

A decrease in arterial O2 pressure (pO2) elicits a peripheral chemosensory reflex that increases ventilation and sympathetic nerve activity. This reflex is initiated at the carotid body glomus (Type 1) cells where the reduced pO2 alters the activities of various ion channels and causes cell depolarization. The depolarization opens the voltage-dependent Ca2+ channel, leading to elevation of intracellular [Ca2+] ([Ca2+]i) and increased transmitter secretion from glomus cells. The transmitters such as acetylcholine and ATP then alter the carotid sinus afferent nerve activity and the respiratory brainstem neurons to adjust ventilation and help restore arterial pO2. This general sequence of events is widely accepted by researchers studying carotid body chemosensory reflex mechanisms.1-3 However, the cellular and molecular details of how O2 is sensed and what signals modulate various ion channels within the carotid body are not well defined.
The glomus cell depolarization that initiates the chemosensory reflex mechanism is thought to arise as a result of inhibition of the outward K+ current via a signal generated by an O2 sensor. Hemeproteins in mitochondria such as cytochrome oxidase are generally considered to mediate O2 sensing, but the signal(s) generated thereon remains largely unknown. Several signals (i.e., carbon monoxide, hydrogen sulfide, nitric oxide, ATP, AMP-activated kinase) have been proposed, suggesting that multiple signals could be involved. Identifying the hypoxic signals that inhibit the K+ current may help to understand the nature of the O2 sensor itself.
Studies in isolated glomus cells from various animal species show that hypoxia inhibits voltage-gated (Kv), two-pore domain (K2P) and large-conductance (BK or maxiK) K+ channels. In isolated rat and mouse glomus cells, TASK1/3 (K2P3/9) is highly active at rest and is strongly inhibited by hypoxia. Mitochondrial inhibitors and uncouplers similarly inhibit TASK1/3. Metabolic inhibition is therefore associated with a reduction of TASK1/3 activity, but the intermediate signals involved have not been identified. Hypoxia has also been reported to inhibit BK, but BK is generally inactive at rest in isolated glomus cells and BK inhibitors have no effect on cell membrane potential (Em). Nevertheless, if BK were active in vivo, its inhibition by hypoxia would produce depolarization. At present, the role of BK in O2 sensing remains controversial.4 Kv current is relatively small in rat glomus cells, and Kv channels are probably closed near the resting Em. Therefore, TASK1/3 is likely to be the main K+ channel responsible for the initiation of depolarization from the resting state in response to acute hypoxia.
The depolarization in any cell is mainly caused by a shift in the balance between Na+ influx and K+ efflux. In glomus cells, hypoxia is thought to inhibit K+ efflux, tilting the balance in favor of Na+ influx. It is generally thought that Na+ influx is via a voltage-independent, background Na+ channel. An interesting new player in O2 sensing has emerged from our recent study on Na+ permeable channels in isolated rat glomus cells. In cell-attached patches, hypoxia elicited two ionic events that were temporally related. An initial inhibition of TASK1/3 was quickly followed by activation of a non-selective cation channel with a single channel conductance of ~20-pS.5 Studies of these events showed that the initial depolarization caused by K+ channel inhibition and the early rise in [Ca2+]i due to opening of the voltage-dependent Ca2+ channel were responsible for the activation of the non-selective cation channel. The 20-pS channel was directly activated by application of 10 μM Ca2+ to inside-out patches, indicating that the non-selective cation channel is a Ca2+-sensitive ion channel. Biophysical analysis showed that the 20-pS channel is permeable to monovalent cations such as Na+ and Cs+, and has a reversal potential of ~-28 mV. Thus, the 20-pS channel shares properties similar to those of Ca2+-activated monovalent cation channels such as TRPM4 and TRPM5. In addition to Ca2+, other as yet undiscovered cellular signals could modulate the 20-pS channel and change its Ca2+ sensitivity.
Other depolarizing stimuli such as high external KCl, low external pH and Ca2+channel agonist activated the 20-pS channel and this was also due to the increase in [Ca2+]i, as activation did not occur in Ca2+-free external solution. The relationships among cell Em, [Ca2+]i and channel activity showed that the 20-pS channel became active when [Ca2+]i increased to levels above ~200 nM from a basal level of ~100 nM, and when the cell Em reached ~-40 mV from a resting level of ~-60 mV. As O2 levels below 5% elicits a rise in [Ca2+]i above 200 nM in these experiments, recruitment of the 20-pS channel may occur at moderate to severe hypoxia (< 5% O2). In addition to hypoxia, mitochondrial inhibitors and uncouplers that elevate [Ca2+]i are predicted to activate the 20-pS channel. Transmitters such as acetylcholine and angiotensin II that act on Gq-coupled receptors and elevate [Ca2+]i via release of Ca2+ from internal stores are also predicted to activate the non-selective cation channel.
An updated model of O2 sensing is presented in Figure 1 that describes the “helper” role of the 20-pS channel, and incorporates the importance of both K+- and Na+-permeable channels in the depolarization of glomus cells during hypoxia. Ca2+-activated Na+ influx via the 20-pS channel provides an additional means to depolarize the glomus cell and further enhance Ca2+ influx. The increase in [Ca2+]i would enhance Na+ influx, at least until the cell Em reaches -28 mV. Therefore, an increase in inward current composed of Na+ and Ca2+ currents along with a decrease in outward K+ current are expected to occur in response to acute hypoxia. Although not obvious in normal whole-cell recording, the inward current was unmasked in ~50% of cells after blocking a major part of the outward K+ current with tetraethylammonium, iberiotoxin and 4-aminopyridine.5 The recruitment of the 20-pS channel by the rise in [Ca2+]i in response to hypoxia may help to sustain an optimal level of depolarization during moderate to severe hypoxia and keep the chemosensory reflex active. Thus, inhibition of the 20-pS channel is predicted to reduce the level of depolarization during hypoxia. A large increase in [Ca2+]i and depolarization beyond ~-30 mV should activate BK and oppose over-excitation. Future identification of a selective inhibitor of the 20-pS channel and the gene that encodes the channel should help to better understand the role of this interesting channel in hypoxia-induced depolarization and the chemosensory reflex.
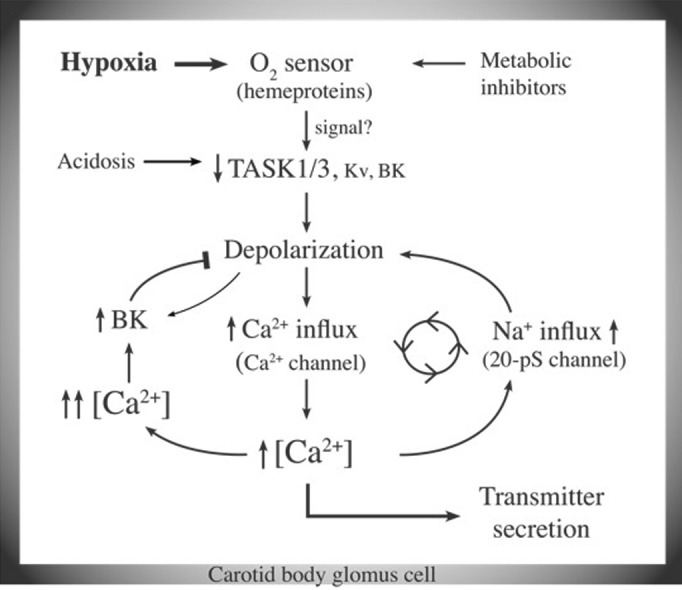
Figure 1. An updated model of O2 sensing by carotid body glomus cells. Hypoxia inhibits K+ channels such as TASK via an O2 sensor. The depolarization opens voltage-gated Ca2+ channels and elevates [Ca2+]i. Rise of [Ca2+]i begins to increase Na+ influx via the 20-pS channel, and initiates a feed-forward mechanism to maintain an optimal level of [Ca2+]i during moderate to severe hypoxia. Further increase in [Ca2+]i is prevented by activation of BK that opposes depolarization.
Reference
- 1.Buckler KJ. . A novel oxygen-sensitive potassium current in rat carotid body type I cells. J Physiol 1997; 498:649 - 62; PMID: 9051577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar P. . Sensing hypoxia in the carotid body: from stimulus to response. Essays Biochem 2007; 43:43 - 60; http://dx.doi.org/ 10.1042/BSE0430043; PMID: 17705792 [DOI] [PubMed] [Google Scholar]
- 3.López-Barneo J, Ortega-Sáenz P, Pardal R, Pascual A, Piruat JI. . Carotid body oxygen sensing. Eur Respir J 2008; 32:1386 - 98; http://dx.doi.org/ 10.1183/09031936.00056408; PMID: 18978138 [DOI] [PubMed] [Google Scholar]
- 4.Peers C, Wyatt CN. . The role of maxiK channels in carotid body chemotransduction. Respir Physiol Neurobiol 2007; 157:75 - 82; http://dx.doi.org/ 10.1016/j.resp.2006.10.010; PMID: 17157084 [DOI] [PubMed] [Google Scholar]
- 5.Kang D, Wang J, Hogan JO, Vennekens R, Freichel M, White C, Kim D. . Increase in cytosolic Ca2+ produced by hypoxia and other depolarizing stimuli activates a non-selective cation channel in chemoreceptor cells of rat carotid body. J Physiol 2014; 592:1975 - 92; PMID: 24591572 [DOI] [PMC free article] [PubMed] [Google Scholar]