

Abstract
Background
Compared to uninfected adults, HIV‐infected adults on antiretroviral therapy are at increased risk of cardiovascular disease. Given the increase in T‐cell dysfunction, inflammation, and coagulation in HIV infection, microvascular dysfunction is thought to contribute to this excess cardiovascular risk. However, the relationships between these variables remain undefined.
Methods and Results
This was a cross‐sectional study of 358 HIV‐infected adults from the SCOPE cohort. Macrovascular endothelial function was assessed using flow‐mediated dilation of the brachial artery and microvascular function by reactive hyperemia. T‐cell phenotype was determined by flow cytometry. Plasma markers of inflammation (tumor necrosis factor‐α, interleukin‐6, high‐sensitivity C‐reactive protein, sCD14) and coagulation (fibrinogen, D‐dimer) were also measured. In all HIV+ subjects, markers of inflammation (tumor necrosis factor‐α, high‐sensitivity C‐reactive protein), coagulation (D‐dimer) and T‐cell activation (CD8+PD1+, CD4+interferon+cytomegalovirus‐specific) were associated with worse reactive hyperemia after adjusting for traditional cardiovascular risk factors and co‐infections. In treated and suppressed subjects, tumor necrosis factor‐α and CD8+PD1+ cells remained associated with worse reactive hyperemia after adjustment. Compared to the untreated subjects, CD8+PD1+ cells were increased in the virally suppressed group. Reactive hyperemia was predictive of flow‐mediated dilation.
Conclusions
CD8+PD1+ cells and tumor necrosis factor‐α were associated with microvascular dysfunction in all HIV+ subjects and the treated and suppressed group. Additionally, D‐dimer, high‐sensitivity C‐reactive protein, sCD‐14, and interleukin‐6 were associated with microvascular dysfunction in all HIV+ subjects. Although T‐cell dysfunction, inflammation, and microvascular dysfunction are thought to play a role in cardiovascular disease in HIV, this study is the first to look at which T‐cell and inflammatory markers are associated with microvascular dysfunction in HIV‐infected individuals.
Keywords: coagulation, HIV, immune system, inflammation, microcirculation
Subject Categories: Coronary Artery Disease, Endothelium/Vascular Type/Nitric Oxide, Inflammation, Vascular Biology, Pathophysiology
Introduction
HIV‐infected individuals have high rates of cardiovascular disease including acute myocardial infarction, heart failure, and arrhythmias.1, 2, 3, 4, 5, 6 The mechanism underlying this excess risk remains largely unknown but likely includes an interplay between traditional risk factors, antiretroviral therapy (ART), along with other HIV‐related features.7, 8, 9
A hallmark of HIV infection is increased inflammation that is present even in the setting of treated and suppressed HIV disease.10 This chronic inflammation is thought to underlie non‐AIDS events including cardiovascular disease.11 In the SMART study, inflammatory and coagulation markers were strongly predictive of mortality and cardiovascular disease independent of viral load or CD4+ count.12, 13, 14
Several different pathways contribute to this chronic inflammatory state in HIV.15, 16 Ongoing activation of the immune system in virally suppressed individuals occurs through persistent low‐level viral replication in protected cells and tissue, translocation of gut microbial products due to breakdown of gut mucosal defenses, and reactivation of chronic infections such as cytomegalovirus.17, 18, 19, 20
Both innate and adaptive immunity play an important role in the pathogenesis of atherosclerosis in HIV‐uninfected individuals.21 Antigens such as oxidized low‐density lipoprotein are recognized as pathogens by dendritic cells, a part of the innate immune system. The dendritic cells present these antigens to naïve T cells, leading to T‐cell activation.22 Activated T cells are defined by markers including CD38, HLA‐DR, CCR5, and PD‐1.23 These activated T cells are significantly increased in HIV‐infected individuals and correlate with viral load and disease stage.23 Some studies have shown that T cell and monocyte activation are independently associated with subclinical atherosclerosis in HIV‐infected individuals on ART as measured by carotid intimal medial thickness or carotid artery stiffness.24, 25, 26, 27, 28, 29, 30, 31 However, other studies have not found a relationship between T‐cell activation and carotid intimal medial thickness or subsequent cardiovascular events in HIV‐infected individuals.32, 33
The vast majority of cardiovascular disease studies in HIV‐infected adults have focused on clinical end points or measures of large‐vessel (macrovascular) function. These include studies of flow‐mediated dilation (FMD), coronary calcification, and carotid intimal medial thickness progression. FMD, a measure of endothelial dysfunction, assesses key steps in the initiation, perpetuation, and clinical manifestations of atherogenesis.34, 35 However, the consistent findings that systemic markers of inflammation and hypercoagulability are elevated in HIV disease and strongly associated with cardiovascular disease suggest that microvascular disease may in fact be more prevalent in this patient population.10, 36 Reactive hyperemia (RH) in the brachial artery is an assessment of microvascular function and serves as the stimulus for FMD. It is measured by Doppler and can be determined along with FMD during brachial artery reactivity studies.37 Prior studies have shown that both RH and FMD are lower in HIV‐infected individuals compared to healthy controls.38, 39
The purpose of this study was to elucidate the role of inflammation, hypercoagulability, and T‐cell immunity as potential determinants of endothelial function, particularly microvascular function, in HIV‐infected individuals. In assessing endothelial function, we measured the response of the microvasculature and the macrovasculature using RH and FMD, respectively. Endothelial dysfunction occurs early on in atherosclerosis and precedes the development of morphological changes in the vasculature.34 Thus, determining whether inflammation, hypercoagulability, and T‐cell dysfunction are associated with endothelial dysfunction in HIV‐infected individuals is an important step in understanding the mechanism underlying HIV‐associated atherosclerosis and heightened cardiovascular risk in the setting of HIV.
Methods
Patient Selection
This was a cross‐sectional study of HIV‐infected adults from the SCOPE cohort. SCOPE is an observational prospective cohort based on the HIV/AIDS clinics at the San Francisco General Hospital and the San Francisco Veterans Affairs Medical Center. Each participant undergoes an in‐depth questionnaire interview detailing their HIV disease history and overall medical history including cardiac risk factors. Participants in the SCOPE cohort are seen every 4 months for blood tests and are given questionnaires regarding medications, health‐related behaviors, and symptoms. The University of California, San Francisco Committee on Human Research approved the study and all participants provided written informed consent.
With respect to cardiac risk factors, hypertension and diabetes mellitus (DM) were defined using standard classifications. Total cholesterol, high‐density lipoprotein cholesterol, low‐density lipoprotein cholesterol, and triglycerides were measured after fasting for 12 hours. For HIV‐specific factors, we measured current as well as nadir CD4 counts. Given that CD4/CD8 ratio is emerging as a prognostic marker and is independently associated with non‐AIDS events or death, current CD8 levels were also measured.40 There were a total of 358 participants in this study, with 250 on ART with suppressed HIV RNA levels (treated suppressed). There were 61 participants off ART with detectable HIV RNA (untreated noncontrollers), 25 participants off ART with a suppressed viral load (elite controllers), and 22 participants on ART with an elevated viral load. Total duration of ART including the duration for nucleoside reverse transcriptase inhibitors, non‐nucleoside reverse transcriptase inhibitors, and protease inhibitors was measured for each individual.
Measurement of Plasma Biomarkers
Soluble markers of inflammation (interleukin‐6, tumor necrosis factor [TNF]‐α, high‐sensitivity C‐reactive protein, sCD14), coagulation (D‐dimer, fibrinogen), macrophage activation (sCD163), and prior cytomegalovirus exposure (cytomegalovirus immunoglobulin G [IgG]) were measured in cryopreserved plasma samples using a multiplex electrochemiluminescence assay (Meso Scale Discovery, MD).
Measurement of T‐Cell Phenotype
T‐cell phenotypes were measured by immunophenotyping performed at the University of California, San Francisco, Core Immunology Laboratory, using methods that have been optimized and validated for frozen peripheral blood mononuclear cells.30 Briefly, cryopreserved peripheral blood mononuclear cells were rapidly thawed in warm media, counted on Accuri C6 (Biosciences) with the Viacount assay (Millipore), washed, and stained with Aqua Amine Reactive Dye to discriminate dead cells. Cells were then stained with the following fluorescently conjugated monoclonal antibodies: CD3‐Pacific Blue, CD4‐PE Texas Red, CD8‐QDot 605, CD38‐PE, HLA‐DR‐FITC, CCR5‐PECY5, and PD‐1‐Alexa647. Stained cells were washed, fixed in 0.5% formaldehyde, and held at 4°C until analysis where they were run on a customized BD LSR II (BD Bioscience). A total of 100,000 lymphocytes were collected for each sample. Data were compensated and analyzed using FlowJo (Tree Star) to determine the proportion of CD4+ and CD8+ T cells expressing each of the T‐cell markers (CD38, HLA‐DR, CCR5, and/or PD‐1). Combinations of markers were calculated in FlowJo, using the Boolean gate function. Cytomegalovirus‐specific T‐cell responses were determined by measuring the proportion of CD4+ and CD8+ T cells that expressed interferon (IFN)‐γ after exposure to cytomegalovirus pp65 peptides using flow cytometry.
Measurement of Endothelial Function
High‐resolution 10‐MHz linear array ultrasound probe and the GE Vivid 7 Imaging Console were used to measure endothelial function in the right brachial artery immediately proximal to the antecubital fossa. Participants were asked not to consume any food, alcohol, caffeine, or nicotine 12 hours prior to the study. Measurements were done in a dark and quiet room while the participant was lying supine. FMD was quantified by inflating a forearm cuff to a suprasystolic pressure for 5 minutes in order to create an ischemic stimulus. Subsequently, the change in brachial artery diameter was measured every 15 s using B‐mode ultrasound 30 to 120 s following cuff deflation. FMD was calculated as the percent change between the maximum post cuff release brachial artery diameter and the baseline diameter. RH was quantified using spectral Doppler images for the first 15 s following cuff release. Maximal RH was measured as the Doppler mean velocity‐time integral of the first 3 complete beats after cuff release. Analysis of digitized images was performed using dedicated software (Medical Imaging Applications, LLC, Coralville, IA). The level of reproducibility for both FMD and RH is excellent in our laboratory as described in previous studies.41 On repeat assessments of FMD, the coefficient of variation has been 2.7% and the intraclass correlation coefficient has been 0.98, indicating high reproducibility. Similarly, on repeat measurements of RH, the coefficient of variation has been 0.65% and the intraclass correlation coefficient has been 0.99.
Statistical Analyses
For all HIV groups, continuous variables were expressed as the median (and interquartile range). Categorical variables were expressed as percent. We used both unadjusted and multivariable adjusted generalized linear regression (using a log link function)42 in order to examine relationships of T‐cell phenotypes and inflammatory markers with endothelial function, in separate models for FMD and RH. Multivariable models were adjusted for demographics (age, sex, race/ethnicity), traditional cardiac risk factors (DM, hypertension, hyperlipidemia, and smoking), and co‐infections (hepatitis C virus, opportunistic infections). Each T‐cell marker and plasma marker was included individually and not simultaneously in the models. Hepatitis B virus and concurrent neoplasms were also identified but given their low prevalence in the cohort, they were not incorporated into the multivariable models. We constructed models using all HIV+ subjects, and also using the treated suppressed group only. Similar models were constructed to determine the association between HIV‐related factors and endothelial function. Each HIV‐related factor was included individually and not simultaneously in the models. Measures that were right‐skewed such as T‐cell phenotypes and inflammatory markers were log transformed in order to normalize their distributions. We calculated the standardized regression coefficients for T‐cell phenotypes, inflammatory markers, and HIV‐related continuous variables to reflect the effects with 1 SD increment. Since FMD and RH also showed right‐skewed distributions, each outcome was log‐transformed for analysis; results were back‐transformed to produce estimated percentage differences.
We also sought to determine whether inflammatory markers were important mediators of the relationship between T‐cell phenotypes and endothelial dysfunction in the treated and suppressed group.42 An inflammatory marker was considered a mediator if it was significantly associated with the T‐cell marker and endothelial dysfunction (at the 0.05 level in multivariable analysis). Mediation models were constructed for that T‐cell marker if it showed a significant association with endothelial dysfunction (also at the 0.05 level in multivariable analysis). The coefficients estimating the associations between the T‐cell phenotype and endothelial dysfunction were compared in models before and after adjustment for the inflammatory marker. Diminishing coefficient would indicate complete or partial mediation effect by inflammatory markers. Additionally, we tested for interactions between T‐cell phenotypes and inflammatory markers in order to assess the presence of moderated mediation. All analyses were performed using the SAS system, version 9.4 (SAS Institute, Inc, Cary, NC).
Results
Clinical Characteristics
We studied 358 HIV‐infected individuals. A large majority, 250 subjects, were in the treated and suppressed group. About 24% of the subjects were untreated and only 6% were on antiretroviral therapy but still had a detectable viral load. As shown in Table 1, the median age was 50 years and 84% were male. Sixty‐one percent were white, 22% black, and 11% Hispanic. The median duration of HIV infection was 15 years and the median CD4 T‐cell count was 542 cells/mm3. While many of the individuals had cardiovascular risk factors such as hypertension (42%) and smoking (31%), relatively few had DM (8%), and the median total cholesterol was within the normal range. The traditional risk factor profile was similar among the treated and suppressed group as compared to the other HIV‐infected individuals. Nearly all of the HIV‐infected participants were cytomegalovirus seropositive (98%).
Table 1.
Summary of Demographics and Clinical Characteristics
| All Groups N=358 | Treated and Suppressed N=250 | Untreated and Unsuppressed N=61 | |
|---|---|---|---|
| Demographics | |||
| Age, ya | 50 (43, 56) | 51 (44, 58) | 44 (39, 54) |
| White | 61% | 65% | 52% |
| Black | 22% | 18% | 31% |
| Hispanic | 11% | 12% | 10% |
| Other | 6% | 5% | 7% |
| Male | 84% | 88% | 77% |
| Cardiovascular risk factors | |||
| Hypertension | 42% | 40% | 41% |
| Diabetes mellitus | 8% | 8% | 7% |
| Current smoking | 31% | 25% | 46% |
| Smoking pack yearsa | 2 (0, 15) | 1 (0, 15) | 2 (0, 13) |
| LDL, mg/dLa | 102 (83, 123) | 103 (86, 123) | 102 (80, 127) |
| HDL, mg/dLa | 45 (37, 53) | 45 (37, 55) | 44 (36, 50) |
| Total cholesterol, mg/dLa | 177 (154, 201) | 179 (156, 204) | 171 (148, 198) |
| Triglycerides, mg/dLa | 116 (84, 192) | 122 (89, 202) | 100 (69, 127) |
| HIV‐related factors | |||
| Current CD4, cells/mm3 a | 542 (339, 720) | 535 (319, 709) | 536 (370, 701) |
| Nadir CD4, cells/mm3 a | 188 (45, 315) | 130 (29, 234) | 350 (272, 458) |
| CD4/CD8 ratioa | 0.58 (0.38, 0.92) | 0.62 (0.40, 0.98) | 0.50 (0.32, 0.76) |
| CD8, cells/mm3 a | 892 (666, 1224) | 852 (632, 1105) | 1069 (752, 1393) |
| HIV duration, ya | 15 (7, 20) | 16 (8, 21) | 9 (4, 20) |
| NNRTI use | 51% | 62% | 10% |
| NRTI use | 80% | 100% | 16% |
| Protease inhibitor use | 62% | 78% | 8% |
| Hepatitis C infection | 18% | 15% | 16% |
| Hepatitis B infection | 5% | 4% | 5% |
| Opportunistic infection | 13% | 12% | 8% |
| Neoplasms (non‐skin cancer) | 4% | 5% | 2% |
| Disease categories | |||
| Off ART, VL <75 copies/mL | 7% | — | — |
| Off ART, VL >75 copies/mL | 17% | — | 100% |
| On ART, VL <75 copies/mL | 70% | 100% | — |
| On ART, VL >75 copies/mL | 6% | — | — |
| Endothelial function | |||
| Reactive hyperemia (%)a | 61.5 (46.3, 83.3) | 59.2 (44.9, 82.2) | 66.9 (51.9, 85.6) |
| Flow‐mediated dilation (%)a | 4.0 (2.6, 5.5) | 3.9 (2.6, 5.4) | 4.2 (2.6, 5.6) |
ART indicates antiretroviral therapy; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; NNRTI, non‐nucleoside reverse transcriptase inhibitors; NRTI, nucleoside reverse transcriptase inhibitors; VL, viral load.
Continuous variables were summarized as median (interquartile range).
Microvascular and Macrovascular Function Are Strongly Correlated
We first examined the relationship between RH and FMD. In unadjusted analysis for all HIV+ subjects, RH was positively and strongly associated with FMD (25% increase in FMD per doubling of RH, 95% CI [15%, 37%], P<0.01). This relationship remained significant after multivariable adjustment for demographics and traditional cardiovascular disease risk factors (26% increase in FMD per doubling of RH, 95% CI [16%, 37%], P<0.01). Similarly, in the treated suppressed group, there was a 23% increase in FMD per doubling of RH after adjustment (95% CI [12%, 35%], P<0.01).
Expression of T‐Cell Markers Is Increased in the Treated Suppressed Group
Compared to subjects who were untreated with an elevated viral load, those in the treated and suppressed group had significantly increased expression of multiple different T‐cell markers (Table 2). This was true for both CD4+ and CD8+ T cells expressing PD1. However, there was no difference between the 2 groups in the CD4+ IFN+ cytomegalovirus specific T‐cell subset.
Table 2.
Expression of T‐Cell Markers in Treated and Suppressed Versus Untreated Subjects
| T Cells | Treated and Suppressed % (Median, IQR) | Untreated % (Median, IQR) | P Value |
|---|---|---|---|
| CD4+ CD38+ DR+ | 0.7 (0.4, 1.5) | 0.4 (0.2, 1.1) | 0.02 |
| CD4+ PD1+ | 0.8 (0.4, 1.6) | 0.3 (0.2, 0.6) | <0.01 |
| CD4+ CCR5+ CD38+ DR+ PD1+ | 1.1 (0.6, 2.3) | 0.5 (0.3, 0.6) | <0.01 |
| CD8+ CD38+ DR+ | 3.9 (1.9, 5.4) | 3.4 (2.0, 4.3) | 0.23 |
| CD8+ PD1+ | 1.9 (1.1, 3.8) | 1.2 (0.7, 1.6) | <0.01 |
| CD8+ CCR5+ CD38+ DR+ PD1+ | 4.6 (2.8, 7.9) | 1.7 (1.0, 3.6) | <0.01 |
| CD4+ IFN+ CMV‐specific | 0.4 (0.2, 1.1) | 0.3 (0.2, 1.2) | 0.66 |
| CD8+ IFN+ CMV‐specific | 5.7 (2.6, 9.9) | 3.2 (2.5, 4.6) | 0.04 |
CMV indicates cytomegalovirus; IFN, interferon; IQR, interquartile range.
CD8+PD1+ T Cells Are Associated With Microvascular Dysfunction
We examined the association between T‐cell phenotypes and microvascular dysfunction as measured by RH (Table 3). Among all HIV+ subjects, after adjustment for demographics, cardiovascular risk factors, and co‐infections, CD8+PD1+ and CD4+IFN+cytomegalovirus‐specific subsets were significantly associated with worse RH. Findings were similar and directionally consistent when we restricted the analysis to those who were treated and suppressed, although only the CD8+PD1+ subset remained statistically significant.
Table 3.
Association of T‐Cell Markers With Reactive Hyperemia and FMD
| HIV+All Groups | HIV+Treated Suppressed | |||
|---|---|---|---|---|
| Parameter (Per Increase in SD) | FMD %Estimatea (95% CI) | Reactive Hyperemia %Estimate (95% CI) | FMD %Estimate (95% CI) | Reactive Hyperemia %Estimate (95% CI) |
| CD4+ CD38 +DR+ |
1.4 (−6.7, 10.2) P=0.75 |
−1.1 (−8.0, 6.3) P=0.76 |
−7.8 (−17.4, 2.8) P=0.14 |
−1.7 (−10.6, 8.1) P=0.72 |
| CD4+ PD1+ |
2.8 (−6.9, 13.6) P=0.59 |
−2.3 (−9.1, 5.1) P=0.54 |
−8.0 (−16.8, 1.6) P=0.10 |
−4.1 (−13.7, 6.5) P=0.43 |
| CD4+ CCR5+ CD38+ DR+ PD1+ |
−4.7 (−13.0, 4.3) P=0.30 |
−5.4 (−13.4, 3.3) P=0.21 |
−9.2 (−19.2, 2.2) P=0.11 |
−7.9 (−18.5, 4.1) P=0.19 |
| CD8+ CD38+ DR+ |
1.4 (−7.3, 10.8) P=0.77 |
−1.1 (−8.1, 6.4) P=0.76 |
−6.8 (−16.4, 3.9) P=0.21 |
−2.7 (−13.1, 9.0) P=0.64 |
| CD8+ PD1+ |
2.2 (−7.6, 13.1) P=0.67 |
−9.0 (−16.4, −1.0) P=0.03 |
0.4 (−10.1, 12.1) P=0.95 |
−11.2 (−19.5, −2.0) P=0.02 |
| CD8+ CCR5+ CD38+ DR+ PD1+ |
−1.6 (−10.3, 7.8) P=0.73 |
−6.2 (−13.1, 1.2) P=0.10 |
−4.4 (−13.5, 5.6) P=0.38 |
−7.5 (−17.3, 3.5) P=0.21 |
| CD4+ IFN+CMV‐specific |
−9.4 (−19.0, 1.4) P=0.09 |
−12.3 (−19.6, −4.3) P<0.01 |
−11.0 (−22.7, 2.4) P=0.10 |
−6.2 (−14.2, 2.6) P=0.16 |
| CD8+ IFN+CMV‐specific |
0.3 (−7.0, 8.2) P=0.94 |
−4.3 (−9.6, 1.3) P=0.13 |
−7.4 (−18.7, 5.6) P=0.25 |
−6.3 (−14.6, 2.9) P=0.17 |
CMV indicates cytomegalovirus; FMD, flow‐mediated dilation; IFN, interferon.
Percent estimate represents estimated percentage difference in reactive hyperemia or FMD associated with a doubling of each T‐cell marker. Each factor of interest was adjusted for age, sex, race, hypertension, diabetes mellitus, hyperlipidemia, smoking, hepatitis C virus, and opportunistic infections. Markers are included individually, not simultaneously.
T‐Cell Dysfunction Is Not Associated With Macrovascular Dysfunction
We then investigated which T‐cell phenotypes were associated with macrovascular dysfunction as measured by FMD. Among all HIV+ subjects, after adjustment for demographics, traditional cardiovascular risk factors, and co‐infections, the T‐cell markers showed little association with FMD. These findings were similar when the analysis was restricted to the treated and suppressed individuals only (Table 3). Thus, CD8+PD1+ T cells were associated with microvascular dysfunction in all HIV+ subjects as well as the treated and suppressed subset, but they had no association with macrovascular dysfunction.
TNF‐α Is Associated With Microvascular Dysfunction
Among all HIV+ subjects, D‐dimer, TNF‐α, high‐sensitivity C‐reactive protein, sCD‐14, and interleukin‐6 were associated with significantly worse RH after adjustment for demographics, cardiovascular risk factors, and co‐infections. Results were similar when the analysis was restricted to the treated suppressed group, although only TNF‐α remained associated with significantly worse RH in adjusted analysis (Table 4).
Table 4.
Association of Plasma Markers With Reactive Hyperemia and FMD
| HIV+All Groups | HIV+Treated Suppressed | |||
|---|---|---|---|---|
| Parameter (Per Increase in SD) | FMD %Estimatea (95% CI) | Reactive Hyperemia %Estimate (95% CI) | FMD %Estimate (95% CI) | Reactive Hyperemia %Estimate (95% CI) |
| IL‐6 |
−3.7 (−11.4, 4.6) P=0.37 |
−6.3 (−12.1, 0.0) P=0.05 |
−5.9 (−14.0, 2.9) P=0.18 |
−5.9 (−13.0, 1.7) P=0.13 |
| TNF‐α |
0.3 (−5.9, 6.9) P=0.93 |
−6.6 (−11.9, −1.0) P=0.02 |
0.7 (−6.2, 8.1) P=0.84 |
−8.2 (−14.1, −2.0) P=0.01 |
| hsCRP |
−4.7 (−10.1, 1.0) P=0.10 |
−5.8 (−10.3, −1.0) P=0.02 |
−3.7 (−10.7, 3.9) P=0.33 |
−4.9 (−11.1, 1.7) P=0.14 |
| D‐Dimer |
−6.0 (−13.3, 2.0) P=0.14 |
−5.8 (−11.0, −0.3) P=0.04 |
−7.7 (−15.7, 1.0) P=0.08 |
−4.6 (−10.3, 1.5) P=0.13 |
| Fibrinogen |
−8.1 (−14.4, −1.3) P=0.02 |
−6.0 (−12.1, 0.6) P=0.07 |
−6.4 (−13.3, 1.1) P=0.09 |
−5.3 (−12.2, 2.1) P=0.16 |
| CMV IgG |
−9.9 (−15.2, −4.3) P<0.01 |
−1.7 (−8.0, 5.0) P=0.61 |
−8.6 (−15.0, −1.6) P=0.02 |
−1.7 (−8.1, 5.1) P=0.61 |
| sCD‐14 |
2.5 (−3.0, 8.4) P=0.37 |
−4.0 (−7.6, −0.3) P=0.04 |
6.8 (−1.4, 15.7) P=0.11 |
4.9 (−1.4, 11.5) P=0.13 |
| sCD‐163 |
2.0 (−5.2, 9.8) P=0.60 |
−1.3 (−7.4, 5.2) P=0.68 |
−0.4 (−7.6, 7.3) P=0.91 |
−1.8 (−8.3, 5.2) P=0.60 |
CMV indicates cytomegalovirus; FMD, flow‐mediated dilation; hsCRP, high‐sensitivity C‐reactive protein; IgG, immunoglobulin G; IL‐6, interleukin‐6; TNF‐α, tumor necrosis factor‐α.
Percent estimate represents estimated percentage difference in reactive hyperemia or FMD associated with a doubling of each plasma marker. Each factor of interest was adjusted for age, sex, race, hypertension, diabetes mellitus, hyperlipidemia, smoking, hepatitis C virus, and opportunistic infections. Markers are included individually, not simultaneously.
Cytomegalovirus IgG Is Associated With Macrovascular Dysfunction
In all HIV+ subjects, cytomegalovirus IgG and fibrinogen were significantly associated with worse FMD after adjustment for demographics, cardiovascular risk factors, and co‐infections. Results were similar when we restricted the analysis to the treated suppressed group, although only cytomegalovirus IgG remained significantly associated with worse FMD (Table 4). Thus, while TNF‐α was associated with microvascular dysfunction, prior infection with cytomegalovirus was associated with macrovascular dysfunction.
Effect of TNF‐α on the Association Between CD8+PD1+ T‐Cell Marker and Microvascular Dysfunction
Given that both TNF‐α and CD8+PD1+ T‐cell marker were significantly associated with microvascular dysfunction in the treated and suppressed group, we evaluated whether TNF‐α was a mediator of the association between CD8+PD1+ T cell and RH. In unadjusted analysis, we found that each 1 SD increase in TNF‐α was associated with a 12.1% decrease in CD8+PD1+ T cells (95% CI: −22.5 to −0.3, P=0.04). After adding TNF‐α to the multivariable adjustment model used for Table 3, the association of CD8+PD1+ T cells with RH weakened from −11.2% (P=0.02) to −8.1% (P=0.09), while TNF‐α remained strongly associated with lower RH (−12.5%, P=0.001). We found that the test for interaction between CD8+PD1+ T cells and TNF‐α was positive and marginally significant (+8.3%, P=0.07), suggesting that the negative effect of CD8+PD1+ T cells on RH was reversed as TNF‐α increased.
Total CD8+ T‐Cell Counts Are Associated With Endothelial Dysfunction
Finally, we assessed the association of traditional markers of HIV disease progression with vascular function. Among all HIV+ subjects, greater CD8+ T‐cell count was associated with worse FMD after adjusting for demographics and traditional cardiovascular risk factors (Table 5). In the treated and suppressed cohort, greater CD8+ T‐cell count was associated with worse RH after adjusting for demographics and traditional cardiovascular risk factors. Higher CD4/CD8 ratio was not associated with improved FMD or RH after adjustment in either group.
Table 5.
Association of HIV‐Related Factors With Reactive Hyperemia and FMD
| HIV+All Groups | HIV+Treated Suppressed | |||
|---|---|---|---|---|
| Parameter (Per Increase in SD) | FMD %Estimatea (95% CI) | Reactive Hyperemia %Estimate (95% CI) | FMD %Estimate (95% CI) | Reactive Hyperemia %Estimate (95% CI) |
| CD4 count |
−1.3 (−7.3, 5.0) P=0.67 |
0.4 (−3.7, 4.7) P=0.84 |
−2.4 (−10.1, 5.8) P=0.55 |
−2.0 (−7.7, 4.1) P=0.52 |
| Nadir CD4 count |
−0.8 (−6.9, 5.7) P=0.80 |
−0.9 (−5.8, 4.1) P=0.71 |
−1.5 (−8.5, 6.1) P=0.69 |
−0.8 (−7.1, 5.9) P=0.80 |
| CD8 count |
−6.9 (−12.7, −0.8) P=0.03 |
−2.6 (−7.7, 2.7) P=0.33 |
−5.8 (−12.3, 1.2) P=0.10 |
−6.3 (−12.1, −0.1) P=0.05 |
| CD4/CD8 ratio |
5.8 (−0.7, 12.8) P=0.08 |
2.5 (−2.6, 7.8) P=0.35 |
6.4 (−1.7, 15.2) P=0.12 |
4.2 (−2.5, 11.4) P=0.22 |
| ART duration |
−4.0 (−9.7, 2.0) P=0.19 |
−2.7 (−8.1, 2.9) P=0.34 |
−4.9 (−11.9, 2.5) P=0.19 |
−4.2 (−10.6, 2.7) P=0.22 |
| PI Duration |
−0.9 (−6.6, 5.3) P=0.78 |
−2.2 (−7.2, 3.2) P=0.42 |
0.2 (−6.6, 7.6) P=0.95 |
−2.5 (−8.5, 3.9) P=0.43 |
| NRTI duration |
−5.0 (−10.6, 1.0) P=0.10 |
−3.2 (−8.6, 2.7) P=0.28 |
−6.2 (−12.9, 1.0) P=0.09 |
−5.1 (−11.4, 1.7) P=0.14 |
| NNRTI duration |
−0.1 (−5.7, 5.9) P=0.98 |
−0.6 (−6.3, 5.4) P=0.85 |
−0.1 (−7.1, 7.3) P=0.97 |
−2.1 (−9.0, 5.2) P=0.56 |
| HIV duration |
−3.3 (−8.8, 2.6) P=0.27 |
−2.3 (−7.7, 3.4) P=0.43 |
−1.3 (−7.5, 5.4) P=0.70 |
−1.6 (−8.2, 5.5) P=0.65 |
| Viral load |
1.0 (−4.6, 6.9) P=0.74 |
1.6 (−2.7, 6.2) P=0.47 |
NA | NA |
ART indicates antiretroviral therapy; FMD, flow‐mediated dilation; NA, not applicable; NNRTI, non‐nucleoside reverse transcriptase inhibitors; NRTI, nucleoside reverse transcriptase inhibitors; PI, protease inhibitor.
Percent estimate represents estimated percentage difference in reactive hyperemia or FMD associated with a doubling of each HIV‐related factor. Each factor of interest was adjusted for age, sex, race, hypertension, diabetes mellitus, hyperlipidemia, smoking. Markers are included individually, not simultaneously.
Discussion
In this relatively large cross‐sectional study of treated and untreated HIV‐infected adults, we investigated associations of markers of inflammation, coagulation, and T‐cell phenotype with vascular function. In addition to measuring large‐vessel function (FMD), we measured for the first time small‐vessel function (as assessed by RH). We identified several markers of inflammation and immune phenotype that were associated with microvascular dysfunction, even after controlling for demographic, traditional cardiovascular risk factors, and co‐infections. Among HIV‐infected adults on effective ART (suppressed viremia), the total CD8+ T‐cell count, the frequency of CD8+ T cells that express PD‐1, and the inflammatory cytokine TNF‐α were each associated with worsened microvascular disease. Among a larger cohort of treated and untreated HIV‐infected adults, a broader panel of markers was linked to microvascular dysfunction, including the frequency of CD8+PD1+ and CD4+IFN+cytomegalovirus‐specific responses, as well TNF‐α, high‐sensitivity C‐reactive protein, D‐dimer, interleukin‐6, and sCD‐14. Importantly, we found that macrovascular function (FMD) significantly correlated with microvascular function (RH), and that the major factors associated with FMD were cytomegalovirus IgG and fibrinogen.
We focused specifically on the treated and suppressed subgroup of the cohort for a few reasons. First, studies have demonstrated that endothelial dysfunction persists in the setting of treated HIV infection and these individuals have an increased risk of cardiovascular disease.3, 39 Accordingly, the impact of immune activation and inflammation on endothelial function is likely distinct in the treated and suppressed subgroup. Thus, focusing on this subgroup allows us to better characterize the relationship between immune activation and vascular function in the setting of controlled HIV infection. Second, this subgroup has become the most clinically relevant as initiation of antiretroviral therapy upon the diagnosis of HIV has become the standard of care. Due to the small number of subjects in each of the remaining 3 subgroups, we did not do separate analyses for the treated and unsuppressed, untreated and suppressed, and untreated and unsuppressed subgroups.
While in the general population, RH is predictive of FMD, macrovascular and microvascular functions are relatively independent in patient with rheumatoid arthritis, which suggests differential regulation of endothelial function in these two vascular beds.43 Given that HIV and rheumatoid arthritis are chronic inflammatory diseases, we wanted to evaluate whether RH and FMD are uncoupled in the setting of HIV infection as well. However, we found in our entire cohort as well as the treated suppressed group that RH was strongly and independently associated with FMD after adjusting for cardiovascular risk factors. Impaired RH has recently been identified as more predictive than FMD of cardiovascular events in the general population.44, 45
We showed that specific T‐cell phenotypic markers were associated with endothelial dysfunction in the general HIV population as well as in the treated suppressed group. Interestingly, these T‐cell markers were predictive of worsened RH but showed little or no association with FMD. One explanation for this finding could be that FMD represents the combination of conduit artery endothelial function as well as the hyperemic stimulus.46 Thus, in order to truly differentiate macrovascular from microvascular function, FMD must be adjusted for RH–induced shear stress.46 Therefore, our results suggest that T‐cell activation plays a role in endothelial dysfunction at a microvascular level in HIV‐infected individuals and that poor macrovascular function in HIV infection might be secondary to diseased microvasculature.
The total CD8+ T‐cell count and the frequency of CD8+ PD1+ T cells were associated with worse RH in the treated suppressed group. This is the first study that has reported this association in HIV‐infected or uninfected individuals. The role of the PD‐1 pathway in HIV infection is complex and remains poorly understood. In acute viral infections, CD8+ T cells are activated and undergo massive expansion, resulting in upregulation of PD‐1.47 In chronic infections, the frequency of PD‐1 expressing cells remains elevated and likely contributes to persistent adaptive immune dysfunction. This in turn likely contributes to excess burden of other microbes (eg, cytomegalovirus, microbial translocation) and perhaps excess risk of malignancy.48 Persistent elevations in CD8+ T‐cell count during ART (often measured as a low CD4/CD8 ratio) are associated with chronic inflammation and have been consistent predictors of cardiovascular complications, non‐AIDS morbidity, and overall mortality.40, 49, 50, 51, 52 Expansion of CD8+ T cells are associated with T‐cell activation, senescence, and altered CD4/CD8 ratio in the gut mucosa.40 Given these results, PD‐1 inhibitors could be potential therapeutic targets for treatment of cardiovascular disease in chronic HIV infection.
Cytomegalovirus co‐infection likely contributes to persistent inflammation and expansion in CD8+ T‐cell counts during ART.18, 49, 53, 54 Cytomegalovirus‐associated inflammation may also contribute to cardiovascular disease in HIV‐infected adults.32, 55, 56 In the Heart Outcomes Prevention Evaluation study, patients with positive cytomegalovirus serology had an increased risk of myocardial infarction, stroke, or cardiovascular death.57 Cytomegalovirus has also been associated with subclinical atherosclerosis in the Multi‐Ethnic Study of Atherosclerosis.58 In our current study, we found that the CD4+ IFN+cytomegalovirus‐specific T‐cell activation marker was associated with worse RH in all HIV+ subjects. Comparable nonsignificant trends were observed in treated and suppressed individuals. These data collectively suggest that cytomegalovirus infection contributes to CD8+ T‐cell expansion, persistent activation of potentially harmful inflammatory pathways, microvascular dysfunction, and cardiovascular disease.
In all HIV+ subjects, fibrinogen had a significant negative association with FMD while D‐dimer had a significant negative association with RH. Both associations weakened among the treated and suppressed individuals. Since both fibrinogen and D‐dimer are proinflammatory markers in the coagulation cascade, these results indicate that a prothrombotic state contributes to microvascular and macrovascular dysfunction.
As discussed, both TNF‐α and CD8+PD1+ T cells had a negative association with RH in the treated and suppressed group after adjusting for potential confounding factors. However, TNF‐α also had a significant negative association with CD8+PD1+ T cells. But when TNF‐α was added to the multivariable adjustment model, the association between CD8+PD1+ T cells and RH weakened. This finding may reflect the effect of an unmeasured confounding variable as opposed to a true mediation effect of TNF‐α, given the negative association between TNF‐α and CD8+PD1+ T cells. Alternatively, this may represent moderated mediation, where the effect of CD8+PD1+ T cells on RH differs depending on the level of inflammation. Our analysis found that CD8+PD1+ T cells were associated with worsening RH in the absence of TNF‐α, but this relationship appeared to reverse with increasing levels of TNF‐α. This reversal in relationship (ie, CD8+PD1+ T cells become protective) is supported by the fact that activation of the PD1 pathway in T cells can have an inhibitory effect and lead to decreased production of cytokines such as TNF‐α.47 Thus, the negative effect of CD8+PD1+ T cells on RH we have shown here indicates a separate mechanism (Figure).
Figure 1.
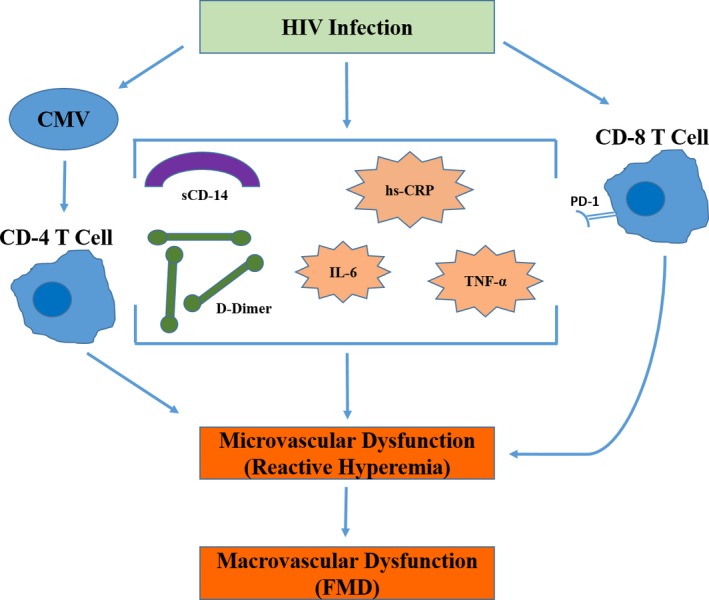
The role of T‐cell dysfunction, inflammation, and coagulation in microvascular and macrovascular dysfunction in HIV‐infected adults. CMV indicates cytomegalovirus; FMD, flow‐mediated dilation; hsCRP, high‐sensitivity C‐reactive protein; IL‐6, interleukin‐6; TNF‐α, tumor necrosis factor‐α.
The main limitation of our study is its cross‐sectional design. Endothelial dysfunction might reflect a cumulative effect of various factors, whereas the T cell and the inflammatory marker levels were measured only at a single time point. Therefore, we performed multivariable regression analysis to control for traditional cardiovascular factors in order to determine whether T‐cell activation and the plasma markers remained predictive of endothelial dysfunction. However, there could be other confounding factors in this population that we did not take into account.
In conclusion, we evaluated the interplay between T‐cell phenotypes, markers of inflammation/coagulation, cytomegalovirus, and endothelial dysfunction as measured by RH and FMD in HIV‐infected individuals. Our findings show that T‐cell dysfunction in HIV is preferentially associated with the microvasculature as assessed by RH. Since RH was strongly coupled to FMD, our study suggests that worse macrovascular function in HIV could be due to the influence of inflammation/immune activation on the microvasculature. We identified markers of inflammation, specifically TNF‐α, coagulation, and measures of CD8+ T‐cell dysfunction (CD8+PD1+) and cytomegalovirus‐associated CD4+ T‐cell dysfunction that, could serve as potential targets for future interventions. Future studies are under way to investigate the impact of lowering inflammation on endothelial function and cardiovascular risk.
Sources of Funding
This study was funded by National Institutes of Health (K24AI112393 to Hsue).
Disclosures
Dr. Hsue has received grant support from Pfizer and honoraria from Gilead, both unrelated to this study.
(J Am Heart Assoc. 2016;5:e004243 doi: 10.1161/JAHA.116.004243)
References
- 1. Lang S, Mary‐Krause M, Cotte L, Gilquin J, Partisani M, Simon A, Boccara F, Bingham A, Costagliola D; French Hospital Database on HIV‐ANRS CO4 . Increased risk of myocardial infarction in HIV‐infected patients in France, relative to the general population. AIDS. 2010;24:1228–1230. [DOI] [PubMed] [Google Scholar]
- 2. Boccara F, Lang S, Meuleman C, Ederhy S, Mary‐Krause M, Costagliola D, Capeau J, Cohen A. HIV and coronary heart disease: time for a better understanding. J Am Coll Cardiol. 2013;61:511–523. [DOI] [PubMed] [Google Scholar]
- 3. Frieberg MS, Chang C, Kuller LH, Skanderson M, Lowy E, Kraemer KL, Butt AA, Bidwell Goetz M, Leaf D, Oursler KA, Rimland D, Rodriguez Barradas M, Brown S, Gilbert C, McGinnis K, Crothers K, Sico J, Crane H, Warner A, Gottlieb S, Gottdiener J, Tracy RP, Budoff M, Watson C, Armah KA, Doebler D, Bryant K, Justice AC. HIV infection and risk of acute myocardial infarction. JAMA Intern Med. 2013;173:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Butt AA, Chang C, Kuller L, Goetz MB, Leaf D, Rimland D, Gilbert CL, Oursler KK, Rodriguez‐Barradas MC, Lim J, Kazis LE, Gottlieb S, Justice AC, Freiberg MS. Risk of heart failure with human immunodeficiency virus in the absence of prior diagnosis of coronary heart disease. Arch Intern Med. 2011;171:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tseng ZH, Secemsky EA, Dowdy D, Vittinghoff E, Moyers B, Wong JK, Havlir DV, Hsue PY. Sudden cardiac death in patients with human immunodeficiency virus infection. J Am Coll Cardiol. 2012;59:1891–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hsu JC, Li Y, Marcus GM, Hsue PY, Scherzer R, Grunfeld C, Shlipak MG. Atrial fibrillation and atrial flutter in human immunodeficiency virus‐infected persons. J Am Coll Cardiol. 2013;61:2288–2295. [DOI] [PubMed] [Google Scholar]
- 7. DAD Study Group , Friis‐Moller N, Reiss P, Sabin CA, Weber R, Monforte Ad, El‐Sadr W, Thiebaut R, De Wit S, Kirk O, Fontas E, Law MG, Phillips A, Lundgren JD. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med. 2007;356:1723–1735. [DOI] [PubMed] [Google Scholar]
- 8. Lang S, Mary‐Krause M, Cotte L, Gilquin J, Partisani M, Simon A, Boccara F, Costagliola D; Clinical Epidemiology Group of the French Hospital Database on HIV . Impact of individual antiretroviral drugs on the risk of myocardial infarction in human immunodeficiency virus‐infected patients: a case‐control study nested within the French Hospital Database on HIV ANRS cohort CO4. Arch Intern Med. 2010;170:1228–1238. [DOI] [PubMed] [Google Scholar]
- 9. Wang T, Yi R, Green LA. Increased cardiovascular disease risk in the HIV‐positive population on ART: potential role of HIV‐Nef and Tat. Cardiovasc Pathol. 2015;24:279–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, Ledergerber B, Lundgren J, Neuhaus J, Nixon D, Paton NI, Neaton JD; INSIGHT SMART Study Group . Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tenorio AR, Zheng Y, Bosch RJ, Krishnan S, Rodriguez B, Hunt PW, Plants J, Seth A, Wilson CC, Deeks SG, Lederman MM, Landay AL. Soluble markers of inflammation and coagulation but not T‐cell activation predict non‐AIDS‐defining morbid events during suppressive antiretroviral treatment. J Infect Dis. 2014;210:1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. SMART Study Group , El‐Sadr WM, Lundgren JD, Neaton JD, Gordin F, Abrams D, Arduino RC, Babiker A, Burman W, Clumeck N, Cohen CJ, Cohn D, Cooper D, Darbyshire J, Emery S, Fatkenheur G, Gazzard B, Grund B, Hoy J, Klingman K, Losso M, Markowitz N, Neuhaus J, Phillips A, Rappoport C. CD4+ count‐guided interruption of antiretroviral treatment. N Engl J Med. 2006;355:2283–2296. [DOI] [PubMed] [Google Scholar]
- 13. Tien PC, Choi AI, Zolopa AR, Benson C, Tracy R, Scherzer R, Bacchetti P, Shlipak M, Grunfeld C. Inflammation and mortality in HIV‐infected adults: analysis of the FRAM study cohort. J Acquir Immune Defic Syndr. 2010;55:316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, Pederson C, Ruxrungtham K, Lewin SR, Emery S, Neaton JD, Brenchley JM, Deeks SG, Sereti I, Douek DC; INSIGHT SMART Study Group . Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. 2011;203:780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bofill M, Mocroft A, Lipman M, Medina E, Borthwick NJ, Sabin CA, Timms A, Winter M, Baptista L, Johnson MA, Lee CA, Phillips AN, Janossy G. Increased numbers of primed activated CD8+CD38+CD45RO+ T cells predict the decline of CD4+ T cells in HIV‐1‐infected patients. AIDS. 1996;10:827–834. [DOI] [PubMed] [Google Scholar]
- 16. Mahalingam M, Peakman M, Davies ET, Pozniak A, McManus TJ, Vergani D. T cell activation and disease severity in HIV infection. Clin Exp Immunol. 1993;93:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. Broadly targeted human cytomegalovirus‐specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hunt PW, Martin JN, Sinclair E, Epling L, Teague J, Jacobson MA, Tracy RP, Corey L, Deeks SG. Valganciclovir reduces T cell activation in HIV‐infected individuals with incomplete CD4+ T cell recovery on antiretroviral therapy. J Infect Dis. 2011;203:1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, Beilman GJ, Khoruts A, Thorkelson A, Schmidt TE, Anderson J, Perkey K, Stevenson M, Perelson AS, Douek DC, Haase AT, Schacker TW. Persistent HIV‐1 replications is associated with lower antiretroviral drug concentrations in lymphatic tissue. Proc Natl Acad Sci USA. 2013;111:2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsue PY, Deeks SG, Hunt PW. Immunologic basis of cardiovascular disease in HIV‐infected adults. J Infect Dis. 2012;205:S375–S382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qiu MK, Wang SC, Dai YX, Wang SQ, Ou JM, Quan ZW. PD‐1 and Tim‐3 pathways regulate CD8+ T cells function in atherosclerosis. PLoS One. 2015;10:e0128523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lichtman AH, Binder CJ, Tsimikas S, Witztum JL. Adaptive immunity in atherogenesis: new insights and therapeutic approaches. J Clin Invest. 2013;123:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Younas M, Psomas C, Reynes J, Corbeau P. Immune activation in the course of HIV‐1 infection: causes, phenotypes and persistence under therapy. HIV Med. 2016;17:89–105. [DOI] [PubMed] [Google Scholar]
- 24. Krikke M, van Lelyveld SFL, Tesselaar K, Arends JE, Hoepelman IM, Visseren FL. The role of T cells in the development of cardiovascular disease in HIV‐infected patients. Atherosclerosis. 2014;237:92–98. [DOI] [PubMed] [Google Scholar]
- 25. Merlini E, Luzi K, Suardi E, Barassi A, Cerrone M, Martinez JS, Bai F, D'Eril GV, Monforte AD, Marchetti G. T‐cell phenotypes, apoptosis and inflammation in HIV+ patients on virologically effective cART with early atherosclerosis. PLoS One. 2012;7:e46073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Longenecker CT, Funderburg NT, Jiang Y, Debanne S, Storer N, Labbato DE, Lederman MM, McComsey GA. Markers of inflammation and CD8 T‐cell activation, but not monocyte activation, are associated with subclinical carotid artery disease in HIV‐infected individuals. HIV Med. 2013;14:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. D'Abramo A, Zingaropoli MA, Oliva A, D'Agostino C, Al Moghazi S, De Luca G, Iannetta M, Mastroianni CM, Vullo V. Immune activation, immunosenescence, and osteoprotegerin as markers of endothelial dysfunction in subclinical HIV‐associated atherosclerosis. Mediators Inflamm. 2014;2014:192594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karim R, Mack WJ, Kono N, Tien PC, Anastos K, Lazar J, Young M, Desai S, Golub ET, Kaplan RC, Hodis HN, Kovacs A. T‐cell activation, both pre‐ and post‐HAART levels, correlates with carotid artery stiffness over 6.5 years among HIV‐infected women in the WIHS. AIDS. 2014;67:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaplan RC, Sinclair E, Landay AL, Lurain N, Sharrett AR, Gange SJ, Xue X, Parrinello CM, Hunt P, Deeks SG, Hodis HN. T cell activation predicts carotid artery stiffness among HIV‐infected women. Atherosclerosis. 2011;217:207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaplan RC, Sinclair E, Landay AL, Lurain N, Sharrett AR, Gange SJ, Xue X, Hunt P, Karim R, Kern DM, Hodis HN, Deeks SG. T cell activation and senescence predict subclinical carotid artery disease in HIV‐infected women. J Infect Dis. 2011;203:452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McKibben RA, Margolick JB, Grinspoon S, Li X, Palella FJ Jr, Kingsley LA, Witt MD, George RT, Jacobson LP, Budoff M, Tracy RP, Brown TT, Post WS. Elevated levels of monocyte activation markers are associated with subclinical atherosclerosis in men with and those without HIV infection. J Infect Dis. 2015;211:1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsue PY, Hunt PW, Sinclair E, Bredt B, Franklin A, Killian M, Hoh R, Martin JN, McCune JM, Waters DD, Deeks SG. Increased carotid intima‐media thickness in HIV patients is associated with increased cytomegalovirus‐specific T‐cell responses. AIDS. 2006;20:2275–2283. [DOI] [PubMed] [Google Scholar]
- 33. Ford ES, Greenwald JH, Richterman AG, Rupert A, Dutcher L, Badralmaa Y, Natarajan V, Rehm C, Hadigan C, Sereti I. Traditional risk factors and D‐dimer predict incident cardiovascular disease events in chronic HIV infection. AIDS. 2009;23:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McLenachan JM, Williams JK, Fish RD, Ganz P, Selwyn AP. Loss of flow‐mediated endothelium‐dependent dilation occurs early in the development of atherosclerosis. Circulation. 1991;84:1272–1277. [DOI] [PubMed] [Google Scholar]
- 35. Charakida M, Masi S, Luscher TF, Kastelein JJ, Deanfield JE. Assessment of atherosclerosis: the role of flow‐mediated dilation. Eur Heart J. 2010;31:2854–2861. [DOI] [PubMed] [Google Scholar]
- 36. Granger DN, Senchenkova E. Inflammation and the Microcirculation. San Rafael, CA: Morgan and Claypool Life Sciences; 2010. [PubMed] [Google Scholar]
- 37. Anderson TJ, Charbonneau F, Title LM, Buithieu J, Rose MS, Conradson H, Hildebrand K, Fung M, Verma S, Lonn EM. Microvascular function predicts cardiovascular events in primary prevention. Circulation. 2011;123:163–169. [DOI] [PubMed] [Google Scholar]
- 38. Arildsen H, Sorensen KE, Ingerslev JM, Ostergaard LJ, Laursen AL. Endothelial dysfunction, increased inflammation, and activated coagulation in HIV‐infected patients improve after initiation of highly active antiretroviral therapy. HIV Med. 2013;14:1–9. [DOI] [PubMed] [Google Scholar]
- 39. Hsue PY, Hunt PW, Wu Y, Schnell A, Ho JE, Hatano H, Xie Y, Martin JN, Ganz P, Deeks SG. Association of abacavir and impaired endothelial function in treated and suppressed HIV‐infected patients. AIDS. 2009;23:2021–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Serrano‐Villa S, Sainz T, Lee SA, Hunt PW, Sinclair E, Shacklett BL, Ferre AL, Hayes TL, Somsouk M, Hsue PY, Van Natta ML, Meinert CL, Lederman MM, Hatano H, Jain V, Huang Y, Hecht FM, Martin JN, McCune JM, Moreno S, Deeks SG. HIV‐infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non‐AIDS morbidity and mortality. PLoS Pathog. 2014;10:e1004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsue PY, Scherzer R, Grunfeld C, Imboden J, Wu Y, Del Puerto G, Nitta E, Shigenaga J, Schnell Heringer A, Ganz P, Graf J. Depletion of B‐cells with rituximab improves endothelial function and reduces inflammation among individuals with rheumatoid arthritis. J Am Heart Assoc. 2014;3:e001267 doi: 10.1161/JAHA.114.001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCullogh P, Nelder J. Generalized Linear Models. 2nd ed Boca Raton: Chapman and Hall/CRC; 1989. [Google Scholar]
- 43. Sandoo A, Carroll D, Metsios GS, Kitas GD, Veldhuijzen van Zanten JJ. The association between microvascular and macrovascular endothelial function in patients with rheumatoid arthritis: a cross‐sectional study. Arthritis Res Ther. 2011;13:R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gokce N, Keaney JF, Hunter LM, Watkins MT, Nedeljkovic ZS, Menzoian JO, Vita JA. Predictive value of noninvasively determined endothelial dysfunction for long‐term cardiovascular events in patients with peripheral vascular disease. J Am Coll Cardiol. 2003;41:1769–1775. [DOI] [PubMed] [Google Scholar]
- 45. Mitchell GF, Parise H, Vita JA, Larson MG, Warner E, Keaney JF Jr, Keyes MJ, Levy D, Vasan RS, Benjamin EJ. Local shear stress and brachial artery flow‐mediated dilation: the Framingham Heart Study. Hypertension. 2004;44:134–139. [DOI] [PubMed] [Google Scholar]
- 46. Padilla J, Johnson BD, Newcomer SC, Wilhite DP, Mickleborough TD, Fly AD, Mather KJ, Wallace JP. Normalization of flow‐mediated dilation to shear stress area under the curve eliminates the impact of variable hyperemic stimulus. Cardiovasc Ultrasound. 2008;6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Velu V, Shetty RD, Larsson M, Shankar EM. Role of PD‐1 co‐inhibitory pathway in HIV infection and potential therapeutic options. Retrovirology. 2015;12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deeks SF, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity. 2013;39:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Freeman ML, Mudd JC, Shive CL, Younes SA, Panigrahi S, Sieg SF, Lee SA, Hunt PW, Calabrese LH, Gianella S, Rodriguez B, Lederman MM. CD8 T‐cell expansion and inflammation linked to CMV coinfection in ART‐treated HIV infection. Clin Infect Dis. 2016;62:392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Helleberg M, Kronborg G, Ullum H, Ryder LP, Obel N, Gerstoft J. Course and clinical significance of CD8+ T‐cell counts in a large cohort of HIV‐infected individuals. J Infect Dis. 2015;211:1726–1734. [DOI] [PubMed] [Google Scholar]
- 51. Badejo OA, Chang CC, So‐Armah KA, Tracy RP, Baker JV, Rimland D, Butt AA, Gordon AJ, Rinaldo CR Jr, Kraemer K, Samet JH, Tindle HA, Goetz MB, Rodriguez‐Barradas MC, Bedimo R, Gibert CL, Leaf DA, Kuller LH, Deeks SG, Justice AC, Freiberg MS. CD8+ T‐cell count in acute myocardial infarction in HIV disease in a predominantly male cohort. Biomed Res Int. 2015;2015:246870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schneider S, Spinner CD, Cassese S, Promny D, Hapfelmeier A, Byrne RA, Baumann M, Jager H, Steinlechner E, Laugwitz KL, Kastrati A. Association of increased CD8+ and persisting C‐reactive protein levels with restenosis in HIV patients after coronary stenting. AIDS. 2016;30:1413–1421. [DOI] [PubMed] [Google Scholar]
- 53. Appay V, Fastenackels S, Katlama C, Ait‐Mohand H, Schneider L, Guihot A, Keller M, Grubeck‐Loebenstein B, Simon A, Lambotte O, Hunt PW, Deeks SG, Costagliola D, Autran B, Sauce D. Old age and anti‐cytomegalovirus immunity are associated with altered T‐cell reconstitution in HIV‐1 infected patients. AIDS. 2011;25:1813–1822. [DOI] [PubMed] [Google Scholar]
- 54. Naeger DM, Martin JN, Sinclar E, Hunt PW, Bangsberg DR, Hecht F, Hsue P, McCune JM, Deeks SG. Cytomegalovirus‐specific T cells persist at very high levels during long‐term antiretroviral treatment of HIV disease. PLoS One. 2010;5:e8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parrinello CM, Sinclair E, Landay AL, Lurain N, Sharrett AR, Gange SJ, Xue X, Hunt PW, Deeks SG, Hodis HN, Kaplan RC. Cytomegalovirus immunoglobulin G antibody is associated with subclinical carotid artery disease among HIV‐infected women. J Infect Dis. 2012;205:1788–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lichtner M, Cicconi P, Vita S, Cozzi‐Lepri A, Galli M, Lo Caputo S, Saracino A, De Luca A, Moioli M, Maggiolo F, Marchetti G, Vullo V, d'Arminio Monforte A; ICONA Foundation Study . Cytomegalovirus coinfection is associated with an increased risk of severe non‐AIDS‐defining events in a large cohort of HIV‐infected patients. J Infect Dis. 2015;211:178–186. [DOI] [PubMed] [Google Scholar]
- 57. Smieja M, Gnarpe J, Lonn E, Gnarpe H, Olsson G, Yi Q, Dzavik V, McQueen M, Yusuf S; HOPE Study Investigators . Multiple infections and subsequent cardiovascular events in the Heart Outcomes Prevention Evaluation (HOPE) Study. Circulation. 2003;107:251–257. [DOI] [PubMed] [Google Scholar]
- 58. Tracy RP, Doyle MF, Olson NC, Huber SA, Jenny NS, Sallam R, Psaty BM, Kronmal RA. T‐helper type 1 bias in healthy people is associated with cytomegalovirus serology and atherosclerosis: the Multi‐Ethnic Study of Atherosclerosis. J Am Heart Assoc. 2013;2:e000117 doi: 10.1161/JAHA.113.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]