collectively, these evidences demonstrate that CAD deletion inhibits the inflammatory response.
Figure 6.
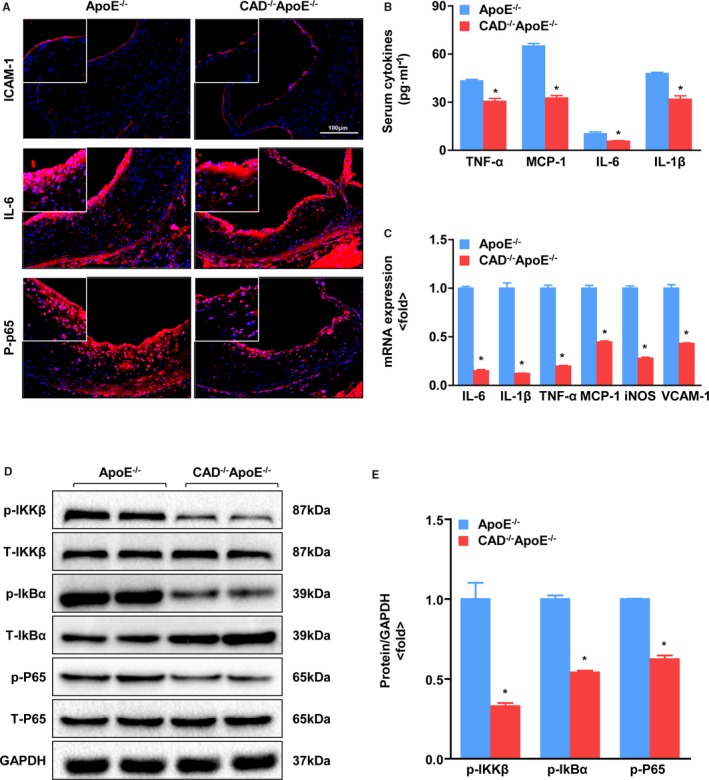
CAD deficiency decreases inflammation. A, Representative images showing immunofluorescence staining of cross‐sections of the aortic sinus for ICAM‐1, IL‐6, and p‐P65. Scale bar=100 μm. B, Serum levels of inflammatory cytokines, TNF‐α, MCP‐1, IL‐6, and IL‐1β, in CAD −/−ApoE−/− mice and ApoE−/− littermates. *P<0.05 versus ApoE−/− mice. C, Real‐time PCR assays were performed to determine mRNA expression of inflammatory mediators and adhesion molecules. *P<0.05 versus ApoE−/− mice. D, Western blot analysis of total (T‐) and phosphorylated (p‐) IKKβ, IkBα, and P65. E, Statistical analysis of the immunoblots. The target protein expression levels were normalized to the levels of GAPDH. *P<0.05 vs ApoE−/− mice. CAD indicates caspase‐activated DNase; ICAM‐1, intercellular adhesion molecule 1; IL, interleukin; IkBα, nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor, alpha; IKKβ, IκB kinase beta; iNOS, inducible nitric oxide synthase; MCP‐1, monocyte chemoattractant protein 1; PCR, polymerase chain reaction; TNF‐α, tumor necrosis factor alpha; VCAM‐1, vascular cell adhesion molecule‐1.
CAD Deficiency Inhibits the Mitogen‐Activated Protein Kinase Kinase/Extracellular Signal‐Regulated Kinase 1 and 2 Signaling Pathway
To define the molecular mechanism underlying the protection effect of CAD ablation on atherosclerosis, we examined the phosphorylated levels of mitogen‐activated protein kinase (MAPK) kinase (MEK1/2), extracellular signal‐regulated kinase 1 and 2 (ERK1/2), c‐Jun N‐terminal kinase 1 and 2 (JNK1/2), and P38 by western blotting. The results indicated that MEK‐ERK1/2 phosphorylation was decreased in CAD−/−ApoE−/− mice, whereas no significant differences in JNK1/2 and P38 were observed between the two groups (Figure 7A and 7B). Moreover, immunofluorescent costaining of ERK phosphorylation and CD68 also revealed a decrease of ERK phosphorylation, mainly in macrophages, in the lesions of the CAD−/−ApoE−/− mice compared with those of the ApoE−/− controls (Figure 7C). Additionally, we pretreated peritoneal macrophage with U0126 (an MEK inhibitor that prevents ERK1/2 phosphorylation) for 1 hour and then treated the cells with Ox‐LDL for 24 hours after infection with adenovirus‐expressing green fluorescent protein (AdGFP) or adenovirus‐expressing CAD (AdCAD) to test the role of CAD‐deficiency on suppression of NF‐κB. We noticed that the increased expression of NF‐κB mediated by overexpression of CAD was dramatically suppressed by pretreatment with U0126 by western blot (Figure 7D), which demonstrated that the detrimental role of CAD in atherosclerosis is largely associated with the role of MEK‐ERK1/2 signaling on NF‐κB activation.
Figure 7.
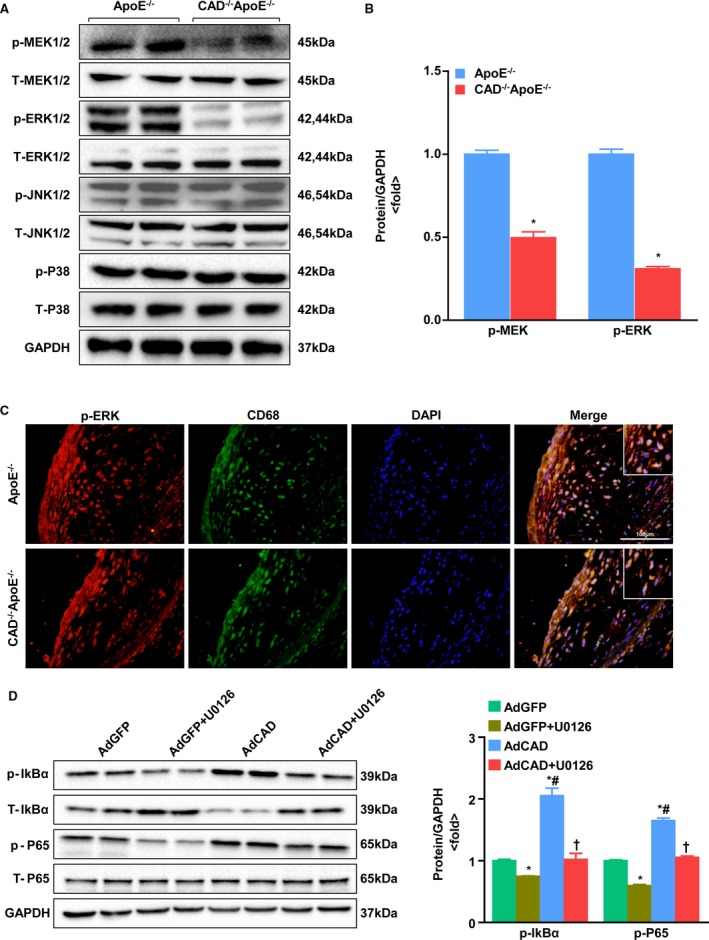
Effect of CAD on the MAPK signaling pathway. A, Representative western blots of total and phosphorylated MEK1/2, ERK1/2, JNK1/2, and P38 in CAD −/−ApoE−/− and ApoE−/− mice. B, Relative protein levels of phosphorylated MEK1/2 and ERK1/2 were quantified after normalization to GAPDH. *P<0.05 versus ApoE−/− mice. C, Representative images of double‐immunofluorescence staining of phosphorylated ERK and CD68. Original magnification, ×40. Scale bar=100 μm. D, (Left) Western blot analysis of phosphorylated and total IkBα, and P65 in the peritoneal macrophages pretreated with U0126 for 1 hour after infection with AdGFP and AdCAD. (Right) Statistical analysis of the immunoblots. Target protein expression levels were normalized to the levels of GAPDH. *P<0.05 versus AdGFP; # P<0.05 versus AdGFP+U0126; † P<0.05 versus AdCAD. AdCAD indicates adenovirus‐expressing CAD; AdGFP, adenovirus‐expressing green fluorescent protein; CAD, caspase‐activated DNase; DAPI, 4’,6‐diamidino‐2‐phenylindole; ERK1/2, extracellular signal‐regulated kinase 1 and 2; IkBα, nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor, alpha; IkBβ, nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor, beta; JNK1/2, c‐Jun N‐terminal kinase 1 and 2; MAPK, mitogen‐activated protein kinase; MEK1/2, MAPK kinase 1 and 2.
Discussion
Atherosclerosis is a chronic and progressive disease that is triggered by lipid accumulation in the arterial wall and is highly correlated with inflammation mediated by macrophages. The infiltration of macrophages differentiated from monocytes plays a crucial role in the severity of atherosclerosis.22 The differentiated macrophages subsequently engulf lipoproteins, particularly modified LDL, resulting in foam cell formation.23 In our study, we demonstrated that CAD was upregulated in the atherosclerotic plaques and was predominantly localized in macrophages. Meanwhile, the BMT assay indicated that the loss of CAD in bone marrow–derived macrophages accounted for decreased atherosclerotic lesions. Furthermore, CAD deficiency inhibited the formation of foam cells, which is perceived as a critical step for the subsequent inflammatory response.24 Increasing evidence suggests that inflammation plays a significant role in all phases of atherosclerosis,25 and macrophages in advanced plaques have been regarded as major contributors to the inflammatory response through the secretion of proinflammatory mediators,26 which promotes highly inflamed and necrotic plaques referred to “vulnerable plaques.”6 Here, we found that the CAD knockout mice exhibited decreased expression levels of proinflammatory cytokines, such as IL‐6, IL‐1β, and TNF‐α, as well as suppressed NF‐κB signaling.27 NF‐κB is a proatherogenic factor, primarily attributed to its regulation of proinflammatory genes correlated with atherosclerosis.28 Its activity is found in many cell types, including adipocytes and macrophages,29 and it is required for macrophage function in atherosclerosis.30
Macrophage apoptosis is complex and occurs throughout the atherosclerotic process.19 In the early phase of atherosclerosis, macrophages in the lesions effectively phagocytosis and clear apoptotic cells, which appears to alleviate atherosclerosis to some extent.19 Nevertheless, in advanced lesions, the increase in apoptotic macrophages gives rise to defective efferocytosis, which promotes inflammatory responses and secondary necrosis of the apoptotic cells through the release of inflammatory mediators.19, 31, 32 This process occurs concomitantly with an increase in the vulnerability of atheromatous plaques, contributing to the amplification of necrotic cores and subsequent plaque rupture.22 Previous studies have stated that CAD is identified as the DNase responsible for apoptotic DNA degradation, which is one of the hallmarks of apoptosis. Cells lacking CAD do not go through apoptotic DNA fragmentation, implying that CAD is solely in charge of this process.7, 33 In proliferating cells, CAD is complexed in the cytosol with its inhibitory partner, ICAD, which functions as a chaperone for CAD synthesis and inhibits its DNase activity.34 When caspases, particularly caspase 3, which is downstream in the cascade, are activated by apoptotic stimuli, CAD is cleaved and dissociated from the CAD/ICAD complex.35 Then, CAD forms a scissors‐like dimer and enters the nucleus to degrade chromosomal DNA.36 In our study, we found that macrophage apoptosis was decreased in CAD−/−ApoE−/− mice. Thus, we concluded that CAD deficiency has an antiapoptotic effect on the development of atherosclerosis, especially in advanced lesions, which were induced by an HFD for 28 weeks. Furthermore, in the current study, smaller necrotic cores of atherosclerotic lesions, but thicker fibrous caps, were observed in CAD−/−ApoE−/− mice than in ApoE−/− controls. Increasing evidence suggests that defective efferocytosis in advanced atheromata is a primary cause of necrotic core formation and thus contributes substantially to the formation of vulnerable plaques.6 In this context, we examined 4 features of plaque stability, and the results showed an increase in the collagen and SMC content and a decrease in the infiltration of macrophages, along with the accumulation of lipids in CAD knockout mice. Multiple factors associated with atherosclerotic plaque vulnerability have been reported, including the thinning of the fibrous cap, expanded necrotic cores, loss of structural proteins, and a highly activated inflammatory response.37 Additionally, vulnerable plaques are prone to rupture and can lead to clinical manifestations, such as myocardial infarction, unstable angina, sudden cardiac death, and stroke.38
A better understanding of the underlying mechanism involved in the pathophysiology of atherosclerosis will be of significance for developing new therapies. A previous study revealed that CAD deficiency protects against cardiac hypertrophy by suppressing the MAPK signaling pathway.10 Additionally, evidence has shown that MAPK plays a pivotal role in the regulation of atherosclerosis,39 and the majority of mammalian MAPK pathways are recruited by stress and inflammatory stimuli, combined with the NF‐κB pathway.40 The MAPK cascade consists of a series of kinases, including ERKs, JNKs, and P38. The prototypic ERK1/2 pathway is mainly responsive to stimulation by growth factors,41 whereas JNK and P38 are induced by physical, chemical, and physiological stressors, such as ultraviolet light, oxidative stress, infection, and cytokines.42 In our study, we determined the protein expression levels of components of the MAPK and NF‐κB signaling pathways. Phosphorylated MEK1/2 and ERK1/2 were decreased in CAD−/−ApoE−/− mice, whereas phosphorylated JNK1/2 and P38 made no difference between the 2 groups. Consistent with the above‐mentioned results, the levels of IKKβ, IκBα, and P65 phosphorylation were downregulated in CAD−/−ApoE−/− mice. Furthermore, we found that phosphorylated ERK was predominantly expressed in the macrophages and was less abundant in the CAD knockout mice compared to the ApoE knockout mice, as determined by immunofluorescence staining. More important, we noticed that the activation of NF‐κB by overexpression of CAD was attenuated by inhibiting ERK1/2 activation. All of the differences in this research have been tested for statistical significance by choosing appropriate statistical analysis methods, it still should be noted that some significant tests might have occurred by chance alone.
In summary, the present study provides the first evidence regarding the role of CAD in atherosclerosis. Our findings indicate that the knockout of CAD globally, or at least in macrophages derived from bone marrow, ameliorates the development of atherosclerosis. This process occurs through an enhancement in the stability of plaques by suppressing foam cell formation and decrease in macrophage apoptosis and inflammation through suppression of the MEK‐ERK1/2 and NF‐κB signaling pathways. Our study suggests a novel therapeutic strategy for the treatment of atherosclerosis and clinical complications.
Sources of Funding
This work was supported by grants from the National Science Fund for Distinguished Young Scholars (No. 81425005), the Key Project of the National Natural Science Foundation (Nos. 81330004 and 81330005), the National Natural Science Foundation of China (Nos. 81370209, 81370365, 81270184, and 31371481), and the National Science and Technology Support Project (Nos. 2013YQ030923‐05, 2014BAI02B01, and 2015BAI08B01).
Disclosures
None.
Supporting information
Table S1. Antibodies for Immunoblot Analyses
Table S2. Antibodies for Immunofluorescence and TUNEL Staining
Table S3. Body Weight and the Levels of Serum Lipids in CAD−/−ApoE−/− Mice and ApoE−/− Mice Fed a High‐Fat Diet for 28 Weeks
Acknowledgments
We thank Cheng Du, Qiaofang Wei, and ShuoXiong for technical assistance.
(J Am Heart Assoc. 2016;5:e004362 doi: 10.1161/JAHA.116.004362)
Contributor Information
Yong Ji, Email: yongji@njmu.edu.cn.
Hongliang Li, Email: lihl@whu.edu.cn.
References
- 1. Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104:503–516. [DOI] [PubMed] [Google Scholar]
- 2. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. [DOI] [PubMed] [Google Scholar]
- 3. Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd‐Jones D, McDermott M, Meigs J, Moy C, Nichol G, O'Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y; American Heart Association Statistics C, Stroke Statistics S . Heart Disease and Stroke Statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–e146. [DOI] [PubMed] [Google Scholar]
- 4. Yamada Y, Doi T, Hamakubo T, Kodama T. Scavenger receptor family proteins: roles for atherosclerosis, host defence and disorders of the central nervous system. Cell Mol Life Sci. 1998;54:628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase‐activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. [DOI] [PubMed] [Google Scholar]
- 8. Mukae N, Enari M, Sakahira H, Fukuda Y, Inazawa J, Toh H, Nagata S. Molecular cloning and characterization of human caspase‐activated DNAse. Proc Natl Acad Sci USA. 1998;95:9123–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Larsen BD, Rampalli S, Burns LE, Brunette S, Dilworth FJ, Megeney LA. Caspase 3/caspase‐activated DNase promote cell differentiation by inducing DNA strand breaks. Proc Natl Acad Sci USA. 2010;107:4230–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao L, Huang K, Jiang DS, Liu X, Huang D, Li H, Zhang XD, Huang K. Novel role for caspase‐activated DNase in the regulation of pathological cardiac hypertrophy. Hypertension. 2015;65:871–881. [DOI] [PubMed] [Google Scholar]
- 11. Cheng WL, Wang PX, Wang T, Zhang Y, Du C, Li H, Ji Y. Regulator of G‐protein signalling 5 protects against atherosclerosis in apolipoprotein E‐deficient mice. Br J Pharmacol. 2015;172:5676–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X, Wang PX, Huang Z, Li H. The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1‐dependent signalling. Nat Commun. 2016;7:11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang PX, Zhang XJ, Luo P, Jiang X, Zhang P, Guo J, Zhao GN, Zhu X, Zhang Y, Yang S, Li H. Hepatocyte TRAF3 promotes liver steatosis and systemic insulin resistance through targeting TAK1‐dependent signalling. Nat Commun. 2016;7:10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y, Zhang P, Jiang X, Gao L, Zhu XY, Zhao Y, Gao L, Yang Q, Zhu XH, Wei X, Pu J, Li H. Suppressor of IKKvarepsilon is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun. 2016;7:11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. [DOI] [PubMed] [Google Scholar]
- 16. Morrison SJ, Qian D, Jerabek L, Thiel BA, Park IK, Ford PS, Kiel MJ, Schork NJ, Weissman IL, Clarke MF. A genetic determinant that specifically regulates the frequency of hematopoietic stem cells. J Immunol. 2002;168:635–642. [DOI] [PubMed] [Google Scholar]
- 17. Schiller NK, Kubo N, Boisvert WA, Curtiss LK. Effect of gamma‐irradiation and bone marrow transplantation on atherosclerosis in LDL receptor‐deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1674–1680. [DOI] [PubMed] [Google Scholar]
- 18. Itabe H. Oxidized low‐density lipoproteins: what is understood and what remains to be clarified. Biol Pharm Bull. 2003;26:1–9. [DOI] [PubMed] [Google Scholar]
- 19. Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. [DOI] [PubMed] [Google Scholar]
- 20. Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. [DOI] [PubMed] [Google Scholar]
- 21. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 22. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Paoli F, Staels B, Chinetti‐Gbaguidi G. Macrophage phenotypes and their modulation in atherosclerosis. Circ J. 2014;78:1775–1781. [DOI] [PubMed] [Google Scholar]
- 25. Siegel D, Devaraj S, Mitra A, Raychaudhuri SP, Raychaudhuri SK, Jialal I. Inflammation, atherosclerosis, and psoriasis. Clin Rev Allergy Immunol. 2013;44:194–204. [DOI] [PubMed] [Google Scholar]
- 26. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor‐kappa B is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xanthoulea S, Curfs DM, Hofker MH, de Winther MP. Nuclear factor kappa B signaling in macrophage function and atherogenesis. Curr Opin Lipidol. 2005;16:536–542. [DOI] [PubMed] [Google Scholar]
- 29. Hayden MS, Ghosh S. Shared principles in NF‐kappaB signaling. Cell. 2008;132:344–362. [DOI] [PubMed] [Google Scholar]
- 30. Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns‐Hendriks I, Fijneman RJ, Clausen BE, Forster I, Kockx MM, Rajewsky K, Kraal G, Hofker MH, de Winther MP. Inhibition of NF‐kappaB activation in macrophages increases atherosclerosis in LDL receptor‐deficient mice. J Clin Invest. 2003;112:1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, Bennett M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. 2007;100:884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin‐1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J. 2011;162:597–605. [DOI] [PubMed] [Google Scholar]
- 33. Kawane K, Fukuyama H, Yoshida H, Nagase H, Ohsawa Y, Uchiyama Y, Okada K, Iida T, Nagata S. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol. 2003;4:138–144. [DOI] [PubMed] [Google Scholar]
- 34. Sakahira H, Takemura Y, Nagata S. Enzymatic active site of caspase‐activated DNase (CAD) and its inhibition by inhibitor of CAD. Arch Biochem Biophys. 2001;388:91–99. [DOI] [PubMed] [Google Scholar]
- 35. McIlroy D, Sakahira H, Talanian RV, Nagata S. Involvement of caspase 3‐activated DNase in internucleosomal DNA cleavage induced by diverse apoptotic stimuli. Oncogene. 1999;18:4401–4408. [DOI] [PubMed] [Google Scholar]
- 36. Woo EJ, Kim YG, Kim MS, Han WD, Shin S, Robinson H, Park SY, Oh BH. Structural mechanism for inactivation and activation of CAD/DFF40 in the apoptotic pathway. Mol Cell. 2004;14:531–539. [DOI] [PubMed] [Google Scholar]
- 37. Clarke M, Bennett M. The emerging role of vascular smooth muscle cell apoptosis in atherosclerosis and plaque stability. Am J Nephrol. 2006;26:531–535. [DOI] [PubMed] [Google Scholar]
- 38. Virmani R, Burke AP, Kolodgie FD, Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol. 2002;15:439–446. [DOI] [PubMed] [Google Scholar]
- 39. Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest. 2009;119:886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10‐year update. Physiol Rev. 2012;92:689–737. [DOI] [PubMed] [Google Scholar]
- 41. Ramos JW. The regulation of extracellular signal‐regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40:2707–2719. [DOI] [PubMed] [Google Scholar]
- 42. Kyriakis JM, Avruch J. Mammalian mitogen‐activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibodies for Immunoblot Analyses
Table S2. Antibodies for Immunofluorescence and TUNEL Staining
Table S3. Body Weight and the Levels of Serum Lipids in CAD−/−ApoE−/− Mice and ApoE−/− Mice Fed a High‐Fat Diet for 28 Weeks