


Abstract
In this work, we developed a database WERAM (http://weram.biocuckoo.org/) for histone acetyltransferases, histone deacetylases, histone methyltransferases, histone demethylases and acetyl- or methyl-binding proteins, which catalyze, remove and recognize histone acetylation and methylation sites as ‘writers’, ‘erasers’ and ‘readers’, and synergistically determine the ‘histone code’. From the scientific literature, we totally collected over 580 experimentally identified histone regulators from eight model organisms, including Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Caenorhabditis elegans, Arabidopsis thaliana, Schizosaccharomyces pombe and Saccharomyces cerevisiae. We also collected ∼900 site-specific regulator-histone relations from the eight species. According to the experimental evidence, known histone regulators were classified into distinct families. To computationally detect more proteins in eukaryotes, we constructed hidden Markov model (HMM) profiles for histone regulator families. For families without HMM profiles, we also conducted orthologous searches. Totally, WERAM database contained more than 20 thousand non-redundant histone regulators from 148 eukaryotes. The detailed annotations and classification information of histone regulators were provided, together with site-specific histone substrates if available.
INTRODUCTION
As an important epigenetic mechanism, post-translational modifications (PTMs) of histones or histone modifications combinatorially and dynamically constitute ‘histone code’, which accurately regulates a variety of DNA-templated processes such as gene transcription, chromosome segregation and DNA repair (1–8). Acetylation and methylation are two predominant types of histone modifications. Histone acetylation neutralizes the positive charges of lysine residues, disrupts the association between histones and DNA, and is a hallmark of open chromatin that can be more accessible to DNA and RNA polymerases as well as transcription factors, resulting in the activation of gene transcription (3,8–11). Histone methylation mainly occurs on arginine and lysine residues, enhances the basicity and hydrophobicity of histone tails, influences the interaction affinity between DNA and transcription factors, and can either activate or repress gene expression (3,8,11–14). For example, the methylation of H3K4 and H3K36 is closely associated with transcriptional activation, whereas methylated H3K9 and H3K27 are highly correlated with gene repression (11,14).
Histone acetylation and methylation are catalyzed, cleared and recognized by histone modifying enzymes (‘writers’), histone demodifying enzymes (‘erasers’), and acetyl- or methyl-binding proteins (‘readers’), respectively (3,15–17). For example, histone acetyltransferases (HATs), histone deacetylases (HDACs) and bromodomains can deposit, remove or interact with specific acetylation sites (2,6,7,9,10,18,19). For methylation, histone methyltransferases (HMTs), histone demethylases (HDMs) and a variety of functional domains such as chromodomain, PHD, ZF-CW and Tudor domains act as writers, erasers and reader proteins of methylation sites (6,7,9,12,18,20). Histone acetylation and methylation are critically and precisely regulated, whereas the aberrances or mutations of histone modifications or regulators are highly associated with memory impairment, developmental defects and cancers (11,13–16,21). In this regard, the identification of histone modifications, regulators and site-specific regulator-histone relations is fundamental for understanding the molecular mechanisms and regulatory roles of the histone code.
Recently, due to the rapid progress in the development of high-throughput techniques such as quantitative mass spectrometry and highly potent antibodies (22,23), a large number of new histone PTMs including acetylation and methylation sites were characterized (1,4,20,24). Thus, the collection of histone modifications together with their cognate regulators has emerged to be a great challenge. Currently, there have been four major databases developed for histones and histone modifications, including HHMD, Hlstome, dbHiMo and HistoneDB (25–28). For example, HHMD collected 43 location-specific histone modifications in humans, focusing on the storage and integration of histone modification datasets (25), while Hlstome contained 55 human histones, 106 site-specific PTMs and 152 histone-modifying enzymes (26). The dbHiMo mainly focused on the identification of histone-modifying enzymes in fungi, whereas readers and site-specific histone modifications were not included (27). HistoneDB maintained histone sequences, structural annotations, for the exploration of histones and their variants (28). These data resources mainly focused on the collection of histone sequences and structures, or histone modifications in human and fungi, whereas an integrative database in eukaryotes is still lacking.
From the scientific literature, we first collected 584 experimentally identified histone regulators, including 72 HATs, 97 HDACs, 116 acetyl-readers, 112 HMTs, 76 HDMs and 156 methyl-readers from eight model organisms including Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Caenorhabditis elegans, Arabidopsis thaliana, Schizosaccharomyces pombe and Saccharomyces cerevisiae (Supplementary Table S1). For a better understanding of the histone code, we also collected 904 known site-specific regulator-histone relations of the eight species. Based on previously established rationales (2,6–10,12,18–20,29), we classified all known HATs, HDACs, acetyl-readers, HMTs, HDMs and methyl-readers into distinct protein families. In our results, there were 15 families for the histone acetylation system, and 32 families for histone methylation regulators. Using HMMER (30), we totally constructed 13 and 30 hidden Markov models (HMMs) for histone acetylation and methylation regulators at the family level, respectively. Then we systematically characterized 1077 HATs, 2408 HDACs, 3901 acetyl-readers, 3990 HMTs, 1610 HDMs and 10 685 methyl-readers from 148 eukaryotic species using the HMM profile of each family. Moreover, for four families without HMMs, the ortholog search was conducted to identify potential histone regulators. The detailed annotations from the Ensembl (31) and UniProt (32) databases were integrated, and the classification information was also provided. Finally, an integrative database of writers, erasers and readers of acetylation and methylation in eukaryotes (WERAM) was developed with 20 033 non-redundant histone regulators, including 1337 HATs, 2504 HDACs, 3901 acetyl-readers, 4409 HMTs, 1610 HDMs and 10 949 methyl-readers.
CONSTRUCTION AND CONTENT
Data collection
Because too many scientific papers were published on histone modifications, here we mainly focused on the curation of the literature published since 2011. For histone acetylation, we used the PubMed Advanced Search Builder and built three filters, including ‘(histone acetyltransferase) NOT (histone acetyltransferase inhibitor)’, ‘((((histone deacetylase [Title/Abstract]) OR HDAC [Title/Abstract]) NOT (histone deacetylase inhibitors) NOT histone deacetylase inhibitor) NOT histone deacetylase inhibition)’, and (histone acetylation) AND (bromodomain or tandem PHD), and retrieved approximately 2500, 2000 and 150 papers, respectively. For histone methylation, we directly searched the PubMed using multiple keywords, such as ‘histone methyltransferase’, ‘histone demethylase’, ‘histone methylation reader’, ‘histone methylation’ and ‘histone modification’, and retrieved over 3000 abstracts in total. In addition, we also considered several important review articles published before 2011 (2,8,11,12,17,19,29). From the abstracts or full-length manuscripts, we manually curated 248 and 336 experimentally identified acetylation and methylation regulators, respectively (Supplementary Table S1).
For the 584 known histone regulators, the information of protein domains was taken from the annotations in UniProt database (32). The functional domains of all writers, erasers and readers were further examined by searching the Pfam database (33). Furthermore, we downloaded the complete proteome sets for 148 eukaryotes including 68 animals, 39 plants and 41 fungi, from Ensembl (31) (release version 84, http://www.ensembl.org/, under the directory of ‘/pub/release-84/fasta’), EnsemblPlants (release version 31, http://plants.ensembl.org/) and EnsemblFungi (release version 31, http://fungi.ensembl.org/), respectively. Because multiple variant nucleotide sequences or peptides can be originated from a single gene, we further used Ensembl Gene ID as the unique accession to eliminate the redundancy. For multiple alternatively splicing isoforms of a single gene, only the longest one was reserved.
The classification of histone regulators
An accurate classification map of known histone regulators is greatly helpful for further identification of new proteins. As previously described (2,6–10,18,19,29), we manually classified collected HATs, HDACs and acetyl-readers into 15 families: (i) HATs have eight families, including p300_CBP, ELP3, GCN5, HAT1, HPA2, GNAT_other, MYST and HAT_other (unclassified HATs); (ii) HDACs contain five families, including Class-I, Class-II, SIR2, Class-IV and HD2; (iii) The acetyl-readers (Ac_Reader) were classified into two families such as Bromodomain, and Tandem-PHD containing at least two PHD domains in proteins (Figure 1).
Figure 1.

The detailed classifications of HATs, HDACs, Ac_Readers, HMTs, HDMs and Me_readers, together with a cut-off value of the proteome-wide identification for each family. The detailed parameters used for the hmmsearch program together with a summary of the testing data set were shown in Supplementary Table S2.
Also, based on experimental evidence (6–9,12,18,20), we manually classified known HMTs, HDMs and methyl-reader into 32 families: (i) HMTs were clustered into 10 families, including SET1, SET2, RIZ, EZ, SUV39, SUV4-20, SMYD, PRMT, nonSET and HMT_other (unclassified HMTs); (ii) HDMs have eight families, including JARID, JHDM1, JHDM2, JHDM3_JMJD2, PHF2_PHF8, UTX_UTY, JmjC_only and LSD1_KDM1. The JmjC_only family contains HDMs with JmjC domains but not to be classified into any other families of the JmjC superfamily; (iii) The methyl-reader proteins (Me_Reader) were grouped into 14 families, including ADD, Ankyrin, BAH, Chromodomain, PHD, PWWP, TTD, Tudor, WD40, ZF-CW, Chromo-Barrel, MBT, DCD and Me_Reader_other (unclassified methyl-readers) (Figure 1).
Genome-wide identification of HATs, HDACs, HMTs, HDMs and reader proteins
For families with ≥3 member genes, the catalytic domain sequences of HATs, HDACs, HMTs and HDMs, or PTM-binding domain sequences of acetyl-readers and methyl-readers were first aligned for each family separately with MUSCLE (http://www.drive5.com/muscle/, version 3.8.31), an extensively used tool for multiple sequence alignment (34). An illustration of aligned domain sequences was shown for four histone regulator families, including HAT/GCN5, HDAC/Class-IV, HMT/SET2 and HDM/JHDM3_JMJD2 (Supplementary Figure S1). HMM models of 569 known histone regulators were then constructed at the family level using the hmmbuild program of the HMMER 3.1b2 package (http://hmmer.org/) (30). We totally constructed 13 and 30 HMM profiles for acetylation and methylation families, respectively.
For the genome-wide identification of histone regulators, the hmmsearch program of HMMER 3.1b2 was used (30). The hmmsearch calculates both the log-odds likelihood scores and E-values for given protein sequences. Because E-values depend on the database size and generate inconsistent results when the database is changed, here realistic constant log-odds likelihood scores were adopted as the threshold values. To evaluate the prediction performance of the HMM identification and specify the cut-off value for each family, the 569 collected histone regulators used for constructing HMM models were taken as the testing dataset. For each family, the known annotated proteins were regarded as positive data (P), while all proteins in other families were taken as negative data (N) (Supplementary Table S2). Two measurements of sensitivity (Sn) and specificity (Sp) were defined and calculated as shown below:
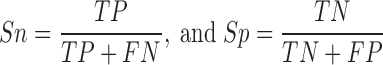 |
First, the self-consistency validation was performed directly with the positive data and negative data for each family. To further evaluate the prediction robustness, the leave-one-out validation was also carried out. As an example, the receiver operating characteristic (ROC) curves were drawn and area under ROC values were calculated for eight families of acetylation regulators (Supplementary Figure S2A), and eight families of methylation regulators (Supplementary Figure S2B), respectively. Our results demonstrated that HMM predictions are both accurate and robust (Supplementary Figure S2). To promise that all known histone regulators can be correctly identified and classified in leave-one-out validations (Sn = 100%), we manually selected a log-odds likelihood score as the threshold for each family (Figure 1). The statistics of the testing data set together with the detailed parameters used for hmmsearch were shown (Supplementary Table S2). Then we used hmmsearch to search potential histone regulators in 148 eukaryotes. Because multiple variant peptides can be generated from a single gene, here we used the Ensembl Gene ID as the unique accession to avoid any redundancy. For a given gene, only the protein with the highest log-odds likelihood score was retained as the representative sequence.
For the four families without HMM profiles (Other of HATs, HD2 of HDACs, Other of HMTs and Other of methyl-readers), we further performed orthologous searches to identify 260 HATs, 96 HDACs, 419 HMTs and 264 methyl-readers from 148 eukaryotic organisms, respectively. As previously described (35), the strategy of Reciprocal Best Hits (RBH) was chosen, and the blastall program in the BLAST package was utilized (36). All protein sequences and HMM profiles of histone regulators can be freely downloaded at: http://weram.biocuckoo.org/download.php.
In total, we identified and integrated 20 033 histone regulators, including 1337 HATs, 2504 HDACs, 3901 acetyl-readers, 4409 HMTs, 1610 HDMs, 10 949 methyl-readers from 148 eukaryotic species (Supplementary Table S3), whereas a heatmap of the results was visualized using the HemI program (http://hemi.biocuckoo.org/) (Supplementary Figure S3) (37). Averagely, there were 90.4 PTM enzymes and 161.0 readers per animal, 73.4 enzymes and 75.9 readers per plant, and 20.7 enzymes and 23.0 readers per fungus (Supplementary Figure S3). Although the numbers of PTM enzymes and readers are quite similar in plants and fungi, animals generally contain more reader proteins than writers and erasers. We also compared WERAM to an existing database dbHiMo, which mainly focused on the identification of histone-modifying enzymes but not reader proteins in fungi (27). For eight eukaryotic organisms, we totally identified 1260 histone regulators, whereas dbHiMo only contained 168 histone-modifying enzymes (Supplementary Table S4).
USAGE
The WERAM database was developed so as to be conveniently used. Here we took a human HMT, Histone-lysine N-methyltransferase 2E (KMT2E), as an example to illustrate the effective usage of WERAM. In order to easily look through the data in WERAM, two approaches were implemented for the browse option: by species or by classification (Figure 2). In the option of ‘Browse by species’, the left tree represents the Ensembl (31) taxonomy categories, including primates, rodents, laurasiatheria and so on, whereas the right tree represents the phylogenetic relationship of the eukaryotic species in Ensembl (Figure 2A). By clicking on the ‘Homo sapiens’ button, the families of HATs, HDACs, HMTs, HDMs and reader proteins in H. sapiens can be visualized (Figure 2A). Since the SET1 family belongs to HMTs, users can click on the ‘HMT’ button to view the family information (Figure 2A). Also, WERAM can be further browsed by classification (Figure 2B). The left tree represents the hierarchical classification, whereas a representative 3D structure of the catalytic or PTM-binding domain was taken from the Pfam database (33) and presented on the right for each family, if available (Figure 2B). Users can click on the ‘SET1’ button to visualize the family information across 148 eukaryotes (Figure 2B). By either clicking on the ‘SET1’ button in the HMT group page (Figure 2A) or the ‘Homo sapiens’ button in the SET1 page (Figure 2B), the members in human SET1 family can be viewed, while a brief description of SET1 functions and regulatory roles is also available (Figure 2C). To organize the database, we used WERAM IDs for the identified HATs, HDACs, HMTs, HDMs and reader proteins, respectively. The Ensembl Gene ID was adopted as the secondary accession (Figure 2C). The users can click on the ‘WERAM-HOS-0004′ button to view the detailed information of human KMT2E (Figure 2D). In addition, WERAM can be queried in an easy-to-use manner with a number of search and advance options (Supplementary Figure S4).
Figure 2.

The browse option of WERAM. Two approaches were provided for browsing the database: (A) By species; (B) By classifications. (C) For a family of histone regulators, a brief description and the associated genes are shown. (D) Detailed information of human KMT2E.
DISCUSSION
Acetylation and methylation are two types of greatly important and ubiquitous PTMs in proteins. Besides histones, acetylation and methylation also occur in thousands of non-histone proteins, and participate in the regulation of a broad spectrum of biological processes such as cell metabolism, mitosis and autophagy (12,38,39). Thus, the identification of acetylation and methylation sites in non-histone proteins is helpful for understanding the molecular mechanisms of the two types of PTMs, beyond epigenetic regulation of gene expression. Due to the data accumulation, a number of data resources have been developed. For example, we recently developed a database of compendium of protein lysine modification (CPLM) for up to 12 types of protein lysine modifications, containing 58 563 acetylation in 20 088 proteins and 1144 methylation sites in 745 substrates (40). More recently, we further collected and integrated 5421 methyllysines and methylarginines of 2592 proteins from the literature, and designed a computational tool of GPS-MSP for the prediction of methylation types of modified lysine and arginine residues (41). In addition, dbPTM (42), HPRD (43), PHOSIDA (44), PhosphositePlus (45) and UniProt (32) also maintained and annotated known protein acetylation and/or methylation sites.
Although more and more non-histone proteins have been identified to be acetylated or methylated, histone modifications remain to be further analyzed because the histone code is still far from fully understood. Recently, nearly 20 types of PTMs including both well-studied and new PTMs, such as phosphorylation, ubiquitination, crotonylation and succinylation, were discovered in histone variants (1,4,5). However, the identification of HATs, HDACs, HMTs, HDMs, acetyl- or methyl-binding proteins and site-specific regulator-histones relations is still an attractive challenge, since acetylation and methylation are two most predominant PTMs of histones (3,8–14,17,18,20). In 2007, 96 known histone modifying and demodifying enzymes together with 161 site-specific enzyme-histone relations were summarized and shown (17). Among four core histones, there were 56 acetylation and methylation regulators with 89 enzyme-specific sites in H. sapiens (17). In our study, besides the integration and identification of 20 033 histone regulators from 148 eukaryotes, we also collected 904 known site-specific regulator-histone relations of eight model organisms for acetylation and methylation. For H. sapiens, there have been 413 site-specific relations together with 168 histone regulators in the four core histone proteins (Figure 3). Most of these regulations were newly identified after 2007. For example, in 2009, it was reported that two human HATs, CBP and p300, acetylate histone H3 on lysine 56 (H3K56), which can be deacetylated by two human HDACs including SIRT1 and SIRT2. Such a regulatory mechanism is conserved, while Drosophila CBP/Nejire and Sir2 carry out acetylation and deacetylation of H3K56 in D. melanogaster. Acetylation of H3K56 plays an important role in chromatin assembly and DNA repair, and is significantly associated with cancers (46). Later, Tropberger et al. discovered that p300/CBP-mediated acetylation of H3K122 is induced by estrogen signaling and stimulates gene transcription (47). More recently, it was demonstrated that an acetylation reader, the bromodomain and PHD finger-containing protein 3 (BRPF3) forms a complex with a human HAT of HBO1, and specifically acetylates H3K14 to ensure efficient activation of DNA replication (48). Thus, our results greatly expanded the integration of current knowledge on the histone code.
Figure 3.

A snapshot of human histone modifications, containing 413 site-specific regulator-histone relations and 168 histone regulators in four core histones. The starting methionine was not counted when we referred to PTM sites in histones.
Taken together, our study not only generated a helpful data resource for further analysis, but also provided a better understanding of histone regulators, histone modifications and site-specific regulator-histone relations. The WERAM database will be continuously updated and improved, when new histone regulators or modifications are reported. Also, the classification and annotation information is still not integrative for several organisms, due to the relatively poor annotation of their genomes. Moreover, more species will be added when the complete proteome sets are available for the proteome-wide identification. In addition, we will further collect regulator-specific acetylation and methylation sites in non-histone proteins and provide a more useful database for protein acetylation and methylation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: National Basic Research Program (973 project) [2013CB933900]; Natural Science Foundation of China [81272578, 31671360, J1103514]; International Science & Technology Cooperation Program of China [2014DFB30020].
Conflict of interest statement. None declared.
REFERENCES
- 1.Li X., Li X.D. Chemical proteomics approaches to examine novel histone posttranslational modifications. Curr. Opin. Chem. Biol. 2015;24:80–90. doi: 10.1016/j.cbpa.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 2.Lee K.K., Workman J.L. Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 3.Allis C.D., Jenuwein T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 4.Zhao Y., Garcia B.A. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 2015;7:a025064. doi: 10.1101/cshperspect.a025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang H., Sabari B.R., Garcia B.A., Allis C.D., Zhao Y. SnapShot: histone modifications. Cell. 2014;159:458.e1. doi: 10.1016/j.cell.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yun M., Wu J., Workman J.L., Li B. Readers of histone modifications. Cell Res. 2011;21:564–578. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel D.J., Wang Z. Readout of epigenetic modifications. Annu. Rev. Biochem. 2013;82:81–118. doi: 10.1146/annurev-biochem-072711-165700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith B.C., Denu J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta. 2009;1789:45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Musselman C.A., Lalonde M.E., Cote J., Kutateladze T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filippakopoulos P., Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 11.Chi P., Allis C.D., Wang G.G. Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teperino R., Schoonjans K., Auwerx J. Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metab. 2010;12:321–327. doi: 10.1016/j.cmet.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu F., Wang L., Perna F., Nimer S.D. Beyond transcription factors: how oncogenic signalling reshapes the epigenetic landscape. Nat. Rev. Cancer. 2016;16:359–372. doi: 10.1038/nrc.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feinberg A.P., Koldobskiy M.A., Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016;17:284–299. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yi X., Jiang X.J., Li X.Y., Jiang D.S. Histone methyltransferases: novel targets for tumor and developmental defects. Am. J. Transl. Res. 2015;7:2159–2175. [PMC free article] [PubMed] [Google Scholar]
- 17.Kouzarides T. SnapShot: histone-modifying enzymes. Cell. 2007;131:822. doi: 10.1016/j.cell.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Kutateladze T.G. SnapShot: histone readers. Cell. 2011;146:842.e1. doi: 10.1016/j.cell.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang X.J., Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohan M., Herz H.M., Shilatifard A. SnapShot: histone lysine methylase complexes. Cell. 2012;149:498.e1. doi: 10.1016/j.cell.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peleg S., Sananbenesi F., Zovoilis A., Burkhardt S., Bahari-Javan S., Agis-Balboa R.C., Cota P., Wittnam J.L., Gogol-Doering A., Opitz L., et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–756. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 22.Britton L.M., Gonzales-Cope M., Zee B.M., Garcia B.A. Breaking the histone code with quantitative mass spectrometry. Expert Rev. Proteomics. 2011;8:631–643. doi: 10.1586/epr.11.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hattori T., Taft J.M., Swist K.M., Luo H., Witt H., Slattery M., Koide A., Ruthenburg A.J., Krajewski K., Strahl B.D., et al. Recombinant antibodies to histone post-translational modifications. Nat. Methods. 2013;10:992–995. doi: 10.1038/nmeth.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan Z.F., Arnaudo A.M., Garcia B.A. Mass spectrometric analysis of histone proteoforms. Annu. Rev. Anal. Chem. (Palo Alto Calif) 2014;7:113–128. doi: 10.1146/annurev-anchem-071213-015959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Lv J., Liu H., Zhu J., Su J., Wu Q., Qi Y., Wang F., Li X. HHMD: the human histone modification database. Nucleic Acids Res. 2010;38:D149–D154. doi: 10.1093/nar/gkp968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khare S.P., Habib F., Sharma R., Gadewal N., Gupta S., Galande S. HIstome–a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic Acids Res. 2012;40:D337–D342. doi: 10.1093/nar/gkr1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi J., Kim K.T., Huh A., Kwon S., Hong C., Asiegbu F.O., Jeon J., Lee Y.H. dbHiMo: a web-based epigenomics platform for histone-modifying enzymes. Database (Oxford) 2015;2015:bav052. doi: 10.1093/database/bav052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Draizen E.J., Shaytan A.K., Marino-Ramirez L., Talbert P.B., Landsman D., Panchenko A.R. HistoneDB 2.0: a histone database with variants–an integrated resource to explore histones and their variants. Database (Oxford) 2016;2016:baw014. doi: 10.1093/database/baw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang X.J., Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 30.Finn R.D., Clements J., Arndt W., Miller B.L., Wheeler T.J., Schreiber F., Bateman A., Eddy S.R. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43:W30–W38. doi: 10.1093/nar/gkv397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A., et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y., Liu Z., Cheng H., Gao T., Pan Z., Yang Q., Guo A., Xue Y. EKPD: a hierarchical database of eukaryotic protein kinases and protein phosphatases. Nucleic Acids Res. 2014;42:D496–D502. doi: 10.1093/nar/gkt1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boratyn G.M., Camacho C., Cooper P.S., Coulouris G., Fong A., Ma N., Madden T.L., Matten W.T., McGinnis S.D., Merezhuk Y., et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41:W29–W33. doi: 10.1093/nar/gkt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng W., Wang Y., Liu Z., Cheng H., Xue Y. HemI: a toolkit for illustrating heatmaps. PLoS One. 2014;9:e111988. doi: 10.1371/journal.pone.0111988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie Y., Kang R., Sun X., Zhong M., Huang J., Klionsky D.J., Tang D. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11:28–45. doi: 10.4161/15548627.2014.984267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mo F., Zhuang X., Liu X., Yao P.Y., Qin B., Su Z., Zang J., Wang Z., Zhang J., Dou Z., et al. Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nat. Chem. Biol. 2016;12:226–232. doi: 10.1038/nchembio.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Z., Wang Y., Gao T., Pan Z., Cheng H., Yang Q., Cheng Z., Guo A., Ren J., Xue Y. CPLM: a database of protein lysine modifications. Nucleic Acids Res. 2014;42:D531–D536. doi: 10.1093/nar/gkt1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng W., Wang Y., Ma L., Zhang Y., Ullah S., Xue Y. Computational prediction of methylation types of covalently modified lysine and arginine residues in proteins. Brief. Bioinform. 2016:bbw041. doi: 10.1093/bib/bbw041. [DOI] [PubMed] [Google Scholar]
- 42.Huang K.Y., Su M.G., Kao H.J., Hsieh Y.C., Jhong J.H., Cheng K.H., Huang H.D., Lee T.Y. dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res. 2016;44:D435–D446. doi: 10.1093/nar/gkv1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., et al. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gnad F., Gunawardena J., Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011;39:D253–D260. doi: 10.1093/nar/gkq1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das C., Lucia M.S., Hansen K.C., Tyler J.K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tropberger P., Pott S., Keller C., Kamieniarz-Gdula K., Caron M., Richter F., Li G., Mittler G., Liu E.T., Buhler M., et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. 2013;152:859–872. doi: 10.1016/j.cell.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 48.Feng Y., Vlassis A., Roques C., Lalonde M.E., Gonzalez-Aguilera C., Lambert J.P., Lee S.B., Zhao X., Alabert C., Johansen J.V., et al. BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. EMBO J. 2016;35:176–192. doi: 10.15252/embj.201591293. [DOI] [PMC free article] [PubMed] [Google Scholar]