

Abstract
Background
Few studies have evaluated the association between secondhand smoke (SHS) and subclinical cardiovascular disease among ethnically diverse populations. This study assesses the impact of SHS on inflammation and atherosclerosis (carotid intima‐media thickness, coronary artery calcification, and peripheral arterial disease).
Methods and Results
We examined 5032 nonsmoking adults aged 45 to 84 years without prior cardiovascular disease participating in the Multi‐Ethnic Study of Atherosclerosis (MESA) from 2000 to 2002. SHS exposure was determined by self‐report, and urinary cotinine was measured in a representative subset (n=2893). The multi‐adjusted geometric mean ratios (95% CIs) for high‐sensitivity C‐reactive protein and interleukin‐6 comparing 407 participants with SHS ≥12 h/wk versus 3035 unexposed participants were 1.13 (1.02–1.26) and 1.04 (0.98–1.11), respectively. The multi‐adjusted geometric mean ratio for carotid intima‐media thickness was 1.02 (0.97–1.07). Fibrinogen and coronary artery calcification were not associated with SHS. The prevalence of peripheral arterial disease (ankle‐brachial index ≤0.9 or ≥1.4) was associated with detectable urinary cotinine (odds ratio, 2.10; 95% CI, 1.09–4.04) but not with self‐reported SHS. Urinary cotinine was not associated with inflammation or carotid intima‐media thickness.
Conclusions
Despite limited exposure assessment, this study supports the association of SHS exposure with inflammation and peripheral arterial disease.
Keywords: ankle‐brachial index, atherosclerosis, carotid intima‐media thickness, coronary artery calcium, inflammation, peripheral artery disease, secondhand smoke, smoking
Subject Categories: Cardiovascular Disease, Epidemiology, Risk Factors, Lifestyle, Primary Prevention
Introduction
Secondhand smoke (SHS) exposure is a global cause of morbidity and mortality.1 A third of nonsmoking adults are exposed to SHS worldwide.1 In the United States, 25% of the population remains exposed to SHS, disproportionately affecting communities with low income.2 SHS is an established cardiovascular disease (CVD) risk factor.3, 4 Meta‐analyses have estimated that SHS exposure is associated with a 31% increased risk of coronary heart disease3 and 20% to 30% increased risk of stroke.5, 6, 7 The enactment of indoor smoke‐free policies have been followed by important reductions in coronary heart disease hospitalizations,8 providing additional support for the potential cardiovascular benefits of reducing SHS exposure. The 2014 Surgeon General Report, however, estimated that around 33 000 nonsmokers continue to die every year from SHS‐related coronary heart disease in the United States.6
Possible mechanisms for SHS‐related cardiovascular toxicity include increased platelet aggregability, endothelial dysfunction, inflammation, oxidative stress, arterial stiffness, and atherosclerosis.9, 10 Relatively few studies have evaluated the association between SHS exposure and subclinical CVD among ethnically diverse populations at current levels of exposure. Self‐reported SHS exposure has been associated with carotid intima‐media thickness (cIMT) and coronary artery calcification (CAC) in studies from the United States11, 12, 13, 14, 15 and Europe,16, 17 although most studies were conducted more than 1 to 2 decades ago, when SHS exposure was much higher than it is today. Few studies have evaluated the association between SHS and peripheral arterial disease (PAD), with inconsistent findings.18, 19, 20 With mostly supportive findings, a larger body of evidence is available for the association between SHS exposure and high‐sensitivity C‐reactive protein (hsCRP), including studies among adolescents,21, 22, 23, 24 pregnant women,25 and adults.26, 27, 28, 29, 30, 31, 32, 33, 34 Studies evaluating the association between self‐reported or biomarker‐based SHS exposure and fibrinogen have generally shown consistent positive associations.28, 30, 31, 32, 33, 35 For interleukin‐6 (IL‐6), the evidence is largely null, although most studies are small.26, 27, 30
The Multi‐Ethnic Study of Atherosclerosis (MESA) was specifically designed to assess subclinical CVD and its risk factors among an ethnically diverse cohort from 6 urban communities around the United States. MESA provides a unique opportunity to inform our understanding of mechanistic pathways for CVD at relevant levels of SHS exposure, which can better inform tobacco product regulation. The objective of this study is to examine the cross‐sectional association of SHS exposure with markers of inflammation, subclinical atherosclerosis, and PAD in nonsmoking MESA participants.
Methods
Study Population
MESA is a community‐based prospective cohort study of 6814 white, black, Hispanic, or Chinese American men and women aged 45 to 84 years free of clinically apparent CVD at baseline (2000–2002). Study details have been previously published.36 Participants were enrolled from Forsyth County, NC; New York City, NY; Baltimore, MD; St. Paul, MN; Chicago, IL; and Los Angeles, CA. The race/ethnicity distribution was as follows: 39% non‐Hispanic whites, 28% black, 22% Hispanics, and 12% Chinese Americans. The institutional review boards from all field centers approved the study and all participants provided written informed consent.
SHS exposure was assessed by self‐report in the overall population as well as by urinary cotinine in a random subset. This study was restricted to the baseline visit (2000–2002). We excluded 887 participants who were current smokers based on self‐report, 107 participants with urinary cotinine concentrations above concentrations ≥200 ng/mL (likely current smokers),37 149 participants missing data on self‐reported SHS exposure, and 639 participants missing other variables of interest, leaving 5032 participants for this analysis (Figure 1). Among them, 2983 participants had urinary cotinine available. Urinary cotinine, a specific biomarker of recent SHS exposure,38 was analyzed in a random subsample of MESA participants who were enrolled in the MESA lung substudy (n=3965). Sociodemographic characteristics in our study sample for analyses based on self‐reported SHS exposure (n=5032) and urinary cotinine (n=2982) were similar to the overall noncurrent smoking MESA population (Table S1).
Figure 1.
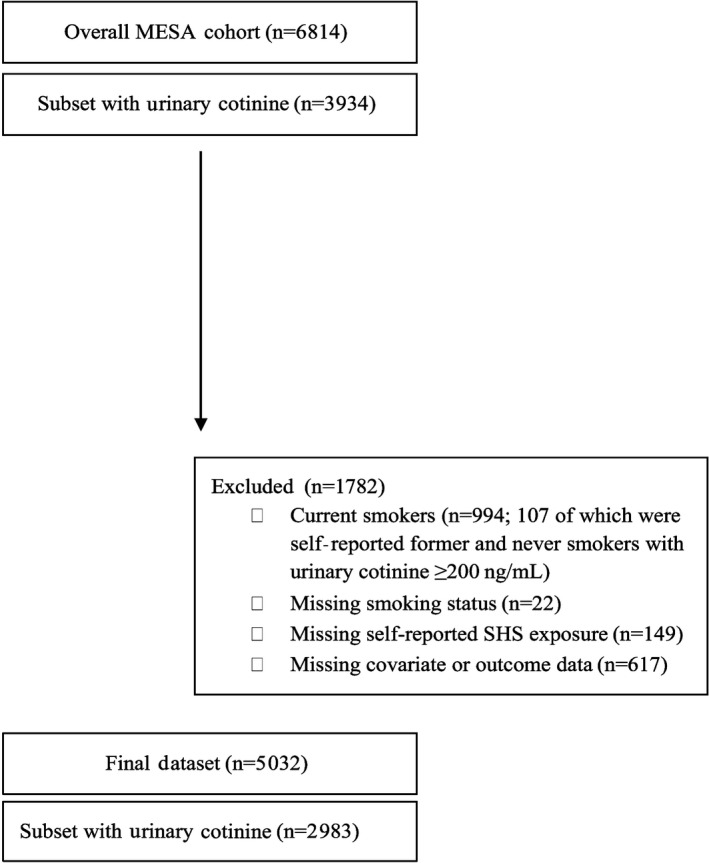
Definition of study population, Multi‐Ethnic Study of Atherosclerosis (MESA), United States, 2000–2002. SHS indicates secondhand smoke.
SHS Exposure
Information on current SHS exposure was obtained during the study visit by asking noncurrent smoking participants the following question: “During the past year about how many hours per week were you in close contact with people when they were smoking? (eg, in your home, in a car, at work or other close quarters).” SHS exposure was categorized as unexposed and as approximate quartiles of hours of SHS exposure per week among the exposed (1, 2–3, 4–11, and 12 or more hours per week).
Urinary cotinine (ng/mL) was measured by immunoassay LLD (lower detection limit) (Immulite 2000 Nicotine Metabolite Assay; Diagnostic Products Corp., Los Angeles, CA) as part of the MESA Lung Study.37 The average half‐life of urinary cotinine is 16 hours.38 The limit of detection for urinary cotinine was 10 ng/mL. In our study sample (which excluded participants with cotinine >200 ng/mL), 10% (n=299) of participants had detectable urinary cotinine measurements.
Inflammation Markers
Serum hsCRP was measured using a high‐sensitivity assay (N‐High‐Sensitivity CRP; Dade Behring, Deerfield, IL) (intra‐assay coefficient of variation [CV] ranged from 2.3% to 4.4% and the interassay CV ranged from 2.1% to 5.7%). IL‐6 was measured by ultrasensitive enzyme‐linked immunosorbent assay (Quantikine HS Human IL‐6 Immunoassay; R&D Systems, Minneapolis, MN) (analytical CV 6.3%).39 Serum fibrinogen was measured by immunoprecipitation using the BNII nephelometer (N‐Antiserum to Human Fibrinogen; Dade Behring) (intra‐assay and interassay CV as 2.7% and 2.6%, respectively). We evaluated inflammation markers as continuous. We also categorized hsCRP ≥2 mg/L as suggested in a previous study.40
cIMT and CAC
The right and left common and internal carotid arteries and the near and far walls were imaged according to a scanning protocol using high‐resolution B‐mode ultrasound with a Logiq 700 machine (General Electric Medical Systems, Waukesha, WI). Images were digitized and analyzed centrally at the MESA ultrasound reading center (Tufts Medical Center). We defined internal and common cIMT as the mean of the maximum cIMT of the near and far walls on the right and left sides as in previous MESA studies.
CAC was measured using an electron‐beam computed tomography scanner (Imatron C‐150XL; GE‐Imatron, San Francisco, CA) (Imatron C‐150XL; GE‐Imatron, San Francisco, CA) in 3 sites (Chicago, IL; Los Angeles, CA; and New York, NY) and by a multidetector row computed tomography system (Lightspeed, General Electric Medical Systems, Waukesha, WI; or Volume Zoom, Siemens, Erlanger, Germany) in 3 sites (Baltimore, MD; Winston‐Salem, NC; and St. Paul, MN).41 Images were centrally read at the MESA CT reading center (Harbor–University of California, Los Angeles). The scanning protocol for MESA has been previously published.42 For each scan, the total phantom‐adjusted Agatston score, defined as the sum of calcium measures from the left anterior descending, circumflex, and left and right coronary arteries, was calculated; the mean score was used in these analyses. We analyzed CAC as two binary measures: (1) present (CAC >0) versus absent, or (2) <75th versus >75th percentile for the entire MESA population.
Peripheral Arterial Disease
Ankle‐brachial index (ABI) measurements were obtained after the patient rested in the supine position for 5 minutes using a specific protocol to measure systolic blood pressure in each posterior tibial and dorsalis pedis artery in both legs and in the brachial artery in both arms with a continuous‐wave Doppler ultrasound probe. For each leg, the ABI was calculated as the higher of the posterior tibial or dorsalis pedis systolic pressures in each leg divided by the higher of the 2 systolic blood pressure measurements in both arms. For this study, we analyzed ABI as 3 binary measurements: (1) ABI ≤0.9 (excluding participants with ABI ≥1.4), (2) ABI ≥1.4 (excluding participants with ABI ≤0.9), and (3) ABI ≤0.9 or ABI ≥1.4 in accordance with previous MESA studies showing both low and high ABIs were associated with CVD events.43
Other Variables
Standardized questionnaires were used to obtain sociodemographic information (education, family income), current alcohol and tobacco use, medical history, medication use, and family history of CVD. Body mass index (BMI) was calculated as measured weight in kilograms divided by measured height in meters squared. Systolic and diastolic resting blood pressures were measured in the seated position using the Critikon Dinamap Pro 100 monitor (Critikon, Tampa, FL). Hypertension was defined as a systolic blood pressure ≥140 mm Hg, a diastolic blood pressure ≥90 mm Hg, or the use of medications for hypertension.44
Lipids including total and high‐density lipoprotein cholesterol, triglycerides, and glucose levels were measured from fasting plasma samples in a central laboratory (University of Vermont, Burlington, VT).45 Low‐density lipoprotein cholesterol (LDL‐C) was calculated by the Friedewald equation among participants with a triglyceride value <400 mg/dL.46 Diabetes mellitus was defined by the use of insulin or oral hypoglycemic medication or a fasting blood glucose of ≥126 mg/dL.47
Physical activity was measured by the MESA Typical Week Physical Activity Survey.48 Minutes of activity were summed for each discrete activity type and multiplied by metabolic equivalent (MET) level. For this analysis, we used a summary variable for physical activity defined as the sum of moderate and vigorous physical activity (MVPA) in MET minutes per day. Urine creatinine was determined using a Jaffe rate reaction measured with the Vitros 950IRC instrument (Ortho‐Clinical Diagnostics, Rochester, NY).
Statistical Analysis
ANOVA and chi‐square tests were used to describe sociodemographic and cardiovascular risk factors by categories of SHS exposure at baseline. hsCRP, IL‐6, fibrinogen, and cIMT were log‐transformed (natural logarithm). Multivariable linear regression models on log‐transformed hsCRP, IL‐6, fibrinogen, and cIMT were used to estimate ratios of geometric means comparing CVD marker levels by SHS exposure category. The geometric mean ratios were obtained by exponentiating the β coefficients from the above models. For dichotomous outcomes (hsCRP ≥2, CAC >0, CAC >75th percentile, ABI ≤0.9, ABI ≥1.4, and ABI ≤0.9 or ABI ≥1.4), we calculated prevalence odds ratios (ORs) by SHS exposure using multivariate logistic regression. Hours of SHS exposure per week were modeled as categorical with 5 categories and 0 hours of self‐reported SHS exposure per week as the reference category. Models were adjusted for covariates in a progressive manner. Model 1 adjusted for age, sex, race/ethnicity, study site, education (high school or less, some college but no degree/technical school certificate, associate's degree/bachelor's degree/graduate degree), and income (<$25 000/y or ≥$25 000/y). Model 2 included model 1 variables plus hypertension medication (yes or no), systolic blood pressure (mm Hg), diabetes mellitus (yes or no), LDL‐C (mg/dL), treatment for dyslipidemia (yes or no), physical activity (MET, h/wk), and smoking status (never or former). Model 3 included model 2 variables and BMI (kg/m2). BMI was adjusted in a separate model because a previous study found that the association between SHS and hsCRP was attenuated after adjustment for BMI,49 and BMI could be either a confounder or a mediator of the association. For all analyses, P values for trend were obtained by including a continuous variable with the medians corresponding to each quartile of the SHS exposure distribution in the regression model.50
We evaluated effect modification of the fully adjusted association between SHS exposure and continuous subclinical CVD markers (log‐transformed hsCRP, IL‐6, internal cIMT, and common cIMT) by categories of sex, age, race/ethnicity, study site, education, and smoking status in interaction models of SHS exposure (≥12 hours of SHS exposure per week to unexposed) times participant subgroups of interest. Estimated 2‐sided P values for the interactions between SHS exposure and the characteristics evaluated were computed using the Wald test. We did not evaluate effect measure modification of dichotomous outcomes due to limited power.
In the subsample of participants with urinary cotinine available (n=2983), we evaluated the association of urinary cotinine with inflammation, cIMT, CAC, and PAD. Urinary cotinine was modeled as detectable versus nondetectable urinary cotinine. All models for urinary cotinine were additionally adjusted for urinary creatinine (log‐transformed) to account for urine dilution in spot urine samples.
We also conducted several sensitivity analyses. First, we repeated models 2 and 3 for each outcome adjusting for alcohol use (n=3962), family history of CHD (n=4713), and heart rate (n=4999). Second, we ran all analyses evaluating the association between SHS exposure and all study outcomes defining SHS as binary (exposed or unexposed). Third, all analyses were performed based on self‐reported SHS exposure only, without using cotinine to exclude potential current smokers. Fourth, all analyses for inflammatory outcomes were further adjusted for self‐reported asthma and self‐reported infections in the past 2 weeks (flu, fever, urinary infection, sinus infection, tooth infection, arthritis flare‐up, gout flare‐up, and pneumonia). For all sensitivity analyses, we observed similar patterns and inference to those in the main analysis (data not shown).
Statistical analyses were performed with Stata version 13.0 (StataCorp, College Station, TX) and graphical displays were created using R version 3.03 (R Foundation for Statistical Computing, www.r-project.org, Vienna, Austria). All statistical tests were 2‐sided, and P values <0.05 were considered statistically significant.
Results
Participant Characteristics
A total of 3035 participants (60%) were unexposed to SHS in the past year. Among participants reporting any SHS exposure in the past year, the median (interquartile range) was 3 (1–10) hours of SHS exposure per week. Forty percent (n=1997) of participants self‐reported ≥1 hour SHS exposure per week and 8.1% (n=407) reported ≥12 hours of SHS exposure per week. Participants with higher SHS exposure were more likely to have lower income and education, be former smokers, and have higher BMI (Table 1). They also tended to have higher physical activity levels. Participants in the highest SHS exposure category were more likely to have hypertension and higher hsCRP and IL‐6 levels. Self‐reported hours of SHS exposure were also positively associated with concentrations of urinary cotinine (Table 1). The Spearman correlation coefficient was 0.30.
Table 1.
Characteristics of 5032 MESA Participants by SHS Exposure, 2000–2002
| Overall | Unexposed | 1 h/wk | 2–3 h/wk | 4–11 h/wk | ≥12 h/wk | P Value | |
|---|---|---|---|---|---|---|---|
| No. | 5032 | 3035 | 682 | 428 | 480 | 407 | |
| Sociodemographic factors | |||||||
| Men | 46.0 | 44.1 | 50.0 | 47.4 | 53.3 | 43.2 | <0.001 |
| Age, y | 62.5 (10.3) | 63.9 (10.4) | 59.8 (9.8) | 61.1 (10) | 60.5 (9.6) | 60.5 (9.3) | <0.001 |
| Race/ethnicity | <0.001 | ||||||
| White | 39.5 | 35.3 | 50.6 | 43.7 | 44.2 | 41.5 | |
| Black | 24.1 | 21.0 | 24.1 | 29.4 | 31.0 | 33.7 | |
| Chinese American | 14.1 | 17.9 | 10.0 | 10.1 | 7.3 | 5.2 | |
| Hispanic | 22.4 | 25.9 | 15.4 | 16.8 | 17.5 | 19.7 | |
| Education | <0.001 | ||||||
| High school or less | 35.0 | 38.1 | 22.0 | 30.4 | 33.8 | 39.8 | |
| Some college but no degree/technical school certificate | 22.1 | 19.8 | 23.0 | 25.7 | 28.1 | 26.3 | |
| Associate's degree, bachelor's degree, or graduate school | 43.0 | 42.1 | 55.0 | 43.9 | 38.1 | 33.9 | |
| Less than $25 000/y | 31.3 | 36.8 | 18.5 | 22.4 | 26.3 | 27.3 | <0.001 |
| CVD risk factors | |||||||
| Family history of CHD | 41.2 | 39.7 | 43.5 | 44.3 | 44.9 | 42.2 | 0.14 |
| Current alcohol usea | 68.4 | 65.1 | 74.0 | 74.7 | 69.0 | 72.5 | <0.001 |
| Former smokers | 40.9 | 37.4 | 45.9 | 44.2 | 49.0 | 46.4 | <0.001 |
| BMI, kg/m2 | 28.2 (5.4) | 27.7 (5.4) | 28.2 (5.2) | 28.5 (5.3) | 29.1 (5.6) | 30.0 (5.7) | <0.001 |
| Physical activity (MET, h/wk) | 94.6 (97.9) | 82.1 (80.3) | 100.9 (102.8) | 113.4 (131.3) | 130.6 (130.6) | 115.3 (104.3) | <0.001 |
| Hypertension | 44.5 | 45.5 | 39.0 | 43.2 | 43.3 | 48.4 | 0.01 |
| Hypertension medication | 36.9 | 37.8 | 32.3 | 34.8 | 36.9 | 40.3 | 0.04 |
| Systolic blood pressure, mm Hg | 126.4 (21.4) | 127.1 (21.9) | 123.8 (20.1) | 126.1 (20.9) | 124.5 (19.8) | 128.0 (21.5) | <0.001 |
| Diabetes mellitus | 11.8 | 12.3 | 7.6 | 14.0 | 11.7 | 13.3 | 0.005 |
| Fasting glucose, mg/dL | 96.7 (29.1) | 97 (29.5) | 93.3 (21.8) | 96 (27) | 98.2 (33.6) | 98.9 (32) | 0.01 |
| Lipid‐lowering medications | 16.6 | 17.3 | 13.9 | 18.2 | 15.8 | 14.5 | 0.14 |
| Total cholesterol, mg/dL | 194.3 (34.6) | 193.6 (34.2) | 196.2 (35) | 193.2 (34.9) | 195 (36.8) | 196.2 (33.8) | 0.47 |
| LDL‐C, mg/dL | 117.6 (31.3) | 116.9 (30.8) | 119.1 (32.1) | 118 (32.2) | 118.4 (32.7) | 118.3 (30.7) | 0.47 |
| Urinary cotinine,a ng/mL | 10.9 (14.6) | 8.8 (9.7) | 11.2 (16.5) | 9.8 (7.8) | 15.4 (20.8) | 22.4 (27.2) | <0.001 |
| Detectable urinary cotininea | 10.0 | 4.6 | 10.5 | 9.4 | 22.2 | 36.9 | <0.001 |
| Living with a smoker as a childa | 52.6 | 49.2 | 60.3 | 60.4 | 54.5 | 55.7 | <0.001 |
| Living with a smoker as an adulta | 40.0 | 35.4 | 41.7 | 44.9 | 48.0 | 57.6 | <0.001 |
| Inflammation markers | |||||||
| hsCRP, mg/L | 1.8 (0.8–4.0) | 1.6 (0.7–3.8) | 1.8 (0.8–3.8) | 1.8 (0.8–4.5) | 1.9 (0.8–4.2) | 2.4 (1.1–5.0) | 0.04 |
| hsCRP ≥2, mg/L | 45.7 | 43.5 | 45.3 | 48.1 | 48.1 | 57.0 | <0.001 |
| IL‐6, pg/mL | 1.5 (1.2) | 1.5 (1.2) | 1.4 (1) | 1.5 (1.1) | 1.4 (1.1) | 1.6 (1.2) | 0.01 |
| Fibrinogen, mg/dL | 343.9 (72.1) | 346.5 (72.3) | 339.5 (70.5) | 338.2 (76.5) | 335.4 (69.2) | 347.9 (71) | <0.001 |
| Subclincal atherosclerosis markers | |||||||
| Internal cIMT, mm | 1.05 (0.59) | 1.05 (0.59) | 1.00 (0.54) | 1.05 (0.60) | 1.05 (0.57) | 1.10 (0.67) | 0.19 |
| Common cIMT, mm | 0.87 (0.19) | 0.87 (0.20) | 0.85 (0.18) | 0.87 (0.19) | 0.86 (0.19) | 0.88 (0.18) | 0.03 |
| CAC >0 | 49.2 | 51.6 | 41.9 | 47.9 | 48.3 | 45.7 | <0.001 |
| CAC ≥75th percentile | 25.0 | 26.8 | 20.1 | 21.3 | 24.6 | 24.6 | 0.002 |
| PAD markers | |||||||
| ABI ≤0.9 | 2.9 | 3.1 | 1.9 | 3.5 | 2.1 | 3.4 | 0.31 |
| ABI ≥1.4 | 0.6 | 0.6 | 0.3 | 0.7 | 1.3 | 1.0 | 0.27 |
| ABI ≤0.9 or ABI ≥1.4 | 3.52 | 3.62 | 2.20 | 4.21 | 3.33 | 4.42 | 0.27 |
Values are expressed as mean (SD), percentage, or median (interquartile range). P values are the differences between groups using 1‐way ANOVA or chi‐square as appropriate. ABI indicates ankle‐brachial index; BMI, body mass index; CAC, coronary artery calcification; CHD, coronary heart disease; CVD, cardiovascular disease; cIMT, carotid intima‐media thickness; hsCRP, high‐sensitivity C‐reactive protein; IL‐6, interleukin 6; LDL‐C, low‐density lipoprotein cholesterol; MESA, Multi‐Ethnic Study of Atherosclerosis; MET, metabolic equivalent; PAD, peripheral arterial disease; SHS, secondhand smoke.
All values are for the entire study sample except for current alcohol use (n=3962), urinary cotinine concentration (n=2983), living with a smoker as a child (n=2977), and living with a smoker as an adult (n=2979).
SHS and Inflammation
After adjustment for age, sex, race/ethnicity, study site, education, income, hypertension medication, systolic blood pressure, diabetes mellitus, LDL‐C, treatment for dyslipidemia, physical activity, and smoking status, participants with ≥12 hours of SHS exposure per week compared with unexposed showed 24% and 10% higher hsCRP and IL‐6 levels, respectively, and a statistically significant trend was observed across increasing categories of SHS exposure for both inflammatory markers (Table 2, model 2). The associations were markedly attenuated for both hsCRP and IL‐6 after adjustment for BMI (model 3), and only the association with hsCRP remained statistically significant (geometric mean ratio, 1.13; 95% CI, 1.02–1.26). The OR of hsCRP ≥2 mg/L was 1.49 (95% CI, 1.19–1.86) and 1.32 (95% CI, 1.04–1.67) before and after adjustment for BMI. No association was found between SHS exposure and fibrinogen.
Table 2.
Association Between SHS Exposure and Inflammation, MESA, United States, 2000–2002
| Unexposed | 1 h/wk | 2–3 h/wk | 4–11 h/wk | ≥12 h/wk | P Trend | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | 3035 | 682 | 428 | 480 | 407 | |||||
| Value | 95% CI | Value | 95% CI | Value | 95% CI | Value | 95% CI | |||
| GM ratio of hsCRP, mg/La | ||||||||||
| Model 1 | 1 (Ref) | 1.08 | 0.99–1.19 | 1.12b | 1.00–1.25b | 1.12b | 1.01–1.25b | 1.26b | 1.13–1.41b | <0.001 |
| Model 2 | 1 (Ref) | 1.08 | 0.98–1.18 | 1.12b | 1.01–1.25b | 1.13b | 1.02–1.26b | 1.24b | 1.11–1.39b | <0.001 |
| Model 3 | 1 (Ref) | 1.06 | 0.98–1.16 | 1.11b | 1.00–1.23b | 1.08 | 0.98–1.19 | 1.13b | 1.02–1.26b | 0.066 |
| Odds ratio of hsCRP ≥2 mg/L | ||||||||||
| Model 1 | 1 (Ref) | 1.12 | 0.94–1.35 | 1.20 | 0.96–1.48 | 1.20 | 0.97–1.47 | 1.52b | 1.22–1.90b | <0.001 |
| Model 2 | 1 (Ref) | 1.12 | 0.93–1.34 | 1.21 | 0.97–1.51 | 1.22 | 0.99–1.51 | 1.49b | 1.19–1.86b | 0.001 |
| Model 3 | 1 (Ref) | 1.10 | 0.91–1.34 | 1.21 | 0.96–1.52 | 1.13 | 0.90–1.41 | 1.32b | 1.04–1.67b | 0.033 |
| GM ratio of IL‐6, pg/mLa | ||||||||||
| Model 1 | 1 (Ref) | 1.00 | 0.95–1.06 | 1.03 | 0.96–1.10 | 0.99 | 0.93–1.05 | 1.11b | 1.04–1.18b | 0.003 |
| Model 2 | 1 (Ref) | 1.01 | 0.95–1.06 | 1.03 | 0.97–1.10 | 0.99 | 0.94. 1.06 | 1.10b | 1.03–1.18b | 0.005 |
| Model 3 | 1 (Ref) | 1.04 | 0.98–1.11 | 0.97 | 0.91–1.03 | 1.02 | 0.96–1.08 | 1.04 | 0.98–1.11 | 0.213 |
| GM ratio of fibrinogen, mg/dLa | ||||||||||
| Model 1 | 1 (Ref) | 1.00 | 0.99–1.02 | 0.98 | 0.97–1.00 | 0.98 | 0.97–1.00 | 1.01 | 0.99–1.03 | 0.443 |
| Model 2 | 1 (Ref) | 1.00 | 0.98–1.02 | 0.98 | 0.97–1.00 | 0.99 | 0.97–1.01 | 1.01 | 0.99–1.03 | 0.380 |
| Model 3 | 1 (Ref) | 1.00 | 0.99–1.02 | 0.98 | 0.96–1.00 | 0.98 | 0.96–1.00 | 1.00 | 0.98–1.02 | 0.793 |
Model 1 is adjusted for age in years, sex (female [reference]/male), race/ethnicity (white [reference], black, Chinese American, Hispanic), study site (Winston‐Salem [reference], New York, Baltimore, St. Paul, Chicago, Los Angeles), education (high school or less [reference], some college but no degree/technical school certificate, associate's degree/bachelor's degree/graduate degree), and income (<$25 000/y [reference]/≥$25 000/y). Model 2 is adjusted for model 1 variables plus hypertension medication (no [reference]/yes), systolic blood pressure (mm Hg), diabetes mellitus (normal [reference] vs untreated diabetes mellitus/treated diabetes mellitus), LDL‐C (mg/dL), treatment for dyslipidemia (no [reference]/yes), physical activity (metabolic equivalent, h/wk), and smoking status (never [reference]/former). Model 3 is adjusted for model 2 variables plus BMI (kg/m2). All values are expressed as odds ratios or geometric mean (GM) ratios, with 95% CIs. For all quartiles of secondhand smoke exposure, reference category is unexposed (0 hours of SHS exposure per week). BMI indicates body mass index; hsCRP, high‐sensitivity C‐reactive protein; IL‐6, interleukin‐6; LDL‐C, low‐density lipoprotein cholesterol; MESA, Multi‐Ethnic Study of Atherosclerosis; SHS, secondhand smoke.
Log‐transformed.
Significant values (P<0.05).
SHS With Subclinical Atherosclerosis
Before adjustment for BMI, participants with ≥12 hours of SHS exposure per week showed higher internal cIMT (geometric mean ratio, 1.04; 95% CI, 1.00–1.09) compared with unexposed (Table 3, model 2). The association was attenuated and not significant after adjustment for BMI (model 3). Common cIMT was associated with SHS exposure in the model adjusted for sociodemographics but not after further adjustment for CVD risk factors. SHS exposure was not associated with the presence of CAC, with CAC levels higher than the 75th percentile, or with PAD (Table 3).
Table 3.
Association of SHS Exposure With Measures of Atherosclerosis, MESA, United States, 2000–2002
| Unexposed | 1 h/wk | 2–3 h/wk | 4–11 h/wk | ≥12 h/wk | P Trend | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | 3035 | 682 | 428 | 480 | 407 | |||||
| Value | 95% CI | Value | 95% CI | Value | 95% CI | Value | 95% CI | |||
| GM ratio of internal cIMT, mma | ||||||||||
| Model 1 | 1 (Ref) | 1.01 | 0.97–1.05 | 1.02 | 0.98–1.06 | 1.02 | 0.98–1.06 | 1.05b | 1.01–1.10b | 0.03 |
| Model 2 | 1 (Ref) | 1.01 | 0.97–1.04 | 1.01 | 0.97–1.05 | 1.01 | 0.97–1.05 | 1.04 | 1.00–1.09 | 0.07 |
| Model 3 | 1 (Ref) | 1.00 | 0.97–1.04 | 1.00 | 0.96–1.05 | 1.01 | 0.97–1.05 | 1.02 | 0.98–1.07 | 0.11 |
| GM ratio of common cIMT, mma | ||||||||||
| Model 1 | 1 (Ref) | 1.00 | 0.98–1.02 | 1.01 | 0.99–1.03 | 1.01 | 0.99–1.02 | 1.02b | 1.01–1.05b | 0.02 |
| Model 2 | 1 (Ref) | 1.00 | 0.98–1.02 | 1.00 | 0.99–1.02 | 1.00 | 0.98–1.02 | 1.02 | 1.00–1.04 | 0.09 |
| Model 3 | 1 (Ref) | 1.00 | 0.99–1.02 | 1.00 | 0.98–1.02 | 0.99 | 0.98–1.02 | 1.01 | 0.99–1.03 | 0.27 |
| Odds ratio of CAC >0 | ||||||||||
| Model 1 | 1 (Ref) | 0.81b | 0.67–0.99b | 1.05 | 0.83–1.32 | 1.05 | 0.84–1.32 | 1.03 | 0.81–1.32 | 0.58 |
| Model 2 | 1 (Ref) | 0.80b | 0.66–0.98b | 1.00 | 0.78–1.27 | 1.00 | 0.80–1.27 | 0.98 | 0.76–1.26 | 0.90 |
| Model 3 | 1 (Ref) | 0.80b | 0.65–0.98b | 0.99 | 0.78–1.26 | 0.99 | 0.78–1.25 | 0.94 | 0.74–1.21 | 0.85 |
| Odds ratio of CAC ≥75th percentile | ||||||||||
| Model 1 | 1 (Ref) | 0.82 | 0.65–1.05 | 0.84 | 0.64–1.12 | 1.06 | 0.82–1.38 | 1.23 | 0.93–1.62 | 0.08 |
| Model 2 | 1 (Ref) | 0.82 | 0.64–1.04 | 0.78 | 0.59–1.05 | 1.01 | 0.77–1.32 | 1.16 | 0.87–1.55 | 0.16 |
| Model 3 | 1 (Ref) | 0.81 | 0.64–1.04 | 0.78 | 0.59–1.05 | 0.99 | 0.76–1.30 | 1.14 | 0.86–1.51 | 0.22 |
| Odds ratio of ABI ≤0.9 | ||||||||||
| Model 1 | 1 (Ref) | 0.87 | 0.47–1.61 | 1.27 | 0.71–2.26 | 0.75 | 0.38–1.48 | 1.21 | 0.67–2.22 | 0.57 |
| Model 2 | 1 (Ref) | 0.82 | 0.43–1.55 | 1.20 | 0.66–2.17 | 0.77 | 0.38–1.53 | 1.19 | 0.65–2.19 | 0.58 |
| Model 3 | 1 (Ref) | 0.82 | 0.43–1.55 | 1.19 | 0.66–2.16 | 0.79 | 0.39–1.59 | 1.23 | 0.67–2.26 | 0.52 |
| Odds ratio of ABI ≥1.4 | ||||||||||
| Model 1 | 1 (Ref) | 0.42 | 0.09–1.86 | 1.06 | 0.30–3.72 | 1.91 | 0.72–5.04 | 1.72 | 0.55–5.35 | 0.22 |
| Model 2 | 1 (Ref) | 0.43 | 0.10–1.92 | 0.98 | 0.28–3.46 | 1.83 | 0.68–4.92 | 1.72 | 0.55–5.42 | 0.22 |
| Model 3 | 1 (Ref) | 0.43 | 0.10–1.92 | 0.98 | 0.28–3.48 | 1.76 | 0.65–4.74 | 1.66 | 0.52–5.24 | 0.25 |
| Odds ratio of ABI ≤0.9 or ABI ≥1.4 | ||||||||||
| Model 1 | 1 (Ref) | 0.74 | 0.42–1.30 | 1.21 | 0.71–2.04 | 0.95 | 0.54–1.65 | 1.29 | 0.75–2.18 | 0.32 |
| Model 2 | 1 (Ref) | 0.69 | 0.38–1.24 | 1.11 | 0.65–1.91 | 0.95 | 0.54–1.67 | 1.22 | 0.72–2.10 | 0.38 |
| Model 3 | 1 (Ref) | 0.69 | 0.38–1.25 | 1.11 | 0.65–1.91 | 0.97 | 0.55–1.70 | 1.25 | 0.73–2.14 | 0.35 |
Model 1 is adjusted for age in years, sex (female [reference]/male), race/ethnicity (white [reference], black, Chinese American, Hispanic), study site (Winston‐Salem [reference], New York, Baltimore, St. Paul, Chicago, Los Angeles), education (high school or less [reference], some college but no degree/technical school certificate, associate's degree/bachelor's degree/graduate degree), and income (<$25 000/y [reference]/≥$25 000/y). Model 2 is adjusted for model 1 variables plus hypertension medication (no [reference]/yes), systolic blood pressure (mm Hg), diabetes mellitus (normal [reference] vs untreated diabetes mellitus/treated diabetes mellitus), low‐density lipoprotein cholesterol (mg/dL), treatment for dyslipidemia (no [reference]/yes), physical activity (metabolic equivalent, h/wk), and smoking status (never [reference]/former). Model 3 is adjusted for model 2 variables plus BMI (kg/m2). All values are expressed as odds ratios or geometric mean (GM) ratios, with 95% CIs. For all quartiles of secondhand smoke exposure, the reference category is unexposed (0 hours of secondhand smoke exposure per week). ABI indicates ankle‐brachial index; BMI, body mass index; CAC, coronary artery calcification; cIMT, carotid intima‐media thickness; MESA, Multi‐Ethnic Study of Atherosclerosis; SHS, secondhand smoke.
Log‐transformed.
Significant values (P<0.05).
Effect Modification
For fibrinogen, the lack of association with SHS exposure remained consistent across participant subgroups evaluated (data not shown). For hsCRP and IL‐6, we found no evidence of interaction by participant characteristics, except for IL‐6 by study site (P interaction=0.03) (Figure 2). The association between SHS exposure and cIMT (common cIMT and internal cIMT) was also consistent across most subgroups evaluated, except for internal cIMT by age (P interaction=0.01) where we observed stronger associations among younger participants (Figure 3). Associations of SHS exposure with inflammatory markers and subclinical atherosclerosis were similar for never‐ and former smokers (Figures 2 and 3). No evidence was found for BMI as a modifier of the association between SHS and the study outcomes (data not shown).
Figure 2.
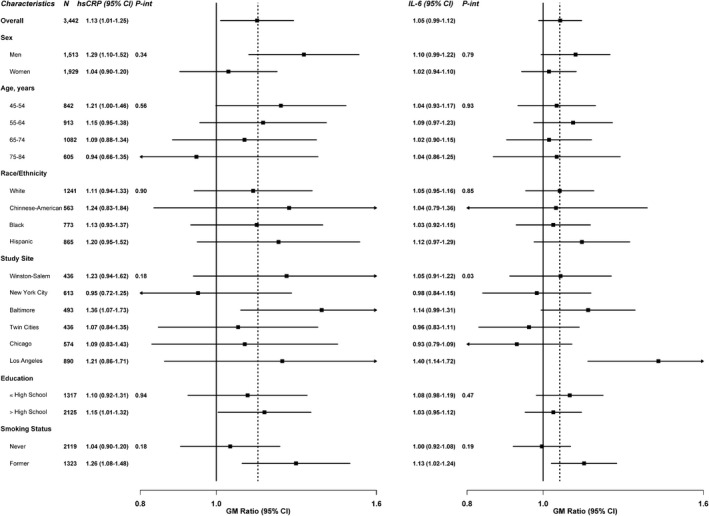
Geometric mean (GM) ratios of high‐sensitivity C‐reactive protein (hsCRP) and interleukin 6 (IL‐6) comparing quartile 4 of secondhand smoke (SHS) to unexposed, stratified by participant characteristics, Multi‐Ethnic Study of Atherosclerosis (MESA), United States, 2000–2002. GM ratios for inflammation comparing the fourth quartile of SHS participants and unexposed SHS (N=3442), by participant characteristics, MESA, 2000–2002. GM ratios were adjusted for age in years, sex, race/ethnicity, study site, education (high school or less [reference], some college but no degree/technical school certificate, associate's degree/bachelor's degree/graduate degree), income (<$25 000/y [reference]/≥$25 000/y), hypertension medication (no [reference]/yes), systolic blood pressure (mm Hg), diabetes mellitus (normal [reference] vs untreated diabetes mellitus/treated diabetes mellitus), low‐density lipoprotein cholesterol (mg/dL), treatment for dyslipidemia (no [reference]/yes), physical activity (metabolic equivalent, h/wk), and smoking status (never [reference]/former), and body mass index (BMI) (kg/m2). Estimated 2‐sided P values for the interaction between SHS exposure with participants’ characteristics were computed using the Wald test.
Figure 3.
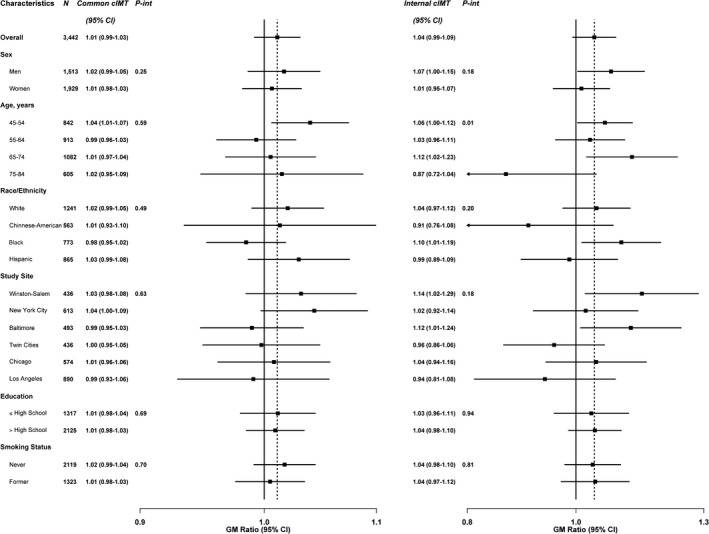
Geometric mean (GM) ratios of common and internal carotid intima‐media thickness (cIMT) comparing quartile 4 of secondhand smoke (SHS) to unexposed, stratified by participant characteristics, Multi‐Ethnic Study of Atherosclerosis (MESA), United States, 2000–2002. GM ratios for subclinical atherosclerosis comparing the fourth quartile of SHS participants and unexposed SHS (N=3442), by participant characteristics, MESA, 2000–2002. GM ratios were adjusted for age in years, sex, race/ethnicity, study site, education (high school or less [reference], some college but no degree/technical school certificate, associate's degree/bachelor's degree/graduate degree), income (<$25 000/y [reference]/≥$25 000/y), hypertension medication (no [reference]/yes), systolic blood pressure (mm Hg), diabetes mellitus (normal [reference] vs untreated diabetes mellitus/treated diabetes mellitus), low‐density lipoprotein cholesterol (mg/dL), treatment for dyslipidemia (no [reference]/yes), physical activity (metabolic equivalent, h/wk), and smoking status (never [reference]/former), and body mass index (BMI) (kg/m2). Estimated 2‐sided P values for the interaction between SHS exposure with participants’ characteristics were computed using the Wald test.
Urinary Cotinine
We found no fully adjusted association between detectable urinary cotinine with inflammatory markers, cIMT or CAC (Table 4). For PAD, the fully adjusted OR for ABI ≤0.9 was 2.21 (95% CI, 1.01–4.83) and for ABI ≤0.9 or ABI ≥1.4 combined was 2.10 (95% CI, 1.09–4.04) (Table 4, model 3).
Table 4.
Association of Detectable Urinary Cotinine With Inflammation and Atherosclerosis, MESA, United States, 2000–2002
| n=2982 | Model 1 | Model 2 | Model 3 | |||
|---|---|---|---|---|---|---|
| Value | 95% CI | Value | 95% CI | Value | 95% CI | |
| Inflammation markers | ||||||
| GM ratio of hsCRP, mg/La | 1.04 | 0.92–1.20 | 1.02 | 0.90–1.16 | 0.97 | 0.86–1.10 |
| Odds ratio of hsCRP ≥2 mg/La | 1.03 | 0.79–1.34 | 0.98 | 0.75–1.28 | 0.90 | 0.68–1.20 |
| GM ratio of IL‐6, pg/mLa | 1.03 | 0.95–1.11 | 1.01 | 0.94–1.10 | 0.99 | 0.92–1.06 |
| GM ratio of fibrinogen, mg/dLa | 0.98 | 0.96–1.00 | 0.98 | 0.96–1.01 | 0.98b | 0.95–1.00b |
| Carotid intima‐media thickness | ||||||
| GM ratio of internal cIMT, mma | 1.07b | 1.01–1.12b | 1.04 | 0.99–1.10 | 1.04 | 0.99–1.09 |
| GM ratio of common cIMT, mma | 1.01 | 0.99–1.03 | 1.00 | 0.98–1.02 | 1.00 | 0.98–1.02 |
| Coronary artery calcification | ||||||
| Odds ratio of CAC >0 | 1.04 | 0.78–1.37 | 0.96 | 0.72–1.28 | 0.94 | 0.70–1.25 |
| Odds ratio of CAC ≥75th percentile | 1.09 | 0.79–1.52 | 0.99 | 0.71–1.39 | 0.98 | 0.69–1.37 |
| Peripheral arterial disease markers | ||||||
| Odds ratio of ABI ≤0.9 | 2.37b | 1.14–4.93b | 2.17b | 1.02–4.62b | 2.21b | 1.01–4.83b |
| Odds ratio of ABI ≥1.4 | 1.70 | 0.51–5.61 | 1.68 | 0.49–5.75 | 1.65 | 0.48–5.64 |
| Odds ratio of ABI ≤0.9 or ABI ≥1.4 | 2.18b | 1.17–4.08b | 2.07b | 1.09–3.92b | 2.10b | 1.09–4.04b |
Model 1 is adjusted for urine creatinine (log‐transformed), age in years, sex (female [reference]/male), race/ethnicity (white [reference], black, Chinese American, Hispanic), study site (Winston‐Salem [reference], New York, Baltimore, St. Paul, Chicago, Los Angeles), education (high school or less [reference], some college but no degree/technical school certificate, associate's degree/bachelor's degree/graduate degree), and income (<$25 000/y [reference]/≥$25 000/y). Model 2 is adjusted for model 1 variables plus hypertension medication (no [reference]/yes), systolic blood pressure (mm Hg), diabetes (normal [reference] vs untreated diabetes/treated diabetes), low‐density lipoprotein cholesterol (mg/dL), treatment for dyslipidemia (no [reference]/yes), physical activity (metabolic equivalent, h/wk), and smoking status (never [reference]/former). Model 3 is adjusted for model 2 variables plus body mass index (kg/m2). All values are expressed as odds ratios or geometric mean (GM) ratios, with 95% CIs. ABI indicates ankle‐brachial index; CAC, coronary artery calcification; cIMT, carotid intima‐media thickness; hsCRP, high‐sensitivity C‐reactive protein; IL‐6, interleukin 6; MESA, Multi‐Ethnic Study of Atherosclerosis.
Log‐transformed.
Significant values (P<0.05).
Discussion
In this ethnically diverse cohort across 6 urban settings in the United States, self‐reported SHS exposure was strongly positively associated with hsCRP and weakly positively associated with IL‐6 and internal cIMT. The associations with hsCRP, IL‐6, and cIMT were markedly attenuated after adjustment for BMI and only the association with hsCRP remained significant after BMI adjustment. SHS exposure in this study was not associated with fibrinogen, common cIMT, CAC, and PAD. In a subset of participants with urinary cotinine available, detectable cotinine was associated with PAD (ABI ≤0.9 and ABI ≤0.9 or ABI ≥1.4), but not with the other subclinical CVD biomarkers evaluated.
hsCRP is an established marker of inflammation that is associated with clinical CVD.51 Several studies have assessed the relationship between SHS and hsCRP. Among adults, SHS exposure was associated with hsCRP levels in most studies,27, 29, 30, 31, 32 although a few studies found no association.26, 28, 33 Among children and adolescents, SHS was positively associated with hsCRP in National Health and Nutrition Examination Survey (NHANES),24 but not in other smaller studies.21, 22, 23 In this study, hours of SHS exposure per week was positively associated with levels of hsCRP; however, urinary cotinine was not associated with hsCRP. The lack of association with urinary cotinine, however, could be related to the use of an assay that had low sensitivity and a large number of undetected samples. The attenuation of the association between SHS and hsCRP after adjustment for BMI may be explained by confounding, as there is a well‐documented positive relationship between hsCRP and BMI.52 Indeed, SHS exposure and obesity disproportionately co‐occur in population groups with low socioeconomic status.2 Alternatively, the attenuation of the association after adjustment for BMI could be related to mediation. The possibility of mediation is supported by prospective evidence showing that SHS exposure is associated with higher adiposity and obesity levels in children53, 54, 55, 56, 57 and adults.58 Randomized animal studies have shown higher weight gain in rats exposed to increasing levels of nicotine in utero,59, 60 but the mechanism has not been fully elucidated.61 Consistent with our findings, the positive association between SHS exposure and hsCRP levels was attenuated after adjustment for BMI in 479 women in the Norwegian Mother and Child Cohort Study.25
For IL‐6, our findings are consistent with other studies also reporting generally null associations.26, 27, 30 Our findings for fibrinogen are mostly inconsistent with other studies, as American,30, 33 Scottish,31 Greek,32 and Japanese35 cohort studies have shown significant positive associations between SHS exposure and fibrinogen. Our study, however, is consistent with another study that found no association between SHS exposure (evaluated by serum cotinine) with fibrinogen among 3221 NHANES (1999–2002) participants aged 20 years and older.28 Currently, a consensus has not been reached on the value of assessing fibrinogen for CVD event prediction.62
There have been studies suggesting an association between SHS with subclinical atherosclerosis and PAD,18, 19, 20 although the studies on PAD have been largely inconsistent. For example, among a cohort of 1209 women in China, a graded (dose‐response) relationship between SHS exposure and prevalent PAD was found,18 while in NHANES 1994–2004 no overall association was found.19 In adult nonsmokers from the Scottish Family Study, exposure to ≥40 hours of SHS per week was significantly and strongly associated with increased risk of prevalent PAD (OR, 5.56; 95% CI, 1.82–17.06).20
CAC is highly predictive of future cardiovascular events.63 Self‐reported SHS exposure was associated with the presence of CAC in a cross‐sectional analysis of 1766 never‐smokers aged 45 to 75 years and free of clinical CVD in Germany, after adjustment for sociodemographic and CVD risk factors (OR, 1.38; 95% CI, 1.03–1.84).17 A similar study found an OR of 1.93 (95% CI, 1.49–2.51) for high versus minimal SHS exposure (based on a calculated score) in 3098 never‐smokers aged 40 to 80 years.14 Several studies have also shown increased cIMT levels with increasing levels of SHS exposure, although these studies were conducted 1 to 2 decades ago.11, 12, 13, 16
Study Strengths and Limitations
The data available to assess SHS exposure in all MESA participants were based on self‐report and referred to a weekly average in the past year. This window of exposure could be more relevant for some outcomes than others. Moreover, the question was subjective and could be interpreted differently by participants. Lifelong SHS exposure or information on living with family members who smoke was not available during the MESA baseline examination. The difficulty in assessing SHS exposure and the importance of accurate measurement of SHS exposure to assess disease risk has been extensively reviewed.4 SHS exposure may be assessed through biomarkers,38 questionnaires,64 or environmental air monitoring.65 Nicotine and its metabolites are commonly used to assess recent SHS exposure. Urinary cotinine is commonly used to differentiate active smoking from SHS exposure, but it can be limited if sensitive methods are not used, as it happened in MESA. Although serum and saliva cotinine are generally preferred to quantify recent SHS exposure compared with urine, they can be costly for large epidemiologic studies. While self‐reported measures of SHS can be affected by substantial measurement error, they provide an important tool to assess the long‐term and short‐term health effects of SHS exposure in large epidemiologic studies.64 In our study, we lacked long‐term information on SHS exposure. Although we adjusted for an important number of sociodemographic and cardiovascular risk factors, residual confounding, for instance by education or socioeconomic status, may remain. Multiple comparisons may also remain a problem. Strengths of this study include the large ethnically diverse modern cohort, rigorous measurements of subclinical CVD outcomes, and the availability of risk factor data.
Our study has several public health implications. Although SHS exposure in the United States has decreased in recent years, cotinine was still detected in 25% of adult nonsmokers and 2 of every 5 children aged 3 to 11 years were exposed to SHS regularly in 2011–2012.2 This still represents ≈100 million American nonsmokers potentially exposed to SHS. Therefore, even a marginal cardiovascular risk with increased SHS exposure is important at the population level. Understanding mechanisms of CVD damage from SHS exposure might better inform tobacco product regulation and issue educational campaigns for the general population. The identification of relevant mechanisms and pathways for SHS exposure can also be useful to inform research for other novel tobacco products for which clinical data would take too long to accrue and for which subclinical data are important. Improvements in SHS exposure assessment are needed in future research. Self‐reported and objective SHS measurements (such as serum cotinine) could be combined to improve exposure assessment in epidemiologic studies. Technological advances in epigenomics and the identification of DNA methylation signatures that are specific for tobacco use also open the possibility to identify signatures of past exposure to SHS.66
Conclusions
This study suggests that SHS exposure may increase the risk of CVD by influencing inflammation and atherosclerosis pathways. The association was stronger for hsCRP than for subclinical measures of atherosclerosis, potentially providing a possible mechanism for the observed short‐term reductions in coronary heart disease admissions following the enactment of smoke‐free laws in many populations around the world.
Sources of Funding
The MESA study, which supplied the data for this analysis, was supported by contracts N01‐HC‐95159 through N01‐HC‐95167, N01‐HC‐95169, and R01‐HL077612 from the National Heart, Lung, and Blood Institute. This analysis was supported by funding from the American Heart Association Tobacco Regulation and Addiction Center (1 P50 HL120163‐01). Dr Jones was supported by a National Research Service Award (T32 CA009314) from the National Cancer Institute. Dr Navas‐Acien was supported by the Flight Attendant Medical Research Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
None.
Supporting information
Table S1. Sociodemographic Characteristics of 5032 Nonsmoking MESA Participants by Smoking Status Exposure Assessment
Acknowledgments
The authors thank the other investigators and the staff of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
(J Am Heart Assoc. 2016;5:e002965 doi: 10.1161/JAHA.115.002965)
References
- 1. Oberg M, Jaakkola MS, Woodward A, Peruga A, Pruss‐Ustun A. Worldwide burden of disease from exposure to second‐hand smoke: a retrospective analysis of data from 192 countries. Lancet. 2011;377:139–146. [DOI] [PubMed] [Google Scholar]
- 2. Homa DM, Neff LJ, King BA, Caraballo RS, Bunnell RE, Babb SD, Garrett BE, Sosnoff CS, Wang L; Centers for Disease Control and Prevention . Vital signs: disparities in nonsmokers’ exposure to secondhand smoke–United States, 1999–2012. MMWR Morb Mortal Wkly Rep. 2015;64:103–108. [PMC free article] [PubMed] [Google Scholar]
- 3. Barnoya J, Glantz SA. Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation. 2005;111:2684–2698. [DOI] [PubMed] [Google Scholar]
- 4. U.S. Department of Health and Human Services . The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2006. [Google Scholar]
- 5. U.S. Department of Health and Human Services . How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking‐Attributable Disease: A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2010. [Google Scholar]
- 6. National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health . Reports of the Surgeon General. The Health Consequences of Smoking‐50 Years of Progress: A Report of the Surgeon General. Atlanta, GA: Centers for Disease Control and Prevention; 2014. [Google Scholar]
- 7. Oono IP, Mackay DF, Pell JP. Meta‐analysis of the association between secondhand smoke exposure and stroke. J Public Health (Oxf). 2011;33:496–502. [DOI] [PubMed] [Google Scholar]
- 8. Jones MR, Barnoya J, Stranges S, Losonczy L, Navas‐Acien A. Cardiovascular events following smoke‐free legislations: an updated systematic review and meta‐analysis. Curr Environ Health Rep. 2014;1:239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. [DOI] [PubMed] [Google Scholar]
- 10. Glantz SA, Parmley WW. Passive smoking and heart disease. Mechanisms and risk. JAMA. 1995;273:1047–1053. [PubMed] [Google Scholar]
- 11. Howard G, Burke GL, Szklo M, Tell GS, Eckfeldt J, Evans G, Heiss G. Active and passive smoking are associated with increased carotid wall thickness. The Atherosclerosis Risk in Communities Study. Arch Intern Med. 1994;154:1277–1282. [PubMed] [Google Scholar]
- 12. Diez‐Roux AV, Nieto FJ, Comstock GW, Howard G, Szklo M. The relationship of active and passive smoking to carotid atherosclerosis 12‐14 years later. Prev Med. 1995;24:48–55. [DOI] [PubMed] [Google Scholar]
- 13. Howard G, Wagenknecht LE, Burke GL, Diez‐Roux A, Evans GW, McGovern P, Nieto FJ, Tell GS. Cigarette smoking and progression of atherosclerosis: the Atherosclerosis Risk in Communities (ARIC) Study. JAMA. 1998;279:119–124. [DOI] [PubMed] [Google Scholar]
- 14. Yankelevitz DF, Henschke CI, Yip R, Boffetta P, Shemesh J, Cham MD, Narula J, Hecht HS; FAMRI‐IELCAP Investigators . Second‐hand tobacco smoke in never smokers is a significant risk factor for coronary artery calcification. JACC Cardiovasc Imaging. 2013;6:651–657. [DOI] [PubMed] [Google Scholar]
- 15. Chen W, Yun M, Fernandez C, Li S, Sun D, Lai CC, Hua Y, Wang F, Zhang T, Srinivasan SR, Johnson CC, Berenson GS. Secondhand smoke exposure is associated with increased carotid artery intima‐media thickness: the Bogalusa Heart Study. Atherosclerosis. 2015;240:374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gall S, Huynh QL, Magnussen CG, Juonala M, Viikari JS, Kähönen M, Dwyer T, Raitakari OT, Venn A. Exposure to parental smoking in childhood or adolescence is associated with increased carotid intima‐media thickness in young adults: evidence from the Cardiovascular Risk in Young Finns study and the Childhood Determinants of Adult Health Study. Eur Heart J. 2014;35:2484–2491. [DOI] [PubMed] [Google Scholar]
- 17. Peinemann F, Moebus S, Dragano N, Möhlenkamp S, Lehmann N, Zeeb H, Erbel R, Jöckel KH, Hoffmann B; Heinz Nixdorf Recall Study Investigative Group . Secondhand smoke exposure and coronary artery calcification among nonsmoking participants of a population‐based cohort. Environ Health Perspect. 2011;119:1556–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He Y, Lam TH, Jiang B, Wang J, Sai X, Fan L, Li X, Qin Y, Hu FB. Passive smoking and risk of peripheral arterial disease and ischemic stroke in Chinese women who never smoked. Circulation. 2008;118:1535–1540. [DOI] [PubMed] [Google Scholar]
- 19. Agarwal S. The association of active and passive smoking with peripheral arterial disease: results from NHANES 1999–2004. Angiology. 2009;60:335–345. [DOI] [PubMed] [Google Scholar]
- 20. Lu L, Mackay DF, Pell JP. Association between level of exposure to secondhand smoke and peripheral arterial disease: cross‐sectional study of 5,686 never smokers. Atherosclerosis. 2013;229:273–276. [DOI] [PubMed] [Google Scholar]
- 21. Groner JA, Huang H, Nagaraja H, Kuck J, Bauer JA. Secondhand smoke exposure and endothelial stress in children and adolescents. Acad Pediatr. 2015;15:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matsunaga Y, Vardavas CI, Plada M, Wärnberg J, Gómez‐Martinez S, Tzatzarakis MN, Tsatsakis AM, Díaz EL, Marcos A, Kafatos AG. The relationship between cotinine concentrations and inflammatory markers among highly secondhand smoke exposed non‐smoking adolescents. Cytokine. 2014;66:17–22. [DOI] [PubMed] [Google Scholar]
- 23. Merghani TH, Saeed A, Alawad A. Changes in plasma IL4, TNFa and CRP in response to regular passive smoking at home among healthy school children in Khartoum, Sudan. Afr Health Sci. 2012;12:41–47. [PMC free article] [PubMed] [Google Scholar]
- 24. Wilkinson JD, Lee DJ, Arheart KL. Secondhand smoke exposure and C‐reactive protein levels in youth. Nicotine Tob Res. 2007;9:305–307. [DOI] [PubMed] [Google Scholar]
- 25. Cupul‐Uicab LA, Skjaerven R, Haug K, Travlos GS, Wilson RE, Eggesbø M, Hoppin JA, Whitworth KW, Longnecker MP. Exposure to tobacco smoke in utero and subsequent plasma lipids, ApoB, and CRP among adult women in the MoBa cohort. Environ Health Perspect. 2012;120:1532–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bonetti PO, Lardi E, Geissmann C, Kuhn MU, Bruesch H, Reinhart WH. Effect of brief secondhand smoke exposure on endothelial function and circulating markers of inflammation. Atherosclerosis. 2011;215:218–222. [DOI] [PubMed] [Google Scholar]
- 27. Chiu YH, Spiegelman D, Dockery DW, Garshick E, Hammond SK, Smith TJ, Hart JE, Laden F. Secondhand smoke exposure and inflammatory markers in nonsmokers in the trucking industry. Environ Health Perspect. 2011;119:1294–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Clark JD III, Wilkinson JD, LeBlanc WG, Dietz NA, Arheart KL, Fleming LE, Lee DJ. Inflammatory markers and secondhand tobacco smoke exposure among U.S. workers. Am J Ind Med. 2008;51:626–632. [DOI] [PubMed] [Google Scholar]
- 29. Hamer M, Stamatakis E, Kivimaki M, Lowe GD, Batty GD. Objectively measured secondhand smoke exposure and risk of cardiovascular disease: what is the mediating role of inflammatory and hemostatic factors? J Am Coll Cardiol. 2010;56:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jefferis BJ, Lowe GD, Welsh P, Rumley A, Lawlor DA, Ebrahim S, Carson C, Doig M, Feyerabend C, McMeekin L, Wannamethee SG, Cook DG, Whincup PH. Secondhand smoke (SHS) exposure is associated with circulating markers of inflammation and endothelial function in adult men and women. Atherosclerosis. 2010;208:550–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu L, Mackay DF, Newby DE, Pell JP. Association between salivary cotinine and cardiovascular biomarkers among nonsmokers and current smokers: cross‐sectional study of 10,081 participants. Eur J Vasc Endovasc Surg. 2014;48:703–710. [DOI] [PubMed] [Google Scholar]
- 32. Panagiotakos DB, Pitsavos C, Chrysohoou C, Skoumas J, Masoura C, Toutouzas P, Stefanadis C; ATTICA study . Effect of exposure to secondhand smoke on markers of inflammation: the ATTICA study. Am J Med. 2004;116:145–150. [DOI] [PubMed] [Google Scholar]
- 33. Venn A, Britton J. Exposure to secondhand smoke and biomarkers of cardiovascular disease risk in never‐smoking adults. Circulation. 2007;115:990–995. [DOI] [PubMed] [Google Scholar]
- 34. Zhang J, Fang SC, Mittleman MA, Christiani DC, Cavallari JM. Secondhand tobacco smoke exposure and heart rate variability and inflammation among non‐smoking construction workers: a repeated measures study. Environ Health. 2013;12:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iso H, Shimamoto T, Sato S, Koike K, Iida M, Komachi Y. Passive smoking and plasma fibrinogen concentrations. Am J Epidemiol. 1996;144:1151–1154. [DOI] [PubMed] [Google Scholar]
- 36. Bild DE, Bluemke DA, Burke GL, Detrano R, Diez‐Roux AV, Folsom AR, Greenland P, Jacob DR Jr, Kronmal R, Liu K, Nelson JC, O'Leary D, Saad MF, Shea S, Szklo M, Tracy RP. Multi‐Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–881. [DOI] [PubMed] [Google Scholar]
- 37. Rodriguez J, Jiang R, Johnson WC, MacKenzie BA, Smith LJ, Barr RG. The association of pipe and cigar use with cotinine levels, lung function, and airflow obstruction: a cross‐sectional study. Ann Intern Med. 2010;152:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Avila‐Tang E, Al‐Delaimy WK, Ashley DL, Benowitz N, Bernert JT, Kim S, Samet JM, Hecht SS. Assessing secondhand smoke using biological markers. Tob Control. 2013;22:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jenny NS, Brown ER, Detrano R, Folsom AR, Saad MF, Shea S, Szklo M, Herrington DM, Jacobs DR Jr. Associations of inflammatory markers with coronary artery calcification: results from the Multi‐Ethnic Study of Atherosclerosis. Atherosclerosis. 2010;209:226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Keenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ; JUPITER Study Group . Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 41. Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, Liu K, Shea S, Szklo M, Bluemke DA, O'Leary DH, Tracy R, Watson K, Wong ND, Kronmal RA. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336–1345. [DOI] [PubMed] [Google Scholar]
- 42. Carr JJ, Nelson JC, Wong ND, McNitt‐Gray M, Arad Y, Jacobs DR Jr, Sidney S, Bild DE, Williams OD, Detrano RC. Calcified coronary artery plaque measurement with cardiac CT in population‐based studies: standardized protocol of Multi‐Ethnic Study of Atherosclerosis (MESA) and Coronary Artery Risk Development in Young Adults (CARDIA) study. Radiology. 2005;234:35–43. [DOI] [PubMed] [Google Scholar]
- 43. Criqui MH, McClelland RL, McDermott MM, Allison MA, Blumenthal RS, Aboyans V, Ix JH, Burke GL, Liu K, Shea S. The ankle‐brachial index and incident cardiovascular events in the Multi‐Ethnic Study of Atherosclerosis (MESA). J Am Coll Cardiol. 2010;56:1506–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, National High Blood Pressure Education Program Coordinating Committee . The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560–2572. [DOI] [PubMed] [Google Scholar]
- 45. Holvoet P, Jenny NS, Schreiner PJ, Tracy RP, Jacobs DR. The relationship between oxidized LDL and other cardiovascular risk factors and subclinical CVD in different ethnic groups: the Multi‐Ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2007;194:245–252. [DOI] [PubMed] [Google Scholar]
- 46. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 47. American Diabetes Association . Diagnosis and classification of diabetes mellitus. Diabetes Care. 2004;27(suppl 1):S5–S10. [DOI] [PubMed] [Google Scholar]
- 48. Storti KL, Arena VC, Barmada MM, Bunker CH, Hanson RL, Laston SL, Yeh JL, Zmuda JM, Howard BV, Kriska AM. Physical activity levels in American‐Indian adults: the Strong Heart Family Study. Am J Prev Med. 2009;37:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cupul‐Uicab LA, Skjaerven R, Haug K, Melve KK, Engel SM, Longnecker MP. In utero exposure to maternal tobacco smoke and subsequent obesity, hypertension, and gestational diabetes among women in the MoBa cohort. Environ Health Perspect. 2012;120:355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Agresti A. Categorical Data Analysis. 2nd ed Hoboken, NJ: Wiley‐Interscience; 2002. [Google Scholar]
- 51. Ridker PM. High‐sensitivity C‐reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation. 2001;103:1813–1818. [DOI] [PubMed] [Google Scholar]
- 52. Brooks GC, Blaha MJ, Blumenthal RS. Relation of C‐reactive protein to abdominal adiposity. Am J Cardiol. 2010;106:56–61. [DOI] [PubMed] [Google Scholar]
- 53. Leary SD, Smith GD, Rogers IS, Reilly JJ, Wells JC, Ness AR. Smoking during pregnancy and offspring fat and lean mass in childhood. Obesity (Silver Spring). 2006;14:2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Raum E, Kupper‐Nybelen J, Lamerz A, Hebebrand J, Herpertz‐Dahlmann B, Brenner H. Tobacco smoke exposure before, during, and after pregnancy and risk of overweight at age 6. Obesity (Silver Spring). 2011;19:2411–2417. [DOI] [PubMed] [Google Scholar]
- 55. Yang S, Decker A, Kramer MS. Exposure to parental smoking and child growth and development: a cohort study. BMC Pediatr. 2013;13:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Riedel C, Schonberger K, Yang S, Koshy G, Chen YC, Gopinath B, Ziebarth S, von Kries R. Parental smoking and childhood obesity: higher effect estimates for maternal smoking in pregnancy compared with paternal smoking–a meta‐analysis. Int J Epidemiol. 2014;43:1593–1606. [DOI] [PubMed] [Google Scholar]
- 57. McConnell R, Shen E, Gilliland FD, Jerrett M, Wolch J, Chang CC, Lurmann F, Berhane K. A longitudinal cohort study of body mass index and childhood exposure to secondhand tobacco smoke and air pollution: the Southern California Children's Health Study. Environ Health Perspect. 2015;123:360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Harris HR, Willett WC, Michels KB. Parental smoking during pregnancy and risk of overweight and obesity in the daughter. Int J Obes. 2013;37:1356–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Oliveira E, Moura EG, Santos‐Silva AP, Fagundes AT, Rios AS, Abreu‐Villaça Y, Nogueira Neto JF, Passos MC, Lisboa PC. Short‐ and long‐term effects of maternal nicotine exposure during lactation on body adiposity, lipid profile, and thyroid function of rat offspring. J Endocrinol. 2009;202:397–405. [DOI] [PubMed] [Google Scholar]
- 60. Somm E, Schwitzgebel VM, Vauthay DM, Camm EJ, Chen CY, Giacobino JP, Sizonenko SV, Aubert ML, Hüppi PS. Prenatal nicotine exposure alters early pancreatic islet and adipose tissue development with consequences on the control of body weight and glucose metabolism later in life. Endocrinology. 2008;149:6289–6299. [DOI] [PubMed] [Google Scholar]
- 61. Sharma AJ, Cogswell ME, Li R. Dose‐response associations between maternal smoking during pregnancy and subsequent childhood obesity: effect modification by maternal race/ethnicity in a low‐income US cohort. Am J Epidemiol. 2008;168:995–1007. [DOI] [PubMed] [Google Scholar]
- 62. Myers GL, Christenson RH, Cushman M, Ballantyne CM, Cooper GR, Pfeiffer CM, Grundy SM, Labarthe DR, Levy D, Rifai N, Wilson PW. National Academy of Clinical Biochemistry Laboratory Medicine Practice guidelines: emerging biomarkers for primary prevention of cardiovascular disease. Clin Chem. 2009;55:378–384. [DOI] [PubMed] [Google Scholar]
- 63. Folsom AR, Kronmal RA, Detrano RC, O'Leary DH, Bild DE, Bluemke DA, Budoff MJ, Liu K, Shea S, Szklo M, Tracy RP, Watson KE, Burke GL. Coronary artery calcification compared with carotid intima‐media thickness in the prediction of cardiovascular disease incidence: the Multi‐Ethnic Study of Atherosclerosis (MESA). Arch Intern Med. 2008;168:1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Avila‐Tang E, Elf JL, Cummings KM, Fong GT, Hovell MF, Klein JD, McMillen R, Winickoff JP, Samet JM. Assessing secondhand smoke exposure with reported measures. Tob Control. 2013;22:156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Apelberg BJ, Hepp LM, Avila‐Tang E, Gundel L, Hammond SK, Hovell MF, Hyland A, Klepeis NE, Madsen CC, Navas‐Acien A, Repace J, Samet JM, Breysse PN. Environmental monitoring of secondhand smoke exposure. Tob Control. 2013;22:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ladd‐Acosta C. Epigenetic signatures as biomarkers of exposure. Curr Environ Health Rep. 2015;2:117–125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sociodemographic Characteristics of 5032 Nonsmoking MESA Participants by Smoking Status Exposure Assessment