

Abstract
Background
MicroRNA miR‐214 has been implicated in many biological cellular functions, but the impact of miR‐214 and its target genes on vascular smooth muscle cell (VSMC) proliferation, migration, and neointima smooth muscle cell hyperplasia is unknown.
Methods and Results
Expression of miR‐214 was closely regulated by different pathogenic stimuli in VSMCs through a transcriptional mechanism and decreased in response to vascular injury. Overexpression of miR‐214 in serum‐starved VSMCs significantly decreased VSMC proliferation and migration, whereas knockdown of miR‐214 dramatically increased VSMC proliferation and migration. Gene and protein biochemical assays, including proteomic analyses, showed that NCK associated protein 1 (NCKAP1)—a major component of the WAVE complex that regulates lamellipodia formation and cell motility—was negatively regulated by miR‐214 in VSMCs. Luciferase assays showed that miR‐214 substantially repressed wild‐type but not the miR‐214 binding site mutated version of NCKAP1 3′ untranslated region luciferase activity in VSMCs. This result confirmed that NCKAP1 is the functional target of miR‐214 in VSMCs. NCKAP1 knockdown in VSMCs recapitulates the inhibitory effects of miR‐214 overexpression on actin polymerization, cell migration, and proliferation. Data from cotransfection experiments also revealed that inhibition of NCKAP1 is required for miR‐214–mediated lamellipodia formation, cell motility, and growth. Importantly, locally enforced expression of miR‐214 in the injured vessels significantly reduced NCKAP1 expression levels, inhibited VSMC proliferation, and prevented neointima smooth muscle cell hyperplasia after injury.
Conclusions
We uncovered an important role of miR‐214 and its target gene NCKAP1 in modulating VSMC functions and neointima hyperplasia. Our findings suggest that miR‐214 represents a potential therapeutic target for vascular diseases.
Keywords: cell migration, cell proliferation, microRNA, miRNA‐214, NCK‐associated protein 1, neointimal hyperplasia, vascular biology, vascular disease, vascular smooth muscle
Subject Categories: Restenosis, Atherosclerosis, Smooth Muscle Proliferation and Differentiation, Vascular Biology, Proteomics
Introduction
Vascular smooth muscle cells (VSMCs) are the major cellular component of the blood vessels and perform versatile functions (eg, maintaining vascular tone and regulating blood volume and pressure in the circulatory system) under normal physiological conditions. They have a profound dual role (propagating and protective) in the development and progression of atherosclerotic lesions. It has been widely known that VSMC proliferation and migration facilitate early lesion development but are equally important for maintaining plaque stability through the maintenance of a protective fibrous cap overlying the thrombotic lipid core of advanced lesions.1 Consequently, investigating key regulators and signaling pathways governing VSMC proliferation and migration within different developmental stages of atherosclerotic lesions will facilitate and inform future strategies to modulate the disease process.
MicroRNAs (miRNAs) are endogenous, highly conserved, single‐strand, short (20–23 nucleotides), noncoding RNAs. They regulate target gene expression at the posttranscription level by interacting with the 3′ untranslated regions (3′UTRs) of the specific mRNAs.2 The fact that a single miRNA can target and regulate multiple mRNAs, and single mRNA can be targeted and regulated by multiple miRNAs makes miRNAs stand out as the key gene regulators; they control many fundamental biological processes including cell proliferation, migration, differentiation, apoptosis, senescence, and aging.3 Unsurprisingly, miRNAs have been extensively implicated in various human diseases.4 A handful miRNAs, including miR‐1,5 miR‐21,6, 7 miR‐34a,8 miR‐133,9 miR‐221/222,10, 11 the miR‐143/145 cluster,12, 13, 14, 15 miR‐638,16 and miR‐663,17 have been recently reported to play roles in modulating the processes of vascular diseases by regulating various VSMC functions; however, the significance and regulatory roles of other miRNAs in regulating VSMC functions and in the development of vascular diseases are still not fully understood.
In particular, a contradictory but pivotal role of miR‐214 (protective and pathological) in cardiovascular disease and cancer has been suggested based on recent studies. miR‐214 is upregulated in response to several factors including cardiac stress, myocardial infarction, and Ca2+ overload.18, 19 It has been reported that miR‐214 protects myocardial cells against excessive Ca2+ uptake during ischemia–reperfusion injury by repressing mRNA‐encoding sodium/calcium exchanger 1 (Ncx1), thus maintaining Ca2+ homeostasis and increasing cell survival.20 In another study, a decrease in miR‐214 levels was associated with an increase in placental growth factor levels and worsening of atherosclerosis, suggesting its role as a protective agent and a promising biomarker for severe coronary artery disease.21 In contrast, antagonizing (inhibiting) miR‐214 was suggested as a potential new therapeutic approach for treating cardiac hypertrophy or heart failure in another study.22 These studies have provided evidence to suggest a role for miR‐214 in cardiac hypertrophy; importantly, in our previous miRNA microarray study, we found that miR‐214 is one of the most upregulated miRNAs during smooth muscle cell (SMC) differentiation from mouse embryonic stem cells.23 Less is known about the functional involvement of miR‐214 and its target gene in regulating VSMC functions and modulating neointima SMC hyperplasia. In this study, we demonstrated that miR‐214 inhibits VSMC proliferation and migration, at least in part, by targeting NCK associated protein 1 (NCKAP1). Consequently, restoring expression of miR‐214 in the diseased blood vessels would be a potential therapeutic approach for treatment of proliferative vascular diseases.
Materials and Methods
Materials
Antibody against NCKAP1 (goat, N‐12, sc‐161124) was purchased from Santa Cruz Biotechnology. The anti‐NCKAP1 antibody used for paraffin‐section staining was purchased from Antibodies‐online GmbH. Antibodies against GAPDH (mouse), Ki‐67 (rabbit), and proliferating cell nuclear antigen (PCNA; rabbit) were from Abcam. All secondary antibodies were from Dako. Other materials including Phalloidin‐FITC (P5282) used in this study were purchased from Sigma‐Aldrich unless specifically indicated.
VSMC Culture and Treatments
Primary murine VSMCs were isolated from mouse aorta and routinely maintained in DMEM supplemented with 10% FBS, as described in our previous study.8, 24 Human aortic SMCs were purchased from PromoCell GmbH (C‐12533) and cultured in SMC growth medium 2 (C‐22062; PromoCell GmbH), according to the manufacturer's instructions. VSMCs between passages 5 and 10 were used in the current study. VSMCs were treated with various atherogenic stimuli, as described in the previous studies.9, 25, 26, 27, 28 Briefly, for platelet‐derived growth factor BB (PDGF‐BB; BioLegend) and serum stimulation, VSMCs were serum starved for 24 to 48 hours (0.5% FBS), followed by an incubation with 20% FBS and 10 ng/mL PDGF‐BB for 3, 6, 12, 24, and 48 hours. For oxidized low‐density lipoprotein component treatments, VSMCs were serum starved for 24 to 48 hours, followed by incubation with 10 μmol/L 4‐hydroxynonenal and 7‐ketocholesterol for 24 hours.
miRNA Mimics and Inhibitor Transfection
miRNA mimics and inhibitor transfection were conducted as described previously with some modifications.8, 23, 29, 30 Briefly, either miRNA mimics or inhibitors and miRNA‐negative controls (25 nmol/L, final concentration) were transfected into VSMCs using TransIT‐X2 transfection reagent (Geneflow Ltd), according to the manufacturer's instructions. VSMCs (1.5×105 per well) were seeded into 6‐well plate 24 hours prior to transfection. Before transfection, cells were washed with 1× PBS once and replenished with 1.75 mL fresh culture medium containing 5% FBS. TransIT‐X2 reagent was warmed to room temperature. Next, 250 µL of serum‐free DMEM was added to a sterile Eppendorf tube, and 5 µL of miR‐214 mimics/inhibitor or their respective control scramble miRNA mimics/inhibitor (10 μmol/L in stock) was added and mixed using a pipette. We added 7.5 µL of TransIT‐X2 reagent and mixed gently. This was left to incubate at room temperature for 15 to 30 minutes to allow the complexes to form. The TransIT‐X2:miRNA complexes were added drop‐wise in circular motions to ensure that all cells were covered by the mixture. The 6‐well plate was rocked back and forth and side to side to evenly distribute the TransIT‐X2 and miRNA mimics/inhibitor complexes onto the cultured cells. The transfected cells were cultured for 16 to 24 hours prior to medium change or serum starvation. All miRNA inhibitors or mimics and respective negative controls were purchased from Sigma‐Aldrich.
Generation of NCKAP1 Short Hairpin RNA Lentivirus and NCKAP1 Stable Knockdown Cell Lines
NCKAP1 short hairpin RNA (shRNA) lentiviral particles were produced using MISSION shRNA NCKAP1 plasmids DNA (SHCLNG‐NM_016965, MISSION shRNA Bacterial Glycerol Stock; Sigma‐Aldrich) according to protocol provided. The shRNA nontargeting control vector (SHC002) was used as a negative control. Briefly, 293T cells were transfected with the lentiviral vector and the packaging plasmids pCMV‐dR8.2 and pCMV‐VSV‐G (both obtained from Addgene) using TransIT‐X2 transfection reagent (Geneflow Ltd), according to the manufacturer's instructions. The supernatant containing the lentivirus was harvested 48 hours later, filtered, aliquoted, and stored at −80°C. The p24 antigen ELISA (Zeptometrix) was used to determine the viral titer. The transducing unit was calculated using the conversion factor recommended by the manufacturer (104 physical particles per pg of p24 and 1 transducing unit per 103 physical particles for a VSV‐G pseudotyped lentiviral vector), with 1 pg of p24 antigen converted to 10 transducing units. shRNA lentiviral infection and NCKAP1 stable knockdown cell line generation were performed as described in our previous studies with some modifications.31, 32, 33 Briefly, VSMCs were plated 24 hours prior to infection in 6‐well plates at 37°C. One transducing unit per cell (or 2–3×105 per well) of shRNA or control virus was added with 10 μg/mL hexadimethrine bromide (H9268; Sigma‐Aldrich). Viral constructs were incubated for 24 hours with the cells before the media was replaced with complete media containing 4 μg/mL puromycin (P9620; Sigma‐Aldrich). For selection of transductants, fresh media containing puromycin was added at 2‐ to 3‐day intervals for 10 days. Stably infected cells were split and frozen for future experiments.
Cotransduction of NCKAP1 shRNA Lentivirus and miR‐214 Inhibitor
For miR‐214 inhibitor and NCKAP1 shRNA lentivirus cotransfection, VSMCs infected with nontarget or NCKAP1 shRNA lentivirus were transfected with miR‐214 inhibitor or control miRNA inhibitor (25 nmol/L), as indicated in the Figure 6, using TransIT‐X2 transfection reagent (Geneflow Ltd), according to the manufacturer's instructions. All procedures were same as the single‐miRNA transfection except that the VSMCs were infected with nontarget or NCKAP1 shRNA virus, as described in the previous section.
Figure 6.
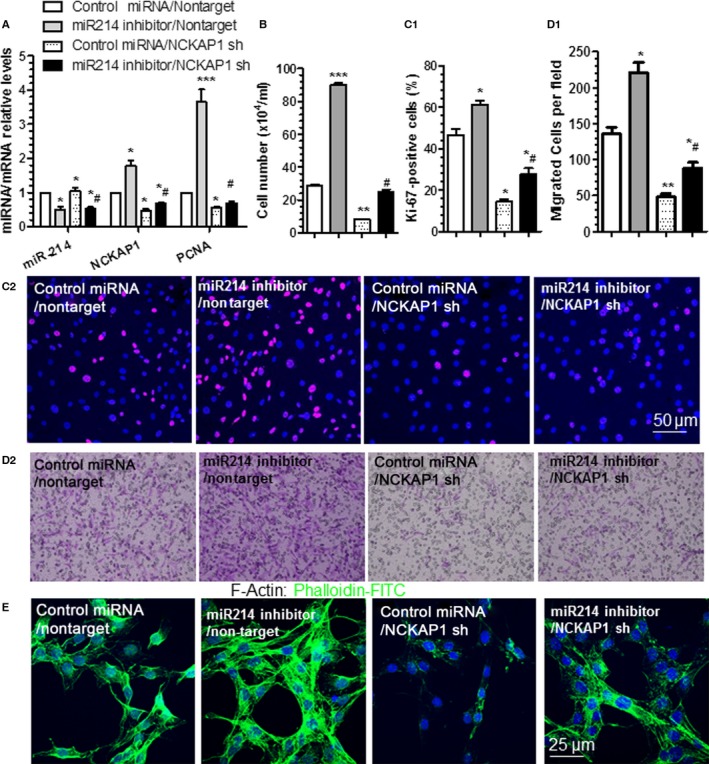
miR‐214 mediates vascular smooth muscle cell (VSMC) growth and motility by modulating NCK associated protein 1 (NCKAP1). VSMCs were cotransduced with miR‐214 inhibitor, NCKAP1 short hairpin RNA (shRNA) lentivirus (NCKAP1 sh), and/or respective controls (control microRNA [miRNA] or nontarget shRNA [nontarget]), as indicated. Transduced cells were serum‐starved for 24 hours and subjected to reverse transcriptase–quantitative PCR analyses to examine miR‐214, NCKAP1, and proliferating cell nuclear antigen (PCNA) expression levels (A), cell counting (B), immunofluorescence staining with antibody against Ki‐67 (C1 and C2), transwell migration (D1 and D2), and F‐actin staining using phalloidin‐FITC (E) in response to serum (A through C and E) or 30 ng/mL platelet‐derived growth factor BB (D) stimulation. The data presented are representative images (C2, D2 and E) or mean+SEM (A, B, C1 and D1) of 3 independent experiments. *<0.05, **<0.01, ***<0.001 (Compared various treatments with double control), #<0.05 (Compared NCKAP1 knockdown with control in VSMCs with miR‐214 inhibition).
NCKAP1 3′UTR Clone and miR‐214 Binding Site Mutation
Reporter vector harboring sequences of the murine NCKAP1 was created using cDNA from VSMCs. The flanking 3′UTR (3605nt–4403nt) of the murine NCKAP1 gene (NM_016965.3) (Figure S1A) was amplified by polymerase chain reaction (PCR) with primers shown in Table S1 and cloned into the Sac I and Hind III sites of the pmiR‐reporter‐basic vector (Thermo Fisher Scientific Inc), designated as pmiR‐Luc‐NCKAP1‐WT.
The miR‐214 binding site 1, 2, or 3 mutations alone or combination were introduced into pmiR‐Luc‐NCKAP1‐WT using the QuikChange site‐directed mutagenesis kit (Agilent Technologies), according to the manufacturer's instructions. These were designated as pmiR‐Luc‐NCKAP1‐BS1mut, pmiR‐Luc‐NCKAP1‐BS2mut, pmiR‐Luc‐NCKAP1‐BS3mut, and pmiR‐Luc‐Notch1‐BS1/2/3mut mutants, respectively.
All vectors were verified by DNA sequencing.
Generation of KLF14 and SMYD5 3′UTR Reporters and Mouse miR‐214 Gene Promoters
Reporter vectors harboring sequences of the murine Krüppel‐like factor 14 (KLF14) and SET and MYND domain containing 5 (SMYD5) were created using cDNA from VSMCs. The flanking 3′UTR of the murine KLF14 gene (NM_001135093; 3′UTR: 1272nt–2970nt) or SMYD5 (NM_144918; 3′UTR: 1281nt–2491nt) gene was amplified by PCR with primers shown in Table S1 and cloned into the Sac I and Hind III sites of the pmiR‐reporter‐basic vector (Thermo Fisher Scientific Inc), designated as pmiR‐Luc‐KLF14 and pmiR‐Luc‐SMYD5, respectively.
The reported functional full length (−640:0) and the truncated (−640:−357) form of miR‐214 gene promoters34 were recovered from mouse genomic DNA by PCR using the respective primers shown in Table S1. Amplified DNA fragments were cloned into the Sac I and Hind III sites of the pGL3‐basic vector (Promega), designated as pGL3‐miR‐214_FL and pGL3‐miR‐214‐short, respectively.
All vectors were verified by DNA sequencing.
Transient Transfection and Luciferase Assay
Luciferase assays for various gene 3′UTR reporters were conducted as described in our previous studies.8, 23, 29, 30, 32 Briefly, VSMCs were cotransfected with an individual reporter gene (pmiR‐Luc‐NCKAP1‐WT, pmiR‐Luc‐NCKAP1‐BS1mut, pmiR‐Luc‐NCKAP1‐BS2mut, pmiR‐Luc‐NCKAP1‐BS3mut, pmiR‐Luc‐Notch1‐BS1/2/3mut, pmiR‐Luc‐KLF14, pmiR‐Luc‐SMYD5, or pmiR‐Luc‐Notch1; 0.15 μg/2.5×104 cells) and control or miR‐214 (or miR‐34a) mimics (25 nmol/L) using TransIT‐X2 transfection reagent (Geneflow Ltd), according to the manufacturer's instructions. pmiR‐Luc‐β‐gal (0.20 μg/2.5×104 cells) or Renilla plasmid (15 ng/well) was included in all transfection assays as an internal control. Luciferase, Renilla, and/or β‐galactosidase activities were detected 48 hours after transfection using a standard protocol. The relative luciferase unit was defined as the ratio of luciferase versus β‐galactosidase or Renilla activity with that of the control (set as 1.0).
VSMC Proliferation Assays
Cell counting
Cell counting was conducted as described previously.8 VSMCs were plated (1×105 per well) and cultured in 6‐well plates precoated with 0.04% gelatin and supplemented with complete culture medium containing DMEM, 10% FBS, and 1% penicillin/streptomycin‐glutamine. The plates were placed in humidified incubators at 37°C and 5% CO2. After culturing for 24 hours, the cells were transfected with single miR‐214 mimics/inhibitor or cotransduced with miR‐214 inhibitor/NCKAP1 shRNA or respective negative control, as indicated in the Figures 2, 3, 5 and 6. After 12 to 16 hours of transfection, the cells were starved by culturing them in the DMEM supplemented with 1% penicillin/streptomycin‐glutamine and 0.5% serum for another 24 hours. After the starvation process, the cells were treated with 20% FBS or PDGF‐BB (10 ng/mL) for 48 hours before trypsinizing and manually counting the cells under a hematocytometer.
Figure 2.
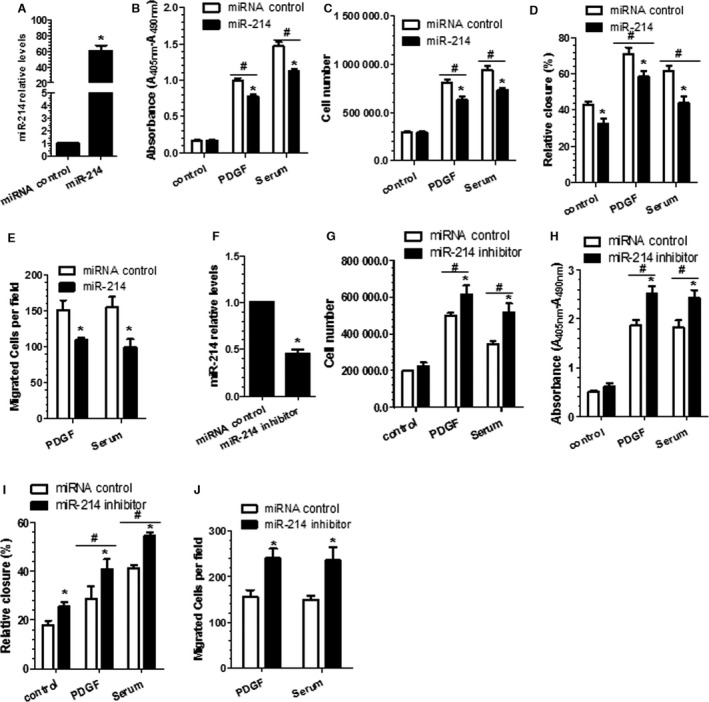
Vascular smooth muscle cell (VSMC) proliferation and migration are modulated by miR‐214. miR‐214 mimics (miR‐214) (A through E) and inhibitor (miR‐214 inhibitor) (F through J) or respective control microRNAs (miRNAs) were transfected into VSMCs, followed by 24 hours of serum starvation. Serum‐starved cells were treated with platelet‐derived growth factor BB (PDGF‐BB) or serum for another 48 hours, followed by bromodeoxyuridine incorporation assays (B and H) or cell counting (C and G). In another setting of experiments, serum‐starved cells were subjected to wound‐healing assays (D and I) or transwell migration analyses (E and J) in response to PDGF‐BB or serum stimulation for another 24 hours, respectively. Meanwhile, cells were harvested to examine the miR‐214 expression levels by reverse transcriptase–quantitative PCR analyses (A and F). Note that in the transwell migration experiments, only a few cells were migrated through the insert without any chemoattractant. In the wound‐healing assays, the percentage of cell closure or migrated area was calculated as described in the Materials and Methods section. Data are presented as mean+SEM of 3 to 4 independent experiments (n=3–4). *<0.05 (miR‐214 mimics or inhibitor vs miRNA control within respective treatment). #<0.05 (Compared PDGF/serum with control, with or without miR‐214 modulation).
Figure 3.
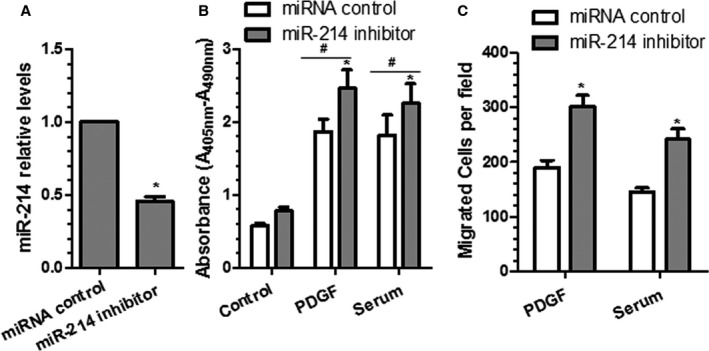
Inhibition of miR‐214 increases human vascular smooth muscle cell (VSMC) proliferation and migration. miR‐214 inhibitor or control microRNA (miRNA) inhibitor were transfected into human aorta smooth muscle cells, followed by 24 hours of serum starvation. Serum‐starved cells were treated with platelet‐derived growth factor BB (PDGF‐BB) or serum for another 48 hours, followed by reverse transcriptase–quantitative PCR analyses to examine the miR‐214 expression levels (A) and bromodeoxyuridine incorporation assays (B), respectively. In another set of experiments, serum‐starved cells were subjected to transwell migration analyses (C) in response to 30 ng/mL PDGF‐BB or 20% serum stimulation for another 24 hours. Mean+SEM of 3 independent experiments (n=3) is presented. *<0.05 (miR‐214 inhibitor vs miRNA control within respective treatment); # <0.05 (PDGF/serum vs control, with or without miR‐214 modulation).
Figure 5.
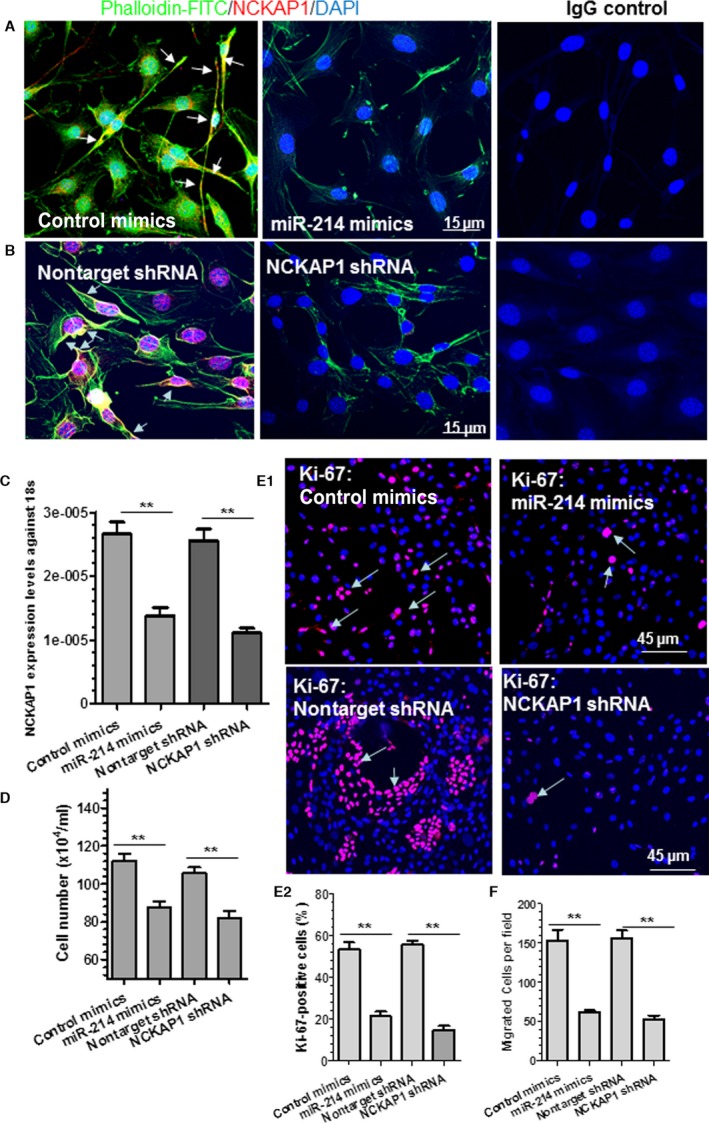
NCK associated protein 1 (NCKAP1) knockdown in vascular smooth muscle cells (VSMCs) recapitulates the effects of miR‐214 overexpression on actin polymerization, cell migration, and proliferation. A, Actin polymerization in VSMCs was inhibited by miR‐214 overexpression. VSMCs were transfected with control or miR‐214 mimics, followed by serum starvation for 24 hours. Serum‐starved cells were stimulated with 20% serum for another 24 hours and subjected to immunofluorescence staining with antibody against NCKAP1 and phalloidin‐FITC (F‐actin staining). B and C, NCKAP1 knockdown decreased actin polymerization in VSMCs. VSMCs were infected with nontarget or NCKAP1 short hairpin RNA (shRNA) virus, followed by similar treatment and analyses, as described in (A). Representative images (A and B) or mean+SEM (C) from 3 independent experiments are presented. White arrows (A and B) indicate that NCKAP1 colocalizes with F‐actin within lamellipodia and membrane ruffles, which face the direction of cell movement. D through F, miR‐214 overexpression or NCKAP1 knockdown inhibited VSMC proliferation (D and E) and migration (F). VSMCs were transfected with control miRNA mimics or miR‐214 mimics or infected with nontarget or NCKAP1 shRNA virus, followed by serum starvation for 24 hours. Serum‐starved cells were stimulated with 20% serum for another 24 hours and subjected to cell counting (D) and immunofluorescence staining with antibody against Ki‐67 (E). E1, representative images; E2, quantitative data (mean+SEM) from 3 independent experiments. In another set of experiments, serum‐starved cells were subjected to transwell migration assays in the presence of 30 ng/mL platelet‐derived growth factor BB (F). Bar graphs show quantitative data (mean+SEM) from 3 independent experiments (n=3). **<0.01 (Compared with control mimics or nontarget shRNA). DAPI, 4′,6‐diamidino‐2‐phenylindole; IgG, immunoglobulin G.
Bromodeoxyuridine incorporation assay
The 5‐bromo‐2′‐deoxy‐uridine (BrdU) incorporation assays were conducted as described previously.8 VSMCs were transfected as described earlier and were recultured (0.75×104 per well) in 96‐well plates overnight, followed by serum starvation for 24 hours. Starved VSMCs were restimulated with 20% FBS or 10 ng/mL PDGF‐BB, respectively, for 48 hours. Cell proliferations were evaluated using the BrdU Labeling and Detection Kit II (Roche), according to the manufacturer's instructions. Briefly, cells were incubated with BrdU at a final concentration of 10 μmol/L for 8 to 12 hours before measurement. After fixation, cellular DNA was digested by nuclease and labeled with a peroxidase‐conjugated BrdU antibody, followed by incubation with the peroxidase substrate. The absorbance of the samples was measured by a microplate reader at 405 nm (OD405) with reference measurement at 490 nm (OD490). Absorbance (A405 nm−A490 nm) values representing cell‐proliferation ability were compared among treatments.
VSMC Migration Assays
Wound healing (scratch model)
Scratch wound healing assays were carried out using a previously described method.8, 35 In brief, VSMCs were cultured on 12‐well plates overnight and transfected with miR‐214 mimics, miR‐214 inhibitor, or respective miRNA‐negative control, as described earlier. After 12 to 16 hours of transfection, the confluent cells were starved by culturing them in the DMEM supplemented with 1% penicillin/streptomycin‐glutamine and 0.5% serum for another 24 hours. After the starvation process, the cells were treated with hydroxyurea (2 mmol/L) to inhibit cell proliferation for 2 hours before subjecting them to 20% FBS or PDGF‐BB (10 ng/mL) treatment. The cells were scratched in a crisscrossed manner and rinsed with PBS or DMEM 3 times to remove cell debris, followed by cultured in DMEM supplemented with 20% FBS or PDGF‐BB (10 ng/mL) in the presence of 2 mmol/L hydroxyurea. The observations were made, and photomicrographic images were taken at 0 and 24 hours. Two experienced investigators blinded to the treatments used ImageJ software (National Institutes of Health) to measure the denuded cell surface of each wound (crisscrossed), and the percentages of cell closures (migrated area) were calculated as the difference in denuded area between hour 0 (A0) and hour 24 (A24) over the denuded area at hour 0 ×100: (A0−A24)/A0×100.
Transwell migration assay
Transwell migration assay was conducted as described previously.8 VSMCs were transfected with miRNA (mimics or inhibitor), NCKAP1 shRNA, and/or their respective control, as indicated in the Figures 2 3, 5 and 6. Transfected cells were cultured in DMEM containing 0.5% serum for 24 hours and harvested by trypsinization. An aliquot (250 000 cells/200 μL) of the cells in serum‐free DMEM was dispensed into the transwell inserts (8‐μm pore size, item number 662638; Greiner Bio‐One Ltd) precoated with 0.5% gelatin (G1393; Sigma‐Aldrich), and DMEM (600 μL) with 20% FBS or 30 ng/mL PDGF‐BB was placed in the lower chamber. The transwell plates were incubated at 37°C in a 5% CO2 incubator for 18 to 24 hours. Nonmigrated cells in the top insert were carefully removed by cotton swab, and the migrated cells in the bottom side were stained with crystal violet dye. Images were captured at 5 fixed locations (right, bottom, left, up, and center), and migrated cells were counted by 2 experienced investigators blinded to the treatments.
Immunoblotting
Cells were harvested and lysed in lysis buffer (50 mmol/L Tris‐Cl, pH 7.5; 150 mmol/L NaCl; 1 mmol/L EDTA, pH 8.0) supplemented with protease inhibitors and 0.5% Triton and sonicated to obtain whole‐cell lysate. Next, 40 µg of protein was separated by SDS‐PAGE with 4% to 20% Tris‐Glycine gel (Invitrogen) and subjected to standard Western blot analysis. In some experiments, the blots were subjected to densitometric analysis with ImageJ software. Relative protein expression level was defined as the ratio of target protein expression level to GAPDH expression level with that of the control sample set as 1.0.
Indirect Immunofluorescent Staining for Cells
For detecting Ki‐67–positive cells, cells subjected to various treatments were fixed and labeled with antibody against Ki‐67 (Abcam) or rabbit isotype immunoglobulin G control and visualized using a donkey anti–rabbit immunoglobulin G antibody conjugated with CF 568 fluorescence (in 1:400 dilution; Sigma‐Aldrich). For phalloidin‐FITC staining alone, another setting of cells with different treatments, as indicated in the Figures 5, 6, was fixed and labeled with phalloidin‐FITC (P5282), according to the manufacturer's instructions. For double‐immunofluorescent staining with NCKAP1 and phalloidin‐FITC, a separate setting of cells with different treatments, as indicated in the Figures 5, 6, was fixed and labeled with anti‐NCKAP1, or rabbit isotype immunoglobulin G control. Next, cells were visualized using a donkey anti–rabbit immunoglobulin G antibody conjugated with CF 568 fluorescence (in 1:400 dilution; Sigma‐Aldrich) and phalloidin‐FITC (P5282), according to the manufacturer's instructions. Cells were counterstained with DAPI (4′,6‐diamidino‐2‐phenylindole; Sigma‐Aldrich) and mounted in Fluoromount‐G (Cytomation; Dako). Images were examined using an SP5 confocal microscope and Leica TCS Sp5 software (Leica) at room temperature and were processed with Photoshop software (Adobe).
Reverse Transcriptase–Quantitative PCR for mRNA and miRNAs
Real‐time quantitative PCR (RT‐qPCR) was performed as described previously.8, 23, 29, 30, 36 Briefly, total RNAs containing small RNAs (miRNAs) were extracted from cells using the mirVana Protein and RNA Isolation System Kit (Thermo Fisher Scientific Inc) or TRI reagent (Sigma‐Aldrich), according to the manufacturer's instructions, and subjected to DNase I (Sigma‐Aldrich) digestion to remove potential DNA contamination. Reverse transcription for long RNA was performed using an Improm‐II RT kit (Promega) with RNase inhibitor (Promega) and Random primers (Promega). The NCode VILO miRNA cDNA Synthesis Kit (A11193‐051; Thermo Fisher Scientific Inc) was used to synthesize poly(A) tails of all the miRNAs followed by cDNA synthesis from the tailed population in a single reaction. The resultant cDNA was diluted to a working concentration of 5 ng/μL and stored at −20°C. NCode EXPRESS SYBR GreenER qPCR SuperMix Universal (Thermo Fisher Scientific Inc) was used in miRNA RT‐qPCR. Relative mRNA or miRNA expression level was defined as the ratio of target gene expression level or miRNA expression level to 18S or U6 small nuclear RNA expression level, respectively, with that of the control sample set as 1.0. In some experiments, relative mRNA or miRNA expression level was defined as the ratio of target gene expression level or miRNA expression level to 18S or U6 small nuclear RNA expression level, respectively. Primers were designed using Primer Express software (Thermo Fisher Scientific Inc); and the sequence for each primer is listed in Table S1.
Proteomics Studies
Sample preparation for proteomic analysis
VSMCs transfected with control or miR‐214 mimics were directly lysed in a urea‐based lysis buffer (8 mol/L urea in 20 mmol/L HEPES, pH 8.0; 1 mmol/L Na3VO4, 1 mmol/L NaF, 0.5 mmol/L β‐glycerol phosphate, and 2.5 mmol/L Na2H2P2O7), and proteins were digested using trypsin, as reported previously.37, 38 The enriched peptides were subjected to the following analyses, as described in our previous study.39, 40
Mass spectrometry
Protein‐derived peptides were analyzed by the LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific Inc) coupled with a nanoAcquity liquid chromatography system (Waters). Peptide separation was performed using solution A (0.1% formic acid in liquid chromatography and mass spectrometry [MS]–grade water) and solution B (0.1% formic acid in liquid chromatography–MS acetonitrile) as mobile phases. Gradient runs from 2% to 30% solution B in 100 minutes and from 30% to 60% in 10 minutes were followed by a final 10‐minute wash at 85% solution B.
Full MS scans were acquired in the Orbitrap mass analyzer over the range m/z 375 to 1500 with a mass resolution of 30 000. Tandem MS (MS/MS) was acquired using top 7 data‐dependent acquisition with high‐energy collision dissociation (40%). The gas‐phase fractionation method was applied to acquire MS/MS scans.
Peptide identification by database search
MS/MS data were converted to Mascot generic format files using Mascot Distiller (version 2.2; Matrix Science) and searched against the 2012_03 databases of UniProt and SwissProt using the Mascot search engine (version 2.2). Significance of peptide identification was assessed by comparing results returned by searches against random and forward databases. Fold discovery rates at several cutoff values of Mascot scores and mass tolerances were used to calculate an empirical value of probability of random identification.
Data analysis and volcano plot analysis
Relative quantification of peptides across experimental conditions was achieved by comparing peak heights of extracted ion chromatograms (automated by Pescal), as described previously.37, 40 The data were normalized to the sum of all intensities derived from a sample (columns). When comparing the effects of miR‐214 overexpression on protein regulation, peptide signals were divided by those of the untreated control samples (control miRNA mimics). The P values of differences across treatments were obtained by means of a t test of log2‐transformed fold changes, and these were adjusted for multiple testing through the Benjamini–Hochberg procedure, as described previously.37 The fold change was transformed using the log2 function, so the data are centered around zero, whereas the Benjamini–Hochberg corrected P value was −log10 transformed for volcano plot analysis.
Mouse Femoral Artery Denudation Injury and miR‐214 Agomir Perivascular Delivery
C57BL/6 mice were anesthetized, and the surgical procedure was similar to that described previously.8, 24, 41, 42 Removal of the endothelium of the femoral arteries was achieved by 3 to 5 passages of a 0.25‐mm angioplasty spring wire (tips of cross‐IT 200× guide wire; Abbott Laboratories). After the vascular injury, the injured femoral arteries were randomly received miR‐214 or Cel‐miR‐67 agomir treatments, as described in our previous study.8 Briefly, after injury, 100 μL of 30% pluronic gel containing chemically modified and cholesterol‐conjugated 2.5 nmol miR‐214 or scramble (Cel‐miR‐67) agomirs was applied perivascularly to injured femoral arteries. The micrON miRNA agomirs were purchased from RiboBio (Guangzhou RiboBio Co, Ltd). The in vivo expression efficiency and stability of such agomirs have been documented extensively by many research groups worldwide.8, 43, 44, 45, 46 Additional femoral arteries were harvested at 3 days (for gene expression) or 14 days (for protein expression) after injury (3–5 femoral arteries from each group were pooled for each independent experiment, and triplicate experiments were conducted). Total RNAs including small RNAs and proteins were extracted for RT‐qPCR or Western blotting analysis of miR‐214, NCKAP1, or PCNA gene/protein expression in injured vessels. Our previous study8 showed that perivascular delivery of 2.5 nmol agomirs into each injured vessel generally resulted in 5 to 10 times higher expression levels compared with control mice (received Cel‐miR‐67 agomirs) or normalized target miRNA expression levels in injured arteries to levels similar to those in normal uninjured vessels. All animal experiments were performed according to the Animals (Scientific Procedures) Act of 1986 (United Kingdom), and all protocols were approved by the Institutional Committee for Use and Care of Laboratory Animals. In addition, the principles governing the care and treatment of animals, as stated in the Guide for the Care and Use of Laboratory Animals published by the National Academy of Sciences (8th ed., 2011), were followed at all times during this study. All mice were euthanized by placing them under deep anesthesia with 100% O2 and 5% isoflurane, followed by decapitation.
Morphometric Analysis and Quantification of Lesion Formation
All procedures used in this study were similar to those described in our previous study.8 Briefly, the femoral arteries (≈1.0 mm from injury site) were harvested 4 weeks after the operation. The specimens were fixed in 4% formaldehyde for hematoxylin and eosin staining. Sections (8 μm) were collected at 100‐μm intervals (10 sections per interval), mounted on slides, and numbered. Six digitized sections with same identification number from 3 segments/intervals (≈0.4, 0.5, and 0.6 mm from injury site) of each animal (eg, IV‐1/2, V‐1/2, VI‐1/2 represent the first and second sections of the fourth, fifth, and sixth segment/interval, respectively) were stained with hematoxylin and eosin for morphometric analysis. The procedure used for lesion quantification was similar to that described previously.8, 24, 41, 42 Briefly, external elastic membrane, internal elastic membrane, lumen, media, and neointimal areas were automatically measured on hematoxylin and eosin–stained cross‐sectional femoral artery segments using a computerized image analysis system (pixel2, a unit of area measurement, AxioVision software; Zeiss) by 2 experienced investigators blinded to the treatments. Six sections were analyzed per vessel sample and averaged.
Statistical Analysis
Results are presented as Mean±SEM. Statistical analysis was performed using GraphPad Prism5 (GraphPad Software). The Shapiro–Wilk normality test was used for checking the normality of the data. Data with a Shapiro–Wilk test P>0.05 was considered to fit a normal distribution. A 2‐tailed unpaired Student t test was used for comparisons between 2 groups, and a 1‐way ANOVA test with a Bonferroni post hoc test was applied when >2 groups were compared if the data displayed a normal distribution. An α=0.05 was chosen as the significance level, and P values of <0.001, <0.01, and <0.05 were considered statistically significant.
Results
miR‐214 Expression in VSMCs is Significantly Downregulated by Various Pathogenic Stimuli
Extensive studies9, 25, 26, 27, 28 have suggested that VSMC proliferation and migration can be promoted by various pathogenic stimuli including high concentration of serum (20%), PDGF‐BB, oxidized low‐density lipoprotein, or its components such as 4‐hydroxynonenal and 7‐ketocholesterol.47 To study whether miR‐214 expression was regulated on such atherogenic stimulation, serum‐starved VSMCs were treated with various stimuli and harvested at different time intervals. As expected, both PDGF‐BB and 20% serum significantly inhibited miR‐214 expression from 3 to 24 hours of treatment compared with expression of untreated cells (at 0 hour) (Figure 1A). Although not reaching a significant level, a decreasing trend was also observed at 48 hours with both treatments (Figure 1A). A similar effect was observed with 4‐hydroxynonenal and 7‐ketocholesterol treatment (Figure 1C), suggesting a role for miR‐214 in VSMC proliferation and migration.
Figure 1.
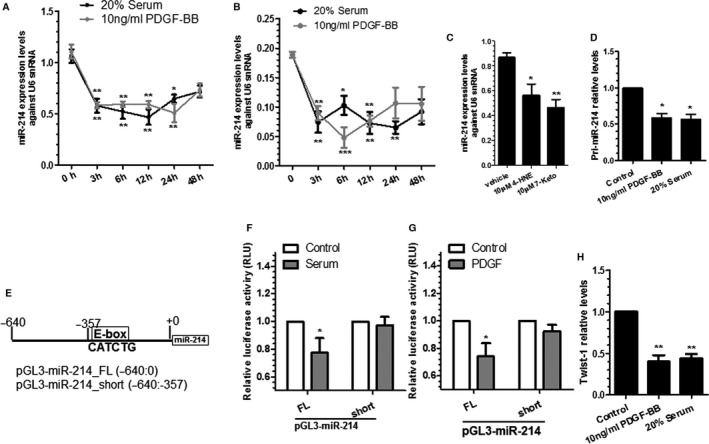
Transcriptional inhibition of miR‐214 by different pathogenic stimuli in vascular smooth muscle cells (VSMCs). A through C, MicroRNA miR‐214 is regulated by various pathogenic stimuli. Murine VSMCs (A and C) and human aorta smooth muscle cells (hAoSMCs) (B) were subjected to serum starvation for 24 hours, followed by different stimuli, as indicated. Total RNAs were harvested at indicated times (10 ng/mL platelet‐derived growth factor BB [PDGF‐BB]/20% serum) (A and B) or at 24 hours for 4‐hydroxynonenal (4‐HNE) and 7‐ketocholesterol (7‐Keto) treatments (C) and subjected to reverse transcriptase–quantitative PCR (RT‐qPCR) analyses. Data are presented as mean±SEM (circles and error bars, respectively) in (A and B) or as mean+SEM (bars plus error bars) in (C) of 5 independent experiments (n=5). *<0.05, **<0.01, and ***<0.001; different treatments were compared with 0 hour (A and B) or vehicle (C). D, Serum and PDGF‐BB downregulate miR‐214 primary transcript. Serum‐starved VSMCs were incubated with serum and PDGF‐BB for 3 hours. Total RNAs were harvested and subjected to RT‐qPCR analyses. E, A schematic diagram of mouse miR‐214 gene promoter reporters. F and G, The E‐box element is required for miR‐214 gene inhibition by serum and PDGF‐BB. VSMCs transfected with respective reporters harboring the full length (pGL3‐miR‐214‐FL; −640:0) or truncated form (pGL3‐miR‐214‐short; −640:−357, lacking E‐box element) of the miR‐214 gene promoter were subjected to serum starvation for 24 hours, followed by incubation with 20% serum (F) or 10 ng/mL PDGF‐BB (G) for 3 hours, respectively. Cell lysate was harvested and subjected to luciferase activity assays. H, Twist‐1 expression is inhibited by serum and PDGF‐BB in VSMCs. cDNA samples generated in (D) were used to examine Twist‐1 expression level by RT‐qPCR. Data are presented as mean+SEM (bars plus error bars) of 3 (D and H) or 6 (F and G) independent experiments (n=3 or n=6). *<0.05, **<0.01, and ***<0.001, compared with vehicle control. FL indicates full length; RLU, relative luciferase unit; snRNA, small nuclear RNA.
To further study whether serum and PDGF‐BB downregulate miR‐214 expression in human VSMCs, similar experiments were conducted in human aortic SMCs. RT‐qPCR data showed that miR‐214 expression levels were significantly downregulated by both serum and PDGF‐BB in human aortic SMCs (Figure 1B), confirming a similar regulatory effect of serum and PDGF‐BB on miR‐214 expression in human VSMCs.
PDGF‐BB and Serum Downregulates miR‐214 in VSMCs By Inhibiting the Transcription Factor Twist‐1
It is well known that miRNA synthesis and maturation is under 2 molecular controls: transcription and biogenesis. To differentiate these 2 mechanisms, we initially conducted RT‐qPCR analyses with specific primers to examine the miR‐214 primary transcript. RT‐qPCR data showed that the expression level of primary miR‐214 was significantly downregulated by serum and PDGF‐BB (Figure 1D), indicating that miR‐214 was regulated by PDGF‐BB and serum at the transcriptional level. It has been reported that miR‐214 expression is regulated by transcription factor Twist‐1 via an E‐box promoter element during development34; therefore, we wondered if a similar mechanism were responsible for miR‐214 inhibition by serum and PDGF‐BB. To this end, we generated 2 mouse miR‐214 gene promoters, pGL3‐miR‐214‐FL and pGL3‐miR‐214‐short (Figure 1E), using a strategy similar to that reported by Lee et al.34 Data from luciferase activity assays showed that the luciferase activity was significantly decreased by serum (Figure 1F) and PDGF‐BB (Figure 1G) in VSMCs transfected with pGL3‐miR‐214‐FL, whereas no such inhibitory effect was observed in VSMCs transfected with pGL3‐miR‐214‐short reporter lacking the E‐box. These data demonstrate that various pathogenic stimuli regulate miR‐214 expression in VSMCs through a transcriptional mechanism, and the E‐box element is required for such regulation. As expected, the expression level of Twist‐1 was dramatically downregulated by serum and PDGF‐BB (Figure 1H), suggesting that these 2 pathogenic stimuli downregulate miR‐214 expression in VSMCs by inhibiting the transcription factor Twist‐1.
VSMC Proliferation and Migration Are Inhibited by miR‐214
VSMC proliferation and growth and accumulation within intima have been recognized as major events during the early stage of atherosclerotic lesion formation and postangioplasty restenosis. We wondered if miR‐214 could play a role in VSMC proliferation. For such a purpose, VSMCs were transfected with miR‐214 mimics and subjected to BrdU incorporation analyses and cell counting, respectively, to evaluate the potential effects of miR‐214 overexpression on VSMC proliferation. As expected, miR‐214 expression in VSMCs was significantly increased by miR‐214 mimics (Figure 2A). Importantly, data from BrdU incorporation assays (Figure 2B) and cell counting (Figure 2C) showed that serum‐ and PDGF‐BB–induced VSMC proliferation was significantly inhibited by miR‐214 overexpression. To further confirm the role of miR‐214 in VSMC growth, miRNA loss‐of‐function experiments were conducted in VSMCs cultured under similar conditions using miR‐214 inhibitor. Data shown in Figure 2F revealed that the endogenous miR‐214 expression level in VSMCs was successfully inhibited by miR‐214 inhibitor. Consistent with the findings from miR‐214 gain‐of‐function experiments, miR‐214 inhibition in VSMCs significantly increased PDGF‐BB– and serum‐induced cell proliferation, respectively (Figure 2G and 2H), suggesting that miR‐214 plays an important role in VSMC growth.
VSMC migration into intima is another key determining factor for atherosclerotic lesion formation and postangioplasty restenosis. To examine whether miR‐214 also plays a role in VSMC migration, VSMCs were transfected with miR‐214 mimics, inhibitor, or their respective negative controls, followed by cell migration assays in response to serum or PDGF‐BB stimulation, respectively. Decreased cell migratory capacity was observed in the VSMCs transfected with miR‐214 mimics in response to both PDGF‐BB and serum stimulation, as demonstrated in wound healing (Figure 2D) and transwell migration assays (Figure 2E), respectively, compared with the control cells. As expected, the migratory ability of cells transfected with miR‐214 inhibitor was clearly greater than that of control cells (Figure 2I and 2J), supporting a role for miR‐214 in regulating VSMC migration.
To investigate whether miR‐214 also plays a pathological role in human VSMCs, miR‐214 inhibitor and control miR inhibitor were transfected into human aortic SMCs. Our data showed that human aortic SMC proliferation and migration were significantly enhanced by miR‐214 inhibition (Figure 3A) in response to serum or PDGF‐BB stimulation, as demonstrated in BrdU incorporation (Figure 3B) and transwell migration (Figure 3C) assays, respectively, inferring a role for miR‐214 in human pathology.
Proteomics Analysis to Uncover the Potential Target Genes of miR‐214 in VSMCs
In searching the potential target genes of miR‐214 in VSMCs, total proteins were harvested from VSMCs transfected with control and miR‐214 mimics and subjected to label‐free quantitative proteomics analyses. miR‐214 overexpression in VSMCs transfected with miR‐214 mimics was confirmed using RT‐qPCR (Figure 4A). By applying 25% of change as the cutoff value, 219 proteins were found to be modulated by miR‐214 overexpression (Table S2). Among them, 59 and 160 proteins were downregulated and upregulated, respectively, by miR‐214. Because it has been widely accepted that miRNAs mainly function in RNA silencing and posttranscriptional regulation of target gene expression, the proteins that were downregulated by miR‐214 overexpression in VSMCs represent the potential direct target genes of miR‐214. Interestingly, Gene Ontology (GO) term enrichment analysis of the downregulated proteins showed that actin filament polymerization was the highest enriched functional/biological process and was inhibited by miR‐214 overexpression in VSMCs (Table S3). More specifically, we observed that most of these downregulated proteins (28 of 59) (yellow‐highlighted proteins in Table S2) are under the GO terms of ‘regulation of cell migration’ (eg, NCKAP1, EMAL1, ARPC2, LIMS2, SPI2), ‘proliferation’ (eg, EMAL1, LIMS2, LTOR3), ‘adhesion’ (eg, SNX5, SNX12, LIMS2), ‘actin filament reorganization and actin polymerization’ (eg, NCKAP1, ARPC2, CAPZB), ‘cell cycle’ (eg, APC7, ARL3, ULA1), and ‘gene expression’ (eg, KLF14, SMYD5, LONM), respectively, further confirming a role of miR‐214 in VSMC proliferation and migration. Importantly, by utilizing several computational algorithmic databases, such as RNA22 (https://cm.jefferson.edu/rna22/), DIANA‐microT‐CDS (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=MicroT_CDS/index), TargetScan7.1 (http://www.targetscan.org/mmu_71/), miRanda‐rel2010 (http://www.microrna.org/microrna/getGeneForm.do), and miRDB4.0 (http://mirdb.org/cgi-bin/search.cgi), we identified ≥1 miR‐214 binding site within the 3′UTR of 39 (of 59) genes with protein expression levels that were decreased by miR‐214 overexpression. As such, these proteins likely represent good candidates as the functional direct target genes of miR‐214 in VSMCs.
Figure 4.
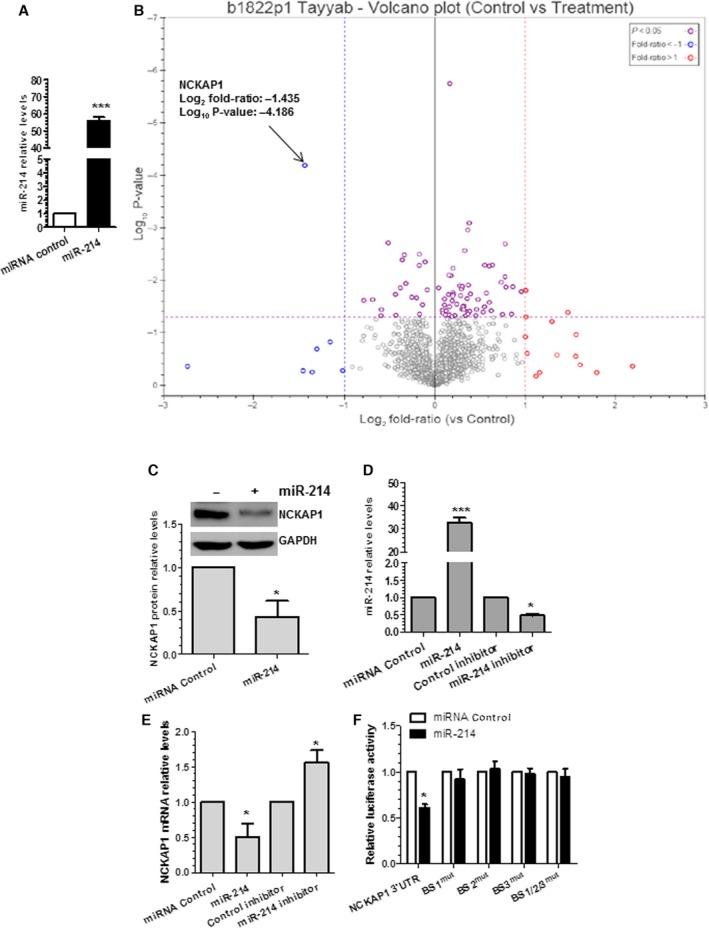
NCK associated protein 1 (NCKAP1) is the target gene of miR‐214 in vascular smooth muscle cells (VSMCs). A through C, NCKAP1 protein level was significantly downregulated by miR‐214 overexpression in VSMCs, as shown in the volcano plot (B), and confirmed by Western blot analyses (C). Reverse transcriptase–quantitative PCR (RT‐qPCR) confirmed miR‐214 overexpression in VSMCs (A). B, Volcano plot showing P values (−log10) vs protein ratio of miR‐214 mimics/control mimics (log2) of all 1594 proteins fulfilling strict quantitation criteria (red, 14 upregulated proteins with 2‐fold changes; blue, 7 downregulated proteins with 2‐fold changes; purple, significantly changed but such change is <2‐fold on miR‐214 overexpression; ANOVA with P<0.05). D and E, Modulations of miR‐214 expression levels in VSMCs negatively regulates NCKAP1 gene expressions. VSMCs were transfected with miR‐214 mimics (miR‐214) or inhibitor or with respective negative controls. Total RNAs were harvested and subjected to RT‐qPCR analyses with indicated primers. F, All 3 binding sites are important for miR‐214–mediated NCKAP1 gene repression. miR‐214 mimics or negative control were cotransfected into VSMCs with wild‐type NCKAP1 3′ untranslated region (3′UTR) reporter or the indicated single/combined binding site mutants (binding sites 1 [BS1mut], 2 [BS2mut], and 3 [BS3mut] or the combinational mutations [BS1/2/3mut]). Luciferase activity was measured at 48 hours after transfection. The data presented are representative (C) or show mean+SEM of 3 to 4 independent experiments (C through F). *<0.05, ***<0.001 Compared with miRNA control.
NCKAP1 Identified as a Functional Target of miR‐214 in VSMCs
It is unrealistic to assess whether all identified 39 downregulated genes with predicted miR‐214 binding sites are direct target genes of miR‐214 in the current study. Consequently, a volcano plot was applied to quickly identify the most meaningful changes in our large proteomics data sets composed of replicate data. As shown in the volcano plot (Figure 4B), with stringent criteria and P<0.05, NCKAP1 (also named as NAP1 or NAP125) was clearly identified as the most meaningful and important target gene of miR‐214 in VSMCs. Data from Western blot analyses (Figure 4C) confirmed the proteomics data that the NCKAP1 protein level was significantly downregulated by miR‐214 overexpression. Unsurprisingly, 3 miR‐214 binding sites (binding site 2 is predicted to be highly conserved across >10 species including human, whereas binding sites 1 and 3 are conserved in mouse and rat) were found within the 3′UTR of NCKAP1 using computational algorithms, DIANA‐microT/RNA22 (Figure S1A). Moreover, a favorable minimum loop‐free energy (dH: −136.5, −143.1 and −150.2 kcal/mol for binding site 1, 2 and 3, respectively) in the formation of the miR‐214:NCKAP1 3′UTR duplex stem loop for all 3 binding sites was observed by using mFold software (http://mfold.rna.albany.edu/?q=DINAMelt/Two-state-melting) (Figure S1B). As expected, NCKAP1 gene expression levels were significantly down‐ or upregulated by overexpression or inhibition, respectively, of miR‐214 in VSMCs (Figure 4D and 4E). These data imply that NCKAP1 is negatively regulated by miR‐214 directly or indirectly. To determine whether miR‐214 can directly regulate NCKAP1, the 3′UTR of NCKAP1 that contained the 3 miR‐214 binding sites was cloned into a luciferase reporter. Data from our miRNA reporter assay showed that the activity of luciferase from construct harboring the wild‐type NCKAP1 3′UTR was significantly inhibited by miR‐214 overexpression (Figure 4F). Importantly, all 3 binding sites are important for NCKAP1 3′UTR reporter activity repression mediated by miR‐214, as demonstrated in luciferase activity assays with miR‐214 binding site single (BS1mut, BS2mut or BS3mut) or combination (BS1/2/3mut) mutant reporters (Figure 4F). Taken together, these data demonstrated that NCKAP1 is a true mRNA target of miR‐214 that is negatively regulated by miR‐214 in VSMCs.
It is well known that 1 miRNA can target multiple target genes. To further assess whether any other candidate genes among the 39 downregulated genes with predicted miR‐214 binding sites are also direct target genes of miR‐214 in the context of VSMCs, the 3′UTRs of KLF14 and SMYD5, which contained 2 and 4 miR‐214 binding sites, respectively, were cloned into a luciferase reporter (pmiR‐Luc). The reason for selecting KLF14 and SMYD5 in our validation experiment is that the KLF14 is an important transcriptional factor, whereas SMYD5 is a powerful epigenetic regulator with methyltransferase activity. The miRNA luciferase activity data showed that the luciferase activity of the construct harboring the SMYD5 3′UTR (Figure S2B), but not of the reporter containing the KLF14 3′UTR (Figure S2A), was significantly inhibited by miR‐214 overexpression, confirming that SMYD5 is another target gene of miR‐214.
We recently demonstrated that another miRNA, miR‐34a, modulates VSMC functions and neointima formation through targeting the Notch1 gene.8 We wondered if there was cross‐talk or overlap between miR‐214 and miR‐34a in terms of target gene regulation. To this end, we conducted cotransfection experiments (miR214 mimics and Notch1 3′UTR reporter; miR34a mimics and NCKAP1 3′UTR reporter) in VSMCs. Data shown in Figure S2C and S2D revealed that neither the Notch1 3′UTR reporter activity nor that of NCKAP1 was regulated by miR‐214 or miR‐34a, respectively, demonstrating that there is not cross‐talk between these 2 miRNAs in the context of VSMCs.
NCKAP1 Knockdown in VSMCs Recapitulates the Effects of miR‐214 Overexpression on Actin Polymerization, Cell Migration, and Proliferation
These data have demonstrated that NCKAP1 is the authentic mRNA target of miR‐214 in VSMCs. Importantly, it has been reported that NCKAP1 is a constitutive and essential component of a WAVE2‐ and Abi‐1–containing complex linking Rac to site‐directed actin assembly and plays an essential role in regulation of cell migration through driving lamellipodia formation.48 It is plausible that miR‐214 could mediate VSMC migration and/or proliferation through modulating actin polymerization and/or lamellipodia formation by targeting NCKAP1. To support such a notion, we examined whether miR‐214 overexpression affects actin polymerization and/or lamellipodia formation in VSMCs. Immunofluorescence staining with NCKAP1 antibody and phalloidin‐FITC (a high‐affinity F‐actin probe conjugated to FITC) showed that typical lamellipodia was formed in VSMCs transfected with control miRNA mimics, whereas such a characteristic was less evident in VSMCs transfected with miR‐214 mimics (Figure 5A). Compared with control cells, a much lower level of actin polymerization was observed in miR‐214–overexpressing VSMCs, as shown with a much weaker F‐actin staining intensity (Figure 5A). As expected, no or very low NCKAP1 expression was observed in VSMCs transfected with miR‐214 mimics, further confirming that NCKAP1 expression is inhibited by miR‐214. An NCKAP1‐stable knockdown cell line was generated using NCKAP1 shRNA lentivirus to further evaluate and confirm a potential role of NCKAP1 in various VSMC functions. More than 60% of NCKAP1 inhibition efficiency was achieved in NCKAP1‐stable knockdown VSMCs, as demonstrated with immunofluorescence staining (Figure 5B) and RT‐qPCR (Figure 5C) assays, respectively. As expected, a similar phenomenon in terms of actin polymerization and/or lamellipodia formation was observed in NCKAP1‐stable knockdown VSMCs (Figure 5B). Interestingly, NCKAP1 was observed to colocalize with F‐actin within lamellipodia and membrane ruffles that face the direction of cell movement in the control cells, and such assembly was disrupted in the cells transfected with miR‐214 mimics (Figure 5A) and infected with NCKAP1 shRNA lentivirus (Figure 5B), respectively. Importantly, compared with control cells VSMCs transfected with miR‐214 mimics or NCKAP1‐stable knockdown VSMCs displayed much lower levels of cell proliferation and migration, as demonstrated in cell counting (Figure 5D), cell proliferation marker (Ki‐67) staining (Figure 5E), and transwell migration (Figure 5F) analyses. Moreover, we observed no significant difference in terms of VSMC proliferation and migration between VSMCs transfected with miR‐214 mimics and infected with NCKAP1 shRNA lentivirus (Figure 5C through 5F), confirming similar effects of miR‐214 overexpression and NCKAP1 silencing on VSMC functions. Overall, these data suggest that NCKAP1 inhibition/repression in VSMCs can recapitulate the effects of miR‐214 overexpression on actin polymerization, cell migration, and proliferation.
NCKAP1 Inhibition Is Required for miR‐214 Mediation of VSMC Growth and Motility
To study whether NCKAP1 knockdown could ablate the promoting effects of miR‐214 inhibition on VSMC functions, cotransduction experiments with miR‐214 inhibitor, NCKAP1 shRNA lentivirus, and/or respective controls, as indicated in the figure 6, were conducted in VSMCs, followed by phalloidin‐FITC staining, VSMC proliferation, and migration assays. RT‐qPCR data showed that both miR‐214 and NCKAP1 were successfully downregulated by respective miR‐214 inhibitor and NCKAP1 shRNA (Figure 6A). Moreover, the expression level of NCKAP1 was upregulated by miR‐214 inhibition, and such induction was abolished by NCKAP1 knockdown in the presence of miR‐214 inhibition (Figure 6A), further confirming that NCKAP1 gene expression is regulated by miR‐214. Importantly, the gene expression levels of PCNA (cell proliferation gene) with indicated treatment showed that miR‐214 inhibition activated but NCKAP1 knockdown inhibited PCNA gene expression and that PCNA gene activation by miR‐214 inhibition was blunted by NCKAP1 shRNA (Figure 6A). Proliferation assay data consistently showed that miR‐214 inhibition or NCKAP1 knockdown alone in the VSMCs significantly increased or decreased VSMC proliferation, as demonstrated in cell counting (Figure 6B) and Ki‐67 staining (Figure 6C1 and 6C2) assays, respectively; however, resuppression of NCKAP1 in the cells with miR‐214 inhibition almost abolished the promoting effects of miR‐214 inhibition on VSMC proliferation (Figure 6B and 6C), suggesting that miR‐214 inhibits VSMC proliferation through repression of NCKAP1. Similar trends were observed in VSMC migration and actin polymerization, as demonstrated in cell transwell migration (Figure 6D1 and 6D2) and phalloidin‐FITC staining (Figure 6E) experiments. These data provide evidence to support the notion that NCKAP1 repression is required for miR‐214–mediated inhibition of VSMC proliferation, migration, and actin polymerization.
Locally Enforced Expression of miR‐214 in the Injured Arteries Inhibited NCKAP1 Expression Levels, Decreased VSMC Proliferation, and Blunted Neointima SMC Hyperplasia After Vascular Injury
To explore the functional implication of miR‐214 in vascular remodeling after angioplasty, femoral arterial wire injuries were conducted, as described in our previous studies,24, 41, 42 and the expression level of miR‐214 was examined by RT‐qPCR assay. Consistent with our in vitro findings (Figure 1), on vascular injury, the expression levels of miR‐214 were substantially downregulated in femoral arteries from day 1 to 14, whereas their expression levels were almost back to normal at 28 days after angioplasty (Figure 7A), suggesting a regulatory role of miR‐214 in neointima formation. To further determine the effects of miR‐214 on VSMC proliferation and neointimal growth in vivo, 100 μL of 30% pluronic gel containing chemically modified and cholesterol‐conjugated 2.5 nmol miR‐214 or Cel‐miR‐67 agomirs (negative control) was applied perivascularly to femoral arteries immediately after injury, as described in our previous study.8 Local transferring miR‐214 agomirs increased vascular miR‐214 levels 72 hours following wire injury to a level comparable to that of uninjured femoral artery, which was significant higher than that of injured vessels treated with Cel‐miR‐67 agomirs (Figure 7B). Compared with the control group, enforced expression of miR‐214 in the injured vessels dramatically decreased NCKAP1 expression levels, as demonstrated in RT‐qPCR (Figure 7B) and Western blotting analyses (Figure 7C and 7D). These data demonstrated an inverse correlation between miR‐214 and NCKAP1 expression in the injured vessels. As expected, a decreased level of PCNA gene (Figure 7B) and protein (Figure 7C and 7D) expression was observed in the injured vessels treated with miR‐214 agomirs, demonstrating that perivascular enforced expression of miR‐214 in the injured vessels inhibits VSMC proliferation. Consequently, local transfection of miR‐214 in the injured vessels resulted in a nearly 42% decrease in neointima formation after angioplasty (Figure 7E and 7F). As expected, a thick neointima was induced by wire injury of the femoral artery after 28 days in the mice treated with Cel‐miR‐67 agomirs (n=11), which significantly reduced the lumen of the vessel. However, such remodeling response was substantially inhibited by treatment with miR‐214 agomirs (n=11). Our data suggest that locally restoring the expression levels of miR‐214 inhibits neointima SMC hyperplasia induced by vascular injury.
Figure 7.
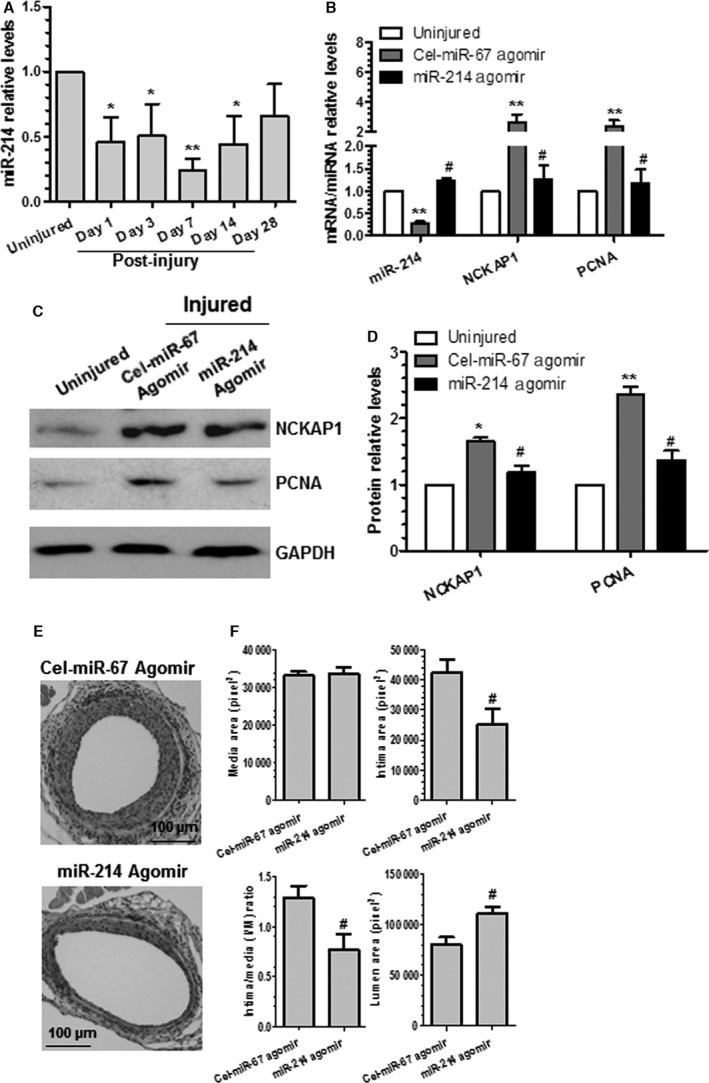
Locally enforced expression of miR‐214 in the injured arteries inhibited NCK associated protein 1 (NCKAP1) expression levels, decreased smooth muscle cell proliferation, and blunted neointima hyperplasia after vascular injury. A, miR‐214 was downregulated after injury. *<0.05, **<0.01 (Compared with uninjured control). B through D, Gene/protein expression levels in the injured vessels were modulated by perivascular delivery of miR‐214 agomirs. After injury, 100 μL of 30% pluronic gel containing 2.5 nmol agomirs per vessel per mouse was immediately applied and packed around the injured vessel. At 3 days (B), 14 days (C and D), or 28 days (E and F) later, injured segments of femoral arteries were harvested and subjected to various studies, as indicated. Total RNAs and proteins were extracted from uninjured and injured vessels (femoral arteries from 3 to 5 mice were pooled for each experiment, n=3 experiments) and subjected to reverse transcriptase–quantitative PCR (B) and Western blotting (C and D) analyses. Representative images (C) and quantitative data (mean+SEM) (B and D) of 3 independent experiments are presented. E and F, Wire injury–induced neointima formation was blunted by miR‐214 overexpression. Paraffin sections from both groups (n=11 mice for each group) were prepared and subjected to hematoxylin and eosin staining analyses. Representative images (E) and quantitative morphological characteristics (mean+SEM) including media area, neointimal area, neointimal/media ratio, and lumen area (F) are presented. #<0.05 (Compared miR‐214 agomirs with Cel‐miR‐67 agomirs [B, D and F]). *<0.05, **<0.01 (Compared with uninjured vessels [B and D]).
Discussion
New investigations into the molecular mechanisms underlying abnormal VSMC functions and the pathogenesis of neointima SMC hyperplasia are still required, and new therapeutic approaches for postangioplasty restenosis are urgently needed. In this study, we expanded our knowledge of the molecular mechanism mediating VSMC proliferation, migration, and neointima formation by uncovering an important role of miR‐214 in modulating VSMC functions in vitro and in vivo. Specifically, we documented that miR‐214 expression in VSMCs was modulated by different atherogenic stimuli (eg, high level of serum, PDGF‐BB, 7‐ketocholesterol, and 4‐hydroxynonenal) and observed a decreased level of miR‐214 during vascular remodeling in response to injury. Regarding cellular functions, our data showed that miR‐214 regulated both VSMC proliferation and migration. Mechanistically, NCKAP1 was identified as the functional target of miR‐214 in the context of VSMC functions. Moreover, our data suggested that actin polymerization and/or lamellipodia formation mediated by NCKAP1 is an underlying molecular mechanism through which miR‐214 regulates VSMC growth and motility. Translationally, documented evidence suggests that locally enforced expression of miR‐214 in the injured arteries can reduce NCKAP1 expression levels, inhibit VSMC proliferation, and thus blunt neointima SMC hyperplasia after vascular injury.
The miRNA miR‐214 is a member of the miR‐199a‐214 cluster encoded by a large noncoding RNA, DNM3os, that is transcribed in the opposite strand of the DNM3 gene.49 Primarily, Dnm3os and the associated miR‐199a‐214 cluster have been suggested to play an indispensable role in normal skeletal development and body growth in mammals,49 probably through regulation of myogenic differentiation from precursor cells.50 Later studies have documented a contradictory role of miR‐214 in cancer progression51, 52 and cardiovascular diseases. It has been reported that genetic deletion of miR‐214 in mice causes loss of cardiac contractility, increased apoptosis, and excessive fibrosis in response to ischemia–reperfusion injury, and the cardioprotective roles of miR‐214 during ischemia–reperfusion injury were attributed to controlling Ca²+ overload and cell death.20 This study provides evidence to support a protective role for miR‐214 in protecting cardiac cells from various insults and maintaining cardiac functions. Other studies, however, proposed an opposite role for miR‐214 in cardiac protection by suggesting that miR‐214 provokes cardiac hypertrophy,22 enhances the cardiac injury of viral myocarditis,53 and mediates cardiac fibroblast proliferation and collagen synthesis.54 The following factors likely contribute to such discrepancies: Different cell lines and animal models were used in these studies, miR‐214 plays a divergent role in various cellular systems, and the functional implication of miR‐214 in different biological processes depends on cell context. miR‐214 has also been implicated in many other cellular functions such as promoting dendritic cell switching from tolerance to immunity,55 regulating mitochondrial morphology and cell cycle,56 suppressing gluconeogenesis,57 controlling skin and hair follicle development,58 inhibiting angiogenesis,59, 60, 61 mediating osteogenic differentiation of myoblast cells,62 and/or impairing mitochondrial fatty acid oxidation.63
Our study provides a new perspective on the role for miR‐214 in VSMC biology and functions. By utilizing miRNA gain‐ and loss‐of‐function analyses, we demonstrated that miR‐214 inhibits both VSMC proliferation and migration, 2 critical cellular events in vascular neointimal lesion formation, in response to higher concentration of serum or PDGF‐BB, supporting a critical role of miR‐214 in these VSMC functions and behaviors. Importantly, by using our well‐established wire injury–induced neointima formation model and perivascular delivery of miR‐214 agomirs into injured vessels, we further demonstrated that miR‐214 modulated VSMC proliferation and inhibited neointima SMC hyperplasia, suggesting that miR‐214 is a potential therapeutic agent in postangioplasty restenosis. Consequently, findings from this study and previous studies indicate that miR‐214 exerts a divergent role in various cellular functions and a wide range of human diseases (particularly cancers and cardiovascular diseases), and more caution should be taken when considering the therapeutic effects of miR‐214 overexpression and/or inhibition on distinct human diseases.
A new finding in this study is that miR‐214 can be modulated by different atherogenic stimuli (eg, high level of serum, PDGF‐BB, 7‐ketocholesterol, and 4‐hydroxynonenal) in both human and murine VSMCs. miR‐214 is upregulated in response to several factors including cardiac stress, myocardial infarction, Ca2+ overload,18, 19 and heart failure.59 It has been reported that miR‐214 is hypoxia‐inducible miRNA and can be regulated in a HIF1α‐dependent and/or HIF1α‐independent manner.63 It is currently unknown whether the abovementioned atherogenic stimuli downregulate miR‐214 expression in VSMCs through an HIF1α‐dependent and/or HIF1α‐independent mechanism; however, it is unlikely that the HIF1α signaling pathway will be involved in miR‐214 modulation by atherogenic stimuli because the VSMCs were cultured under normoxic but not hypoxic conditions in this study. Instead, our data (Figure 1D through 1H) demonstrated that both serum and PDGF‐BB regulate miR‐214 expression through a transcriptional mechanism, an E‐box element within the miR‐214 promoter, that can be bound by the basic helix‐loop‐helix transcription factor Twist‐1, which is required for such regulation, and that both atherogenic stimuli can downregulate Twist‐1 gene expression in VSMCs. Our data are consistent with the previous finding that miR‐214 expression in hepatic stellate cells64 and during development34 is dependent on Twist‐1.
Another major novel finding in this study is that we identified NCKAP1 as the functional target gene of miR‐214 in VSMC functions and related diseases. Although multiple miR‐214 targets have been reported in various cellular contexts and diseases (eg, ITCH,53 TFAP2C,52 Osterix,62 FGFR1,51 PPARδ,63 Quaking,60 Ncx1,20 β‐catenin,55, 58 Mitofusin2,56 and ATF457), we provided evidence to suggest that NCKAP1 is a novel target gene of miR‐214 in VSMCs (Figure 4 and Figure S1) and is required for miR‐214 mediation of VSMC growth and migration (Figures 5 and 6). Cell migration is a highly orchestrated multistep process. It has been elegantly proposed that in response to extracellular signals, a cell initially extends its flat membrane protrusions (lamellipodia) and fingerlike protrusions (filopodia) and forms adhesions at the cell front (or leading edge), followed by retracting its tail to move forward.65 The actin cytoskeleton and associated proteins play a major role in this process. NCKAP1 (or p125Nap1) was primarily shown to associate with NCK (noncatalytic region of tyrosine kinase adaptor protein 1) both in vitro and in intact cells through binding to the Src homology 3 (SH3) domains of NCK66 and was found to localize along the lamellipodia and to mediate contact‐dependent cell migration.67 A later study also suggested a critical developmental role for NCKAP1 because mice lacking this gene will be arrested at midgestation and have defects in morphogenesis of all 3 embryonic germ layers.68 Unsurprisingly, as a constitutive and essential component of a WAVE complex, NCKAP1 has been reported to play a key role in regulation of cell motility and adhesion by driving actin assembly and polymerization and lamellipodia formation.48, 69 Nevertheless, the mechanism through which NCKAP1 is modulated in the context of VSMC functions remains elusive. In the current study, we demonstrated for the first time that NCKAP1 is regulated by miR‐214 in VSMCs. In our proteomics analyses, NCKAP1 was found to be one of the top downregulated proteins with the highest confidence and significance (Figure 4B). Importantly, 3 miR‐214 binding sites were found within the 3′UTR of NCKAP1, all of them with favorable minimum loop‐free energy in the formation of the miR‐214:NCKAP1 3′UTR duplex stem loop (Figure S1). Moreover, our data showed that NCKAP1 is reversely regulated by miR‐214 in VSMCs because miR‐214 overexpression significantly downregulated, whereas miR‐214 inhibition increased, NCKAP1 expression (Figure 4). Luciferase reporter and mutagenesis assays finally confirmed that NCKAP1 is the direct and functional target of miR‐214.
Functionally and mechanistically, we also demonstrated that controlling actin assembly and polymerization and lamellipodia formation in VSMCs through modulation of NCKAP1 expression is a primary mechanism through which miR‐214 mediates VSMC functions and behaviors including growth and movement. We observed a much lower level of F‐actin and lamellipodia formation and a significantly decreased level of NCKAP1 in miR‐214–overexpressing VSMCs (Figure 5A), suggesting that miR‐214 impairs actin assembly and polymerization and lamellipodia formation through inhibition of NCKAP1. Additional data from NCKAP1 knockdown experiments (Figure 5B through 5F) showed that NCKAP1 inhibition can recapitulate the inhibitory effects of miR‐214 overexpression on VSMC functions and behaviors. Finally, evidence from the cotransduction experiments (miR‐214 inhibition and NCKAP1 knockdown) (Figure 6) indicated that NCKAP1 repression is required for miR‐214–mediated inhibition of VSMC proliferation, migration, and actin polymerization. Consistently, our data showed that NCKAP1 is colocalized with F‐actin within lamellipodia at the cell front (Figure 5A and 5B); however, we unexpectedly observed that NCKAP1 can be relocalized inside the nuclei of VSMCs, particularly after the cells were subjected to serum‐starvation treatment (Figure 5B). The functional implication of such unexpected findings warrants further investigation in a separate study. Moreover, we observed a higher level of nuclear localization of NCKAP1 in control VSMCs infected with shRNA lentivirus (Figure 5B) than in VSMCs transfected with control miRNA mimics (Figure 5A). Because these 2 control VSMCs were subjected to different treatments (cells with control miRNA mimics received transient transfection for 24 hours, whereas the control VSMCs with nontarget shRNA were generated from lentivirus infection and a 10‐day puromycin selection) prior to serum starvation, we speculated that such discrepancies were due to the additive or synergistic effects of serum restimulation and lentivirus infection (and/or puromycin selection during cell line generation), which is under investigation in our separate study.
Apart from NCKAP1, which was identified and validated as a bona fide miR‐214 mRNA target in VSMCs in the current study, the abovementioned studies also suggest that multiple other genes can be regulated by miR‐214 in other cellular contexts and related diseases. Interestingly, only 2 reported miR‐214 targets—β‐catenin55, 58 and PCBP270—were slightly repressed by miR‐214 in VSMCs, as demonstrated in our proteomics analyses (Table S2). All other reported miR‐214 target genes failed to pass the threshold setting for our proteomics analyses, likely because VSMCs contain lower amounts or lack of expression of these target proteins or because of low sensitivity in identifying such regulatory proteins in the whole‐cell lysate using proteomics analyses. Despite this limitation, data from our proteomics analysis showed that the majority of the proteins downregulated by miR‐214 are involved in regulation of cell migration, actin filament reorganization and actin polymerization, proliferation, cell cycle, and gene expression, further supporting a role of miR‐214 in VSMC proliferation and migration. The fact that multiple genes were identified as the functional targets of miR‐214 in different studies suggests that miR‐214 plays a divergent role under various physiological and pathological conditions by targeting distinct target genes and/or that miR‐214 regulates its target genes in a cellular context–dependent manner.
Surprisingly, our proteomics data showed that more proteins were upregulated in VSMCs by miR‐214 (160 and 59 for upregulated and downregulated proteins, respectively) (Table S2), which suggests that miR‐214 positively regulates these proteins by suppressing ≥1 major transcription/epigenetic factor. Indeed, we observed that SMYD5, an epigenetic regulator, was significantly downregulated by miR‐214 overexpression. Moreover, data from reporter luciferase assays showed that miR‐214 overexpression significantly repressed SMYD5 3′UTR reporter activity (Figure S2B), providing direct evidence to support SMYD5 as a target gene of miR‐214 in VSMCs. SMYD5 belongs to the class V–like SAM‐binding methyltransferase superfamily, which has been suggested to play an important role in regulation of gene silencing by modulating the methylation of a variety of histone and nonhistone targets.71 It would be plausible that miR‐214 could upregulate a large number of proteins in VSMCs by desuppressing SMYD5; however, the implications of modulated SMYD5 in VSMC functions remain to be further investigated.
It is worth mentioning that in response to injury, lack of reendothelialization represents another major contribution to neointimal formation. Several studies59, 60, 61 have suggested that miR‐214 may play a role in regulating angiogenesis; therefore, the biological effects of miR‐214 on endothelial cells and reendothelialization after injury may also contribute to the miR‐214–mediated effect on vascular remodeling and needs to be investigated in future studies. Nonetheless, in the current study, we successfully uncovered the functional involvements of miR‐214 in VSMC biology and in vascular remodeling after injury. Moreover, the newly identified target NCKAP1 is at least partially responsible for miR‐214–mediated VSMC functions. Taken together, the findings presented in the current study suggest that modulated miR‐214 can be a potential therapeutic target for VSMC‐related diseases such as atherosclerosis and postangioplasty restenosis.
Sources of Funding
This work was supported by British Heart Foundation (FS/09/044/28007, PG/11/40/28891, PG/13/45/30326, PG/15/11/31279, PG/15/86/31723 and PG/16/1/31892 to Xiao); National Natural Science Foundation of China Grant (91339102, 30900571, 81270001, 81570249, 91539103, and 81270180); and Zhejiang Provincial Nature Science Foundation (LR14H020001). This work forms part of the research themes contributing to the translational research portfolio of Barts and the London Cardiovascular Biomedical Research Unit which is supported and funded by the National Institute of Health Research.
Disclosures
None.
Supporting information
Figure S1. miR‐214 binding sites within the 3′ untranslated region (3′UTR) of the NCK associated protein 1 (NCKAP1) gene and the miR‐214:NCKAP1 3′UTR duplex stem loop and respective minimum loop‐free energy. A, Three potential wild‐type binding sites (BS1–BS3) of miR‐214 within NCKAP1 3′UTR, as predicted by RNAhybrid and their mutants (BS1mut, BS2mut, and BS3mut). B, The formation of the miR‐214:NCKAP1 3′UTR (spanning through miR‐214 BS1‐3) duplex stem loop and the minimum loop‐free energy for individual loops (binding sites) were calculated by and extracted from mFold software (http://mfold.rna.albany.edu/?q=DINAMelt/Two-state-melting).
Figure S2. Gene 3′ untranslated region (3′UTR) reporter luciferase activity assays. miR‐214 mimic, miR‐34A mimics or negative control was cotransfected into vascular smooth muscle cells (VSMCs) with respective gene 3′UTR reporter. A, KLF14. B, SMYD5. C, NCKAP1. D, Notch1. Luciferase activity was measured at 48 hours after transfection. The data are presented as representative or as mean±S.E.M. of 4 to 6 independent experiments (n=4–6). *P<0.05 (versus microRNA [miR] control).
Table S1. Primer Sets Used in the Present Study
Table S2. Selected Protein List Regulated by miR‐214 in Vascular Smooth Muscle Cells (VSMCs)
Table S3. Gene Ontology Term Enrichment Analysis of Proteins Downregulated by miR‐214 in Vascular Smooth Muscle Cells (VSMCs)
(J Am Heart Assoc. 2016;5:e004629 doi: 10.1161/JAHA.116.004629)
Contributor Information
Li Zhang, li.zhang.uk@googlemail.com.
Qingzhong Xiao, Email: q.xiao@qmul.ac.uk.
References
- 1. Johnson JL. Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovasc Res. 2014;103:452–460. [DOI] [PubMed] [Google Scholar]
- 2. Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. [DOI] [PubMed] [Google Scholar]
- 3. Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thromb Vasc Biol. 2010;30:1118–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonauer A, Boon RA, Dimmeler S. Vascular microRNAs. Curr Drug Targets. 2010;11:943–949. [DOI] [PubMed] [Google Scholar]
- 5. Chen J, Yin H, Jiang Y, Radhakrishnan SK, Huang ZP, Li J, Shi Z, Kilsdonk EP, Gui Y, Wang DZ, Zheng XL. Induction of microRNA‐1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler Thromb Vasc Biol. 2011;31:368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense‐mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–1588. [DOI] [PubMed] [Google Scholar]
- 7. Wang M, Li W, Chang GQ, Ye CS, Ou JS, Li XX, Liu Y, Cheang TY, Huang XL, Wang SM. MicroRNA‐21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler Thromb Vasc Biol. 2011;31:2044–2053. [DOI] [PubMed] [Google Scholar]
- 8. Chen Q, Yang F, Guo M, Wen G, Zhang C, Luong le A, Zhu J, Xiao Q, Zhang L. miRNA‐34a reduces neointima formation through inhibiting smooth muscle cell proliferation and migration. J Mol Cell Cardiol. 2015;89:75–86. [DOI] [PubMed] [Google Scholar]
- 9. Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA‐133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011;109:880–893. [DOI] [PubMed] [Google Scholar]
- 10. Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR‐221 and miR‐222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Induction of microRNA‐221 by platelet‐derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284:3728–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119:2634–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA‐145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao N, Koenig SN, Trask AJ, Lin CH, Hans CP, Garg V, Lilly B. MicroRNA miR145 regulates TGFBR2 expression and matrix synthesis in vascular smooth muscle cells. Circ Res. 2015;116:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G. The knockout of miR‐143 and ‐145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16:1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li P, Liu Y, Yi B, Wang G, You X, Zhao X, Summer R, Qin Y, Sun J. MicroRNA‐638 is highly expressed in human vascular smooth muscle cells and inhibits PDGF‐BB‐induced cell proliferation and migration through targeting orphan nuclear receptor NOR1. Cardiovasc Res. 2013;99:185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li P, Zhu N, Yi B, Wang N, Chen M, You X, Zhao X, Solomides CC, Qin Y, Sun J. MicroRNA‐663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circ Res. 2013;113:1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress‐responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006;103:18255–18260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR‐29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel‐Duby R, Sadek HA, Olson EN. MicroRNA‐214 protects the mouse heart from ischemic injury by controlling Ca²⁺ overload and cell death. J Clin Invest. 2012;122:1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu HQ, Liang C, He ZQ, Fan M, Wu ZG. Circulating miR‐214 is associated with the severity of coronary artery disease. J Geriatr Cardiol. 2013;10:34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang T, Zhang GF, Chen XF, Gu HH, Fu SZ, Xu HF, Feng Q, Ni YM. MicroRNA‐214 provokes cardiac hypertrophy via repression of EZH2. Biochem Biophys Res Commun. 2013;436:578–584. [DOI] [PubMed] [Google Scholar]
- 23. Yu X, Zhang L, Wen G, Zhao H, Luong LA, Chen Q, Huang Y, Zhu J, Ye S, Xu Q, Wang W, Xiao Q. Upregulated sirtuin 1 by miRNA‐34a is required for smooth muscle cell differentiation from pluripotent stem cells. Cell Death Differ. 2015;22:1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiao Q, Zhang F, Grassia G, Hu Y, Zhang Z, Xing Q, Yin X, Maddaluno M, Drung B, Schmidt B, Maffia P, Ialenti A, Mayr M, Xu Q, Ye S. Matrix metalloproteinase‐8 promotes vascular smooth muscle cell proliferation and neointima formation. Arterioscler Thromb Vasc Biol. 2014;34:90–98. [DOI] [PubMed] [Google Scholar]
- 25. Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, Liu CJ, Zhu Y, Gao Y, Xu Q, Kong W, Wang X. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ Res. 2010;106:514–525. [DOI] [PubMed] [Google Scholar]
- 26. Salmon M, Gomez D, Greene E, Shankman L, Owens GK. Cooperative binding of KLF4, pELK‐1, and HDAC2 to a G/C repressor element in the SM22α promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res. 2012;111:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chahine MN, Blackwood DP, Dibrov E, Richard MN, Pierce GN. Oxidized LDL affects smooth muscle cell growth through MAPK‐mediated actions on nuclear protein import. J Mol Cell Cardiol. 2009;46:431–441. [DOI] [PubMed] [Google Scholar]
- 28. Auge N, Garcia V, Maupas‐Schwalm F, Levade T, Salvayre R, Negre‐Salvayre A. Oxidized LDL‐induced smooth muscle cell proliferation involves the EGF receptor/PI‐3 kinase/Akt and the sphingolipid signaling pathways. Arterioscler Thromb Vasc Biol. 2002;22:1990–1995. [DOI] [PubMed] [Google Scholar]
- 29. Zhao H, Wen G, Huang Y, Yu X, Chen Q, Afzal TA, Luong le A, Zhu J, Ye S, Zhang L, Xiao Q. MicroRNA‐22 regulates smooth muscle cell differentiation from stem cells by targeting methyl CpG‐binding protein 2. Arterioscler Thromb Vasc Biol. 2015;35:918–929. [DOI] [PubMed] [Google Scholar]
- 30. Luo Z, Wen G, Wang G, Pu X, Ye S, Xu Q, Wang W, Xiao Q. MicroRNA‐200C and ‐150 play an important role in endothelial cell differentiation and vasculogenesis by targeting transcription repressor ZEB1. Stem Cells. 2013;31:1749–1762. [DOI] [PubMed] [Google Scholar]
- 31. Xiao Q, Zhang F, Lin L, Fang C, Wen G, Tsai TN, Pu X, Sims D, Zhang Z, Yin X, Thomaszewski B, Schmidt B, Mayr M, Suzuki K, Xu Q, Ye S. Functional role of matrix metalloproteinase‐8 in stem/progenitor cell migration and their recruitment into atherosclerotic lesions. Circ Res. 2013;112:35–47. [DOI] [PubMed] [Google Scholar]
- 32. Huang Y, Lin L, Yu X, Wen G, Pu X, Zhao H, Fang C, Zhu J, Ye S, Zhang L, Xiao Q. Functional involvements of heterogeneous nuclear ribonucleoprotein A1 in smooth muscle differentiation from stem cells in vitro and in vivo. Stem Cells. 2013;31:906–917. [DOI] [PubMed] [Google Scholar]
- 33. Fang C, Wen G, Zhang L, Lin L, Moore A, Wu S, Ye S, Xiao Q. An important role of matrix metalloproteinase‐8 in angiogenesis in vitro and in vivo. Cardiovasc Res. 2013;99:146–155. [DOI] [PubMed] [Google Scholar]
- 34. Lee YB, Bantounas I, Lee DY, Phylactou L, Caldwell MA, Uney JB. Twist‐1 regulates the miR‐199a/214 cluster during development. Nucleic Acids Res. 2009;37:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. [DOI] [PubMed] [Google Scholar]
- 36. Wen G, Zhang C, Chen Q, le Luong A, Mustafa A, Ye S, Xiao Q. A novel role of matrix metalloproteinase‐8 in macrophage differentiation and polarization. J Biol Chem. 2015;290:19158–19172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alcolea MP, Casado P, Rodriguez‐Prados JC, Vanhaesebroeck B, Cutillas PR. Phosphoproteomic analysis of leukemia cells under basal and drug‐treated conditions identifies markers of kinase pathway activation and mechanisms of resistance. Mol Cell Proteomics. 2012;11:453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Montoya A, Beltran L, Casado P, Rodriguez‐Prados JC, Cutillas PR. Characterization of a TiO(2) enrichment method for label‐free quantitative phosphoproteomics. Methods. 2011;54:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rajeeve V, Vendrell I, Wilkes E, Torbett N, Cutillas PR. Cross‐species proteomics reveals specific modulation of signaling in cancer and stromal cells by phosphoinositide 3‐kinase (PI3K) inhibitors. Mol Cell Proteomics. 2014;13:1457–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cutillas PR, Vanhaesebroeck B. Quantitative profile of five murine core proteomes using label‐free functional proteomics. Mol Cell Proteomics. 2007;6:1560–1573. [DOI] [PubMed] [Google Scholar]
- 41. Xiao Q, Zeng L, Zhang Z, Margariti A, Ali ZA, Channon KM, Xu Q, Hu Y. Sca‐1+ progenitors derived from embryonic stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Arterioscler Thromb Vasc Biol. 2006;26:2244–2251. [DOI] [PubMed] [Google Scholar]
- 42. Zeng L, Xiao Q, Margariti A, Zhang Z, Zampetaki A, Patel S, Capogrossi MC, Hu Y, Xu Q. HDAC3 is crucial in shear‐ and VEGF‐induced stem cell differentiation toward endothelial cells. J Cell Biol. 2006;174:1059–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cai J, Yang C, Yang Q, Ding H, Jia J, Guo J, Wang J, Wang Z. Deregulation of let‐7e in epithelial ovarian cancer promotes the development of resistance to cisplatin. Oncogenesis. 2013;2:e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang X, Guo B, Li Q, Peng J, Yang Z, Wang A, Li D, Hou Z, Lv K, Kan G, Cao H, Wu H, Song J, Pan X, Sun Q, Ling S, Li Y, Zhu M, Zhang P, Peng S, Xie X, Tang T, Hong A, Bian Z, Bai Y, Lu A, He F, Zhang G. miR‐214 targets ATF4 to inhibit bone formation. Nat Med. 2013;19:93–100. [DOI] [PubMed] [Google Scholar]
- 45. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. [DOI] [PubMed] [Google Scholar]
- 46. Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, Heim MH, Stoffel M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474:649–653. [DOI] [PubMed] [Google Scholar]
- 47. Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, Leitinger N, Owens GK. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101:792–801. [DOI] [PubMed] [Google Scholar]
- 48. Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Wehland J, Stradal TE. Sra‐1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 2004;23:749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watanabe T, Sato T, Amano T, Kawamura Y, Kawamura N, Kawaguchi H, Yamashita N, Kurihara H, Nakaoka T. Dnm3os, a non‐coding RNA, is required for normal growth and skeletal development in mice. Dev Dyn. 2008;237:3738–3748. [DOI] [PubMed] [Google Scholar]
- 50. Liu J, Luo XJ, Xiong AW, Zhang ZD, Yue S, Zhu MS, Cheng SY. MicroRNA‐214 promotes myogenic differentiation by facilitating exit from mitosis via down‐regulation of proto‐oncogene N‐ras. J Biol Chem. 2010;285:26599–26607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen DL, Wang ZQ, Zeng ZL, Wu WJ, Zhang DS, Luo HY, Wang F, Qiu MZ, Wang DS, Ren C, Wang FH, Chiao LJ, Pelicano H, Huang P, Li YH, Xu RH. Identification of microRNA‐214 as a negative regulator of colorectal cancer liver metastasis by way of regulation of fibroblast growth factor receptor 1 expression. Hepatology. 2014;60:598–609. [DOI] [PubMed] [Google Scholar]
- 52. Penna E, Orso F, Cimino D, Tenaglia E, Lembo A, Quaglino E, Poliseno L, Haimovic A, Osella‐Abate S, De Pitta C, Pinatel E, Stadler MB, Provero P, Bernengo MG, Osman I, Taverna D. MicroRNA‐214 contributes to melanoma tumour progression through suppression of TFAP2C. EMBO J. 2011;30:1990–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen ZG, Liu H, Zhang JB, Zhang SL, Zhao LH, Liang WQ. Upregulated microRNA‐214 enhances cardiac injury by targeting ITCH during coxsackievirus infection. Mol Med Rep. 2015;12:1258–1264. [DOI] [PubMed] [Google Scholar]
- 54. Sun M, Yu H, Zhang Y, Li Z, Gao W. MicroRNA‐214 mediates isoproterenol‐induced proliferation and collagen synthesis in cardiac fibroblasts. Sci Rep. 2015;5:18351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gu C, Zhou XD, Yuan Y, Miao XH, Liu Y, Ru YW, Li KQ, Li G. MicroRNA‐214 induces dendritic cell switching from tolerance to immunity by targeting beta‐catenin signaling. Int J Clin Exp Pathol. 2015;8:10050–10060. [PMC free article] [PubMed] [Google Scholar]
- 56. Bucha S, Mukhopadhyay D, Bhattacharyya NP. Regulation of mitochondrial morphology and cell cycle by microRNA‐214 targeting Mitofusin2. Biochem Biophys Res Commun. 2015;465:797–802. [DOI] [PubMed] [Google Scholar]
- 57. Li K, Zhang J, Yu J, Liu B, Guo Y, Deng J, Chen S, Wang C, Guo F. MicroRNA‐214 suppresses gluconeogenesis by targeting activating transcriptional factor 4. J Biol Chem. 2015;290:8185–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ahmed MI, Alam M, Emelianov VU, Poterlowicz K, Patel A, Sharov AA, Mardaryev AN, Botchkareva NV. MicroRNA‐214 controls skin and hair follicle development by modulating the activity of the Wnt pathway. J Cell Biol. 2014;207:549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Duan Q, Yang L, Gong W, Chaugai S, Wang F, Chen C, Wang P, Zou MH, Wang DW. MicroRNA‐214 is upregulated in heart failure patients and suppresses XBP1‐mediated endothelial cells angiogenesis. J Cell Physiol. 2015;230:1964–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. van Mil A, Grundmann S, Goumans MJ, Lei Z, Oerlemans MI, Jaksani S, Doevendans PA, Sluijter JP. MicroRNA‐214 inhibits angiogenesis by targeting quaking and reducing angiogenic growth factor release. Cardiovasc Res. 2012;93:655–665. [DOI] [PubMed] [Google Scholar]
- 61. Chan LS, Yue PY, Mak NK, Wong RN. Role of microRNA‐214 in ginsenoside‐Rg1‐induced angiogenesis. Eur J Pharm Sci. 2009;38:370–377. [DOI] [PubMed] [Google Scholar]
- 62. Shi K, Lu J, Zhao Y, Wang L, Li J, Qi B, Li H, Ma C. MicroRNA‐214 suppresses osteogenic differentiation of C2C12 myoblast cells by targeting Osterix. Bone. 2013;55:487–494. [DOI] [PubMed] [Google Scholar]
- 63. el Azzouzi H, Leptidis S, Dirkx E, Hoeks J, van Bree B, Brand K, McClellan EA, Poels E, Sluimer JC, van den Hoogenhof MM, Armand AS, Yin X, Langley S, Bourajjaj M, Olieslagers S, Krishnan J, Vooijs M, Kurihara H, Stubbs A, Pinto YM, Krek W, Mayr M, da Costa Martins PA, Schrauwen P, De Windt LJ. The hypoxia‐inducible microRNA cluster miR‐199a approximately 214 targets myocardial PPARδ and impairs mitochondrial fatty acid oxidation. Cell Metab. 2013;18:341–354. [DOI] [PubMed] [Google Scholar]
- 64. Chen L, Chen R, Kemper S, Charrier A, Brigstock DR. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1‐dependent microRNA‐214 expression: role of exosomes in horizontal transfer of Twist1. Am J Physiol Gastrointest Liver Physiol. 2015;309:G491–G499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Le Clainche C, Carlier MF. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 2008;88:489–513. [DOI] [PubMed] [Google Scholar]
- 66. Kitamura T, Kitamura Y, Yonezawa K, Totty NF, Gout I, Hara K, Waterfield MD, Sakaue M, Ogawa W, Kasuga M. Molecular cloning of p125Nap1, a protein that associates with an SH3 domain of Nck. Biochem Biophys Res Commun. 1996;219:509–514. [DOI] [PubMed] [Google Scholar]
- 67. Nakao S, Platek A, Hirano S, Takeichi M. Contact‐dependent promotion of cell migration by the OL‐protocadherin‐Nap1 interaction. J Cell Biol. 2008;182:395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rakeman AS, Anderson KV. Axis specification and morphogenesis in the mouse embryo require Nap1, a regulator of WAVE‐mediated actin branching. Development. 2006;133:3075–3083. [DOI] [PubMed] [Google Scholar]
- 69. Ibarra N, Blagg SL, Vazquez F, Insall RH. Nap1 regulates Dictyostelium cell motility and adhesion through SCAR‐dependent and ‐independent pathways. Curr Biol. 2006;16:717–722. [DOI] [PubMed] [Google Scholar]
- 70. Tang SL, Gao YL, Chen XB. MicroRNA‐214 targets PCBP2 to suppress the proliferation and growth of glioma cells. Int J Clin Exp Pathol. 2015;8:12571–12576. [PMC free article] [PubMed] [Google Scholar]
- 71. Spellmon N, Holcomb J, Trescott L, Sirinupong N, Yang Z. Structure and function of SET and MYND domain‐containing proteins. Int J Mol Sci. 2015;16:1406–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. miR‐214 binding sites within the 3′ untranslated region (3′UTR) of the NCK associated protein 1 (NCKAP1) gene and the miR‐214:NCKAP1 3′UTR duplex stem loop and respective minimum loop‐free energy. A, Three potential wild‐type binding sites (BS1–BS3) of miR‐214 within NCKAP1 3′UTR, as predicted by RNAhybrid and their mutants (BS1mut, BS2mut, and BS3mut). B, The formation of the miR‐214:NCKAP1 3′UTR (spanning through miR‐214 BS1‐3) duplex stem loop and the minimum loop‐free energy for individual loops (binding sites) were calculated by and extracted from mFold software (http://mfold.rna.albany.edu/?q=DINAMelt/Two-state-melting).
Figure S2. Gene 3′ untranslated region (3′UTR) reporter luciferase activity assays. miR‐214 mimic, miR‐34A mimics or negative control was cotransfected into vascular smooth muscle cells (VSMCs) with respective gene 3′UTR reporter. A, KLF14. B, SMYD5. C, NCKAP1. D, Notch1. Luciferase activity was measured at 48 hours after transfection. The data are presented as representative or as mean±S.E.M. of 4 to 6 independent experiments (n=4–6). *P<0.05 (versus microRNA [miR] control).
Table S1. Primer Sets Used in the Present Study
Table S2. Selected Protein List Regulated by miR‐214 in Vascular Smooth Muscle Cells (VSMCs)
Table S3. Gene Ontology Term Enrichment Analysis of Proteins Downregulated by miR‐214 in Vascular Smooth Muscle Cells (VSMCs)