


Abstract
R-loopDB (http://rloop.bii.a-star.edu.sg) was originally constructed as a collection of computationally predicted R-loop forming sequences (RLFSs) in the human genic regions. The renewed R-loopDB provides updates, improvements and new options, including access to recent experimental data. It includes genome-scale prediction of RLFSs for humans, six other animals and yeast. Using the extended quantitative model of RLFSs (QmRLFS), we significantly increased the number of RLFSs predicted in the human genes and identified RLFSs in other organism genomes. R-loopDB allows searching of RLFSs in the genes and in the 2 kb upstream and downstream flanking sequences of any gene. R-loopDB exploits the Ensembl gene annotation system, providing users with chromosome coordinates, sequences, gene and genomic data of the 1 565 795 RLFSs distributed in 121 056 genic or proximal gene regions of the covered organisms. It provides a comprehensive annotation of Ensembl RLFS-positive genes including 93 454 protein coding genes, 12 480 long non-coding RNA and 7 568 small non-coding RNA genes and 7 554 pseudogenes. Using new interface and genome viewers of R-loopDB, users can search the gene(s) in multiple species with keywords in a single query. R-loopDB provides tools to carry out comparative evolution and genome-scale analyses in R-loop biology.
INTRODUCTION
An R-loop is a three-stranded nucleic acid structure comprising nascent RNA hybridized with its corresponding DNA template strand while leaving the non-template DNA single-stranded. R-loops are often formed during transcription. Both in vitro and in vivo, a few short guanine clusters and further G-rich DNA segment in non-template DNA strand, as well as its sequence length, can provide important determinants of the initiation of R-loop formation and its stabilization (1–4). R-loop formation has been observed from bacteria to mammalian species (5,6). In cells these structures have long been considered to result from rare or accidental RNA with DNA hybrids detected in a few specific loci. However, their functional importance was poor understood.
For the last several years, the roles of R-loops in the association of transcription, splicing, chromatin remodeling, telomere maintenance, genome instability, mutagenesis, cell proliferation, differentiation, epigenetic regulation and silencing as well as disease involvement have been demonstrated (7–17). RNA:DNA hybrids as the component of R-loops are being increasingly associated with human diseases, with a major concern that their presence predisposes the locus to chromosomal instability, mutation and breakage (7,8,18).
In 2011, using the first version of our quantitative R-loop forming sequence (RLFS) prediction model (QmRLFS), R-loopDB was originally constructed as a collection of the RLFSs, identified computationally in the genes of the human genome (18). R-loop models collected in R-loopDB had a number of important features, including genic strand RLFS directionality, CpG island abundance, preferential location of the RLFSs in the open chromatin and highly regulated genic regions (e.g. in the vicinity of alternative transcription start sites, RNA polymerase II pause sites, splicing regions, the first exon and first intron), activation-induced cytidine deaminase target sequences, fragile regions and disease critical loci (18). In silico analysis of the human genome identified a high number of RLFS (245 181) in 59% well-annotated genes (18). Consistent with these predictions, several recent genome-wide experimental studies mapped thousands of RNA:DNA hybridization signals distributed along human and mouse genomes and demonstrated that R-loops can be formed not only in the gene body but also in the upstream promoter regions (9–11) and transcription pause sites located downstream of the poly(A) signal (9,10,12,13,19). Formation of these R-loops in proximal promoter regions was shown to be involved in transcription initiation or elongation (9) and the recruitment of key pluripotency regulators (11). R-loops forming in transcription termination sites could promote termination by slowing down the advance of the RNA polymerase II and allowing the recruitment of termination factors (13). Interestingly, such co-localization of R-loops with key regulatory regions was not limited to humans but also found or predicted in many other species (7,20,21).
Previously, we have presented QmRLFS-finder, a tool using an updated quantitative model of RLFS (QmRLFS) model that has predicted the RLFS locations in ∼75% of the human genes (22). This tool allowed a higher accuracy, precision and strand specificity of the QmRLFS-based predictions of the R-loops in the proximal promoter and downstream transcription termination site regions (7,20). Soon after the publication of the R-loopDB, the DNA:RNA immune-precipitation coupled to high-throughput sequencing (DRIP-seq) data became available (9,23). Ginno et al. (9) have improved the initial DRIP-seq method (23) and reported detection of 4181 RNA:DNA hybrid regions. In (22), we reported that RLFS predicted by QmRLFS-finder overlapped with 79.2% of DRIP-seq regions defined by Ginno et al. (9). In a comparison of the frequencies of experimentally defined R-loops with those RLFSs predicted by QmRLFS-finder, our tool showed 91% accuracy, 94% sensitivity and 75% specificity (22).
Recently, new high-throughput technologies for detection of RNA:DNA hybrid formation and new data sets became available, which provide an opportunity to specify prevalence, strand specificity and chromosome distribution of R-loops in the human and mouse genomes, their conservation across cell types and species, their mechanisms of formation and turnover and specify their functions (12,14,24).
The new version of R-loopDB, based on the predictions by QmRLFS-finder (22), includes several key improvements and extensions that facilitate exploration of the RLFS analysis. We updated the identification of RLFSs in the human genome and provided for the first time the genome-scale prediction of RLFSs in seven additional organisms. The updated R-loopDB allows the user to search for gene(s) containing RLFSs not only in genic regions but also in 2 kb upstream and downstream flanking sequence of the gene, termed ‘RLFS-positive genes’. Compared to the previous R-loopDB version, we now use Ensembl gene annotation system that contains information on protein coding genes, pseudogenes, long noncoding RNA genes and short noncoding RNA genes (25). Mapping of RLFSs and other genomic regulatory sequences is graphically visualized in Gbrowse2 (26) and UCSC genome browser (27). In the case of the human and mouse genomes, the experimentally detected RNA:DNA hybrid data (9,12,14,24) on the genome scale were integrated into the genomics viewers. That allows users to carry out comprehensive comparative analyses of the predicted and experimentally detected RLFSs. Based on the provided results, users can narrow down their target and pick the most plausible sequence of the R-loop forming region.
Data sources
We collected all genome assembly data from the UCSC genome browser (https://genome.ucsc.edu/) (27). The human genome hg19 (NCBI build GRCh37) was used as the reference genome in this database. We also analyzed genomic data of selected model organisms including Mus musculus (mm10), Rattus norvegicus (rn5), Pan troglodytes (panTro4), Gallus gallus (galGal4), Xenopus tropicalis (xenTro3), Drosophila melanogaster (dm5) and Saccharomyces cerevisiae (saccer3). Only the reference chromosomes were used in this study. Therefore, we excluded other sequence regions such as unlocalized and unplaced scaffolds, supercontigs, assembly patches and alternate loci (haplotypes). Gene annotation data for these organisms was retrieved directly from MySQL server of BioMart database build 75 (ensembl_mart_75; http://www.ensembl.org/info/data/mysql.html) (25). Genomic information of the gene structures for these organisms (GTF format) was downloaded from Ensembl Release 75 (http://feb2014.archive.ensembl.org/info/data/ftp/; accessed 1 April 2016).
Identification of R-loop structures
We predicted R-loop structures on both strands of genomic DNA using QmRLFS-finder (22). After obtaining RLFSs, we collected RLFSs located in the genic region and 2 kb upstream and downstream flanking regions of each gene using BEDTools (28). RLFSs that overlapped by at least one nucleotide were merged into unique RLFS clusters. When no further RLFSs overlapped the long merged sequence, then this sequence, called an RLFS-merged region, has reached its maximum length. Such regions were reported, counted and made visible in the updated R-loopDB.
RLFSs that are located in any genic region were considered gene-associated RLFS. We determined locations of RLFSs on the 2 kb upstream, genic (gene body) and 2 kb downstream regions. RLFSs that are located simultaneously in any genic combination of these three regions were also identified and counted.
GC skew calculation
The GC skew could favor the thermodynamic stability of R-loop formation (21,24). GC skew values, reflecting the asymmetric distribution of G and C between the leading strand and the lagging strand were calculated according to the following equation
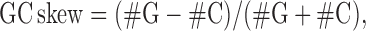 |
(1) |
using the numbers of G and C in 200 bp sliding window and a step size of 10 bp. The GC skew profiles of each organism were stored in BigWig file format.
RLFS, RLFS-merged regions and RLFS-positive genes
R-loopDB currently covers eight organisms, including human, mouse, rat, chimpanzee, chicken, frog, fruit fly and yeast. Supplementary Table S1 provides information on the genome assemblies of these species. Using QmRLFS-finder (22), we significantly extended the list of RLFSs and RLFS-positive genes in the human genome compared to the previous version (18). The summary statistics of RLFSs, RLFS-merged regions and RLFS-positive genes presented in the previous and updated R-loopDBs are shown in Table 1. For instance, 511 651 RLFSs (or 169 222 RLFS-merged regions) were computationally predicted in genic regions and 2 kb upstream and downstream flanking regions of the genes, whereas, in the previous R-loopDB, 245 181 RLFSs (or 140 106 RLFS-merged regions) were found in the UCSC-defined gene regions. In total, R-loopDB provides chromosome coordinates, sequences and genomic data of 1565 795 RLFSs distributed across 121 056 genes and 2 kb upstream and downstream flanking region of the genes of eight organisms.
Table 1. The growth of data hosted by the R-loopDB since the initial release.
| Initial release in 2012 | Up-to-date in 2016 | |
|---|---|---|
| Gene annotation system | UCSC known genes | Ensembl build 75 |
| No. of organisms | 1 species | 8 species |
| No. of transcript IDs | 39 720 | 286 584 |
| No. of gene IDs | 14 954 | 121 056 |
| No. of RLFSs in the human genes | 245 181 | 511 651 |
| No. of RLFS-merged regions in the human genes | 140 106 | 169 222 |
| No. of RLFSs in genes | 245 181 | 1 565 795 |
| No. of RLFS-merged regions in genes | 140 106 | 565 789 |
Remark: the initial release database provides RLFSs only in genic regions, where the updated database provides RLFSs in genic regions and 2 kb up/downstream region of genes.
Using the Ensembl gene annotation system (25) in R-loopDB, RLFS-positive genes can be classified into 93 454 protein-coding genes, 7 554 pseudogenes, 12 480 long non-coding RNA genes and 7 568 short non-coding RNA genes (Table 2).
Table 2. Statistics of RLFS-positive genes according to the Ensembl gene annotation system and ratio of RLFS-positive gene to total number of gene for each gene category in each organism.
| No. of protein coding genes* | No. of pseudogenes* | No. of long noncoding RNA genes* | No. of short noncoding RNA genes* | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Organism | Total | RLFS positive | Ratio | Total | RLFS positive | Ratio | Total | RLFS positive | Ratio | Total | RLFS positive | Ratio |
| human | 23 431 | 17 798 | 0.76 | 15 934 | 4 433 | 0.28 | 14 966 | 9 028 | 0.60 | 9 771 | 2 296 | 0.23 |
| mouse | 22 835 | 18 201 | 0.80 | 6 129 | 2 161 | 0.35 | 4 291 | 3 283 | 0.77 | 5 924 | 1 950 | 0.33 |
| rat | 22 771 | 17 529 | 0.77 | 1 840 | 662 | 0.36 | 79 | 65 | 0.82 | 1 714 | 608 | 0.35 |
| chimpanzee | 18 759 | 15 462 | 0.82 | 572 | 213 | 0.37 | 0 | 0 | n/a | 8 681 | 1 870 | 0.22 |
| chicken | 15 508 | 12 007 | 0.77 | 42 | 26 | 0.62 | 0 | 0 | n/a | 1 558 | 546 | 0.35 |
| frog | 18 442 | 9 932 | 0.54 | 173 | 44 | 0.25 | 0 | 0 | n/a | 1 306 | 268 | 0.21 |
| fruit_fly | 13 863 | 2 423 | 0.17 | 200 | 11 | 0.06 | 830 | 104 | 0.13 | 789 | 30 | 0.04 |
| yeast | 6 692 | 102 | 0.02 | 21 | 4 | 0.19 | 15 | 0 | n/a | 398 | 0 | n/a |
| All organisms | 142 301 | 93 454 | 0.66 | 24 911 | 7 554 | 0.30 | 20 181 | 12 480 | 0.62 | 30 141 | 7 568 | 0.25 |
*The Ensemble gene biotypes were grouped into protein coding genes, pseudogenes, long non-coding RNA genes and short non-coding RNA genes (available at http://asia.ensembl.org/Help/Faq?id=468). Examples of gene biotypes in each group are as follows:
-Protein coding genes: IGC gene, IGD gene, IG gene, IGJ gene, IGLV gene, IGM gene, IGV gene, IGZ gene, nonsense mediated decay, non-translating CDS, non-stop decay, polymorphic pseudogene, TRC gene, TRD gene, TRJ gene.
-Pseudogenes: disrupted domain, IGC pseudogene, IGJ pseudogene, IG pseudogene, IGV pseudogene, processed pseudogene, transcribed processed pseudogene, transcribed unitary pseudogene, transcribed unprocessed pseudogene, translated processed pseudogene, TRJ pseudogene, unprocessed pseudogene.
-Long non-coding RNA genes: 3-prime overlapping ncRNA, ambiguous ORF, antisense, antisense RNA, lincRNA, ncRNA host, processed transcript, sense intronic, sense overlapping.
-Short non-coding RNA genes: miRNA, miRNA pseudogene, miscRNA, miscRNA pseudogene, Mt rRNA, Mt tRNA, rRNA, scRNA, snlRNA, snoRNA, snRNA, tRNA, tRNA pseudogene.
Search for RLFS-positive genes and visualization tools
Visual tools for RLFS search could be used for locating putative R-loops in a specific region of genic or gene-proximal regions for further experimental design and validation of the computational prediction(s). Here, we redesigned the interface of R-loopDB to search genes containing RLFS in multiple organisms with a single search. The genes containing RLFS can be retrieved from the database using several search options, e.g. keywords, Ensembl gene ID or Ensembl transcript ID, gene symbol, gene description, Entrez ID, Refseq ID, gene biotype and synonyms (Figure 1A). Note that the keywords option will search the query input string against all terms in the database. The gene search query returns RLFS-positive genes (Figure 1B). Each Ensembl gene ID of the search result is linked to the result gene page that provides information in two parts ‘Gene Overview’ (Figure 1C) and ‘Genomic Location’ (Figure 2).
Figure 1.

Screenshots of R-loopDB and search results. (A) R-loopDB main search page where a user can perform a search based on keywords or specific gene ID and the resulting page is shown in (B). R-loop forming sequence (RLFS) and other regulatory sequences and their characteristics for NEAT1 (nuclear noncoding RNA gene) are visualized. (C) The user can view the gene result page with RLFS information by clicking on the Ensembl gene ID linked from (B).
Figure 2.

Screenshots of mapping of RLFS, genes, GC skew profile and other annotation data. The genomic mapping can be viewed in the bottom panel of the gene result page.
The ‘Gene Overview’ provides detailed information of the Ensembl gene (Figure 1C.i) and RLFS-merged regions within proximal gene regions (Figure 1C.ii). Here, NEAT1 gene encodes a nuclear long non-coding RNA. According to its annotation, NEAT1 RNA is essential for the formation and maintenance of nuclear structures called paraspeckles and is involved in cell malignancy. The gene information panel (Figure 1C.i) shows the gene annotation of a given Ensembl gene ID retrieved from the BioMart database (build 75), i.e. gene description, gene biotype, chromosome location, Ensembl transcript ID, Entrez ID and RefSeq ID, while gene synonyms were retrieved from the Ensembl database (build 84). In RLFS-merged region information (Figure 1C.ii; also see Figure 2a), this table provides a list of RLFS-merged regions located in the genic region and 2 kb upstream and downstream flanking regions of the particular gene. Each RLFS-merged region has useful information for supporting the R-loop forming potential, such as (i) the number of RLFSs, and (ii) strand and (iii) statistics of the GC skew value. To find RLFS candidates for experimental validation, we recommend that users consider the information in the RLFS-merged region table (Figure 1C.ii). The first one is the ‘number of RLFSs’. A higher number of RLFSs in the RLFS-merged region implies a higher propensity of R-loop initiation and formation in a given locus. The second one is ‘strand’. According to the R-loop definition where a nascent RNA is hybridized with a DNA template, predicted RLFS are located on the same strand as the transcribed gene (Supplementary Figure S1). The third one is ‘statistics of GC skew value’. GC skew value was calculated at the whole genomic scale, as shown in Figure 2c. The ‘statistics of GC skew value’ show statistics of GC skew value in each RLFS-merged region including minimum, maximum, mean and median. GC skew maximum values > 0.1 (for plus strand) or GC skew minimum values < −0.1 (for minus strand) are considered G-rich regions (21). The more G-rich an RLFS-merged region is, the more likely it is to increase the thermodynamic stability of R-loop formation (24). In the case of the human and mouse genomes, ‘DRIP/RDIP support’ and ‘DRIPc support’ columns, which indicate the number of RNA:DNA hybrid detection experiments for each RLFS-merged region, are provided. There are at least nine RNA:DNA hybrid detection experiments including four DRIP-seq studies in Ntera2, one DRIP-seq in K562, one DRIP-seq in primary fibroblast cells (9,12,14), two RDIP-seq studies in IMR-90 and HEK293T cells (24) and one DRIPc-seq in Ntera2. Individually, these experiments showed variations in the sequence overlap region boundaries, their locations and the sequence cluster overlap peak heights, however, collectively they often demonstrate the patterns converged on the theoretical predictions. A combination of computational predictions and biological data sets suggests a role of NEAT1-associated R-loops in an aberrant NEAT1 transcription initiation and/or 3-end elongation isoform syntheses, leading to modulation of the paraspeckle formation and cell malignancy.
The RLFS or RLFS-merged region results can be downloaded into text file, BED file and FASTA file (Figure 1C.iii).
The top panel of the ‘Genomic Location’ result shows gene name, gene location, strand and cytoband information of the particular gene (see in Figure 2). New genomics visualization has been integrated into R-loopDB using embedded Gbrowse2. The genomics viewer provides visualization of RLFS-merged, RLFS, CpG Island, GC skew profile, Ensembl genes, Ensembl transcripts, DRIP-seq profile, RDIP-seq profile and DRIPc-seq profile in a particular genic region as well as in 2 kb upstream and downstream flanking regions of the gene. In Figure 2a–d, the light blue and pink colors indicate the orientation of the sense- and antisense-strand of the annotations, respectively. Clicking on an individual RLFS-merged (Figure 2a) or RLFS (Figure 2b) will link to the detailed information of RLFS-merged (Supplementary Figure S2A) or RLFS (Supplementary Figure S2B), respectively. GC skew profile (Figure 2c) indicates the boundaries between the bias distributions of G-rich regions in a sense- or antisense-strand of the chromosome. Clicking on an individual Ensembl gene or Ensembl transcript (Figure 2d) will result in a close-up view of the particular gene or transcript. In Figure 2e and f, DRIP-seq, RDIP-seq and DRIPc-seq peak regions indicate significant enrichment of DRIP-seq, RDIP-seq and DRIPc-seq signals, respectively, from different studies of experimental RNA:DNA hybrid detection. R-loopDB also provides external browser buttons which are linked to interactive genomics viewers such as UCSC Genome Browser (27) and GBrowse2 (26) (Supplementary Figure S3). Furthermore, users can directly input the gene coordinate or gene symbol on UCSC Genome Browser for searching. Users have the option to turn on/off or re-order the tracks that are displayed on the genome browser.
Comparative analysis of RLFS in genic and proximal gene regions
According to the updated R-loopDB, the largest number of RLFSs is observed in the human genome (664 791 sequences) and the smallest number of RLFSs is found in the yeast genome (78 sequences). Mammalian genomes are highly abundant in RLFSs and RLFS-merged clusters (Supplementary Table S2). The RLFSs are preferentially located in genic and proximal gene regions.
The most abundant are yeast (95%), human (77%), fruit fly (77%) and chicken (68%) gene body and 2 kb proximal genic regions. The fraction of RLFS-positive genes and 2 kb proximal genic regions is relatively lower in chimpanzee (57%), mouse (53%), rat (42%) and frog (33%) genomes.
The numbers and the proportions of RLFSs within genic and proximal gene regions also differ across the organisms. Detailed information is presented in Supplementary Table S2. According to the updated R-loopDB, the upstream and downstream proximal gene regions are highly enriched with unique RLFSs, the boundaries of which can either overlap or not overlap with gene starts (or transcription start sites), gene body and the gene ends. Such data could provide the targets for detailed characterization in future experimental studies. The Venn diagrams of Supplementary Figure S4A and S4B shows these values for the RLFS overlapped with the human genes and transcripts, respectively.
RLFS positions within a gene of different organisms
R-loopDB allows users to search and analyze the RLFS in a gene and in the proximal up-stream and down-stream gene body regions in multiple organisms (Supplementary Figure S5A). For illustration purposes, glycine dehydrogenase (GLDC) gene was used as a query. GLDC protein is a member of the multiple-enzyme glycine cleavage system involved in the degradation of glycine. The deletion in GLDC is a major cause of nonketotic hyperglycinemia, an inborn error of metabolism characterized by the accumulation of glycine in body fluids and various neurological symptoms (29). In the previous paper, we have shown that RLFSs are located at the deleted regions of the human GLDC and may contribute to recombination of this gene (18). In this work, the updated R-loopDB result page shows that five of the eight organisms, including human, chimpanzee, mouse, rat and chicken, contain at least one RLFS in GLDC gene. Supplementary Figure S5B shows the search result of GLDC containing RLFS in five analyzed organisms. Figure 3 illustrates the relative RLFS positions in GLDC gene of the organisms. Interestingly, RLFSs have relative positions within the first exon of GLDC gene between all five organisms (green color box in Figure 3). In addition, there are several other RLFSs showing relative positions within an intron of the GLDC gene, which may characterize the commonness and uniqueness of the human and chimpanzee intron sequences (blue color boxes in Figure 3). These RLFS clusters (in blue color boxes in Figure 3) are not computationally identified in other (non-primate) organisms. This example allows users to investigate several hypotheses regarding the functional roles of the sequence structure and genome location of the predicted RLFSs. Many aspects of comparative evolution and evolutional conservation of RLFSs and the disease-associated functions of the R-loops in GLDC and other genic regions annotated in the R-loopDB may be investigated using the R-loopDB.
Figure 3.

RLFS in different genic and the proximal gene regions and across different organisms. Positions of RLFSs predicted in the GLDC gene in five organisms. Screenshot from Gbrowse2 shows relative positions of RLFSs in the GLDC gene for human, chimpanzee, mouse, rat and chicken. The green color box indicates relatively similar positions of the RLFSs within exon 1 of GLDC across all five organisms. Blue color boxes indicate the relationship of the positions of RLFSs within an intron of GLDC gene between human and chimpanzee.
CONCLUSION AND FUTURE DIRECTIONS
Here, we present a comprehensive database of RLFSs in eight organisms, i.e. human, chimp, mouse, rat, chicken, frog, fruit fly and yeast, that offers benefits for the researchers who study R-loop related human diseases using these species. The combination of several other data resources, such as RLFS strand, GC skew value and experimental R-loop detection data, could provide more information for advanced analyses and predictions. For instance, predicted RLFS and other R-loop formation features with DRIP-seq data and Global-Run-On sequencing was demonstrated as a fruitful strategy in identification of the specific transcriptional features, namely convergence of transcription and polymerase II stalling, as important factors underlying secondary genetic lesions frequently seen in precursor B leukemia (30). RNA:DNA hybrids are being increasingly associated with human diseases, with a major concern that their presence predisposes a locus to chromosomal breakage (7,8).
The updated R-loopDB could be utilized not only as a predictive model resource but also as an analytical tool to predict and validate many RLFS-positive genes (as coding and non-coding for proteins), pseudogenes to test the specific hypotheses in R-loop biology, to improve the existing methods, to design new experimental studies and to provide an adequate interpretation of the experimental and theoretical results. In those contexts, a combination of R-loopDB and QmRLFS-finder web server (identifying RLFSs in non-annotated and artificial targeting sequences) might be useful.
Further development of R-loopDB will be focused on classification of RLFS and R-loops, evolutionary conservation and de novo RLFSs and the RLFS-positive genes in different gene families and subsets, cell types and organisms. We plan to develop and integrate the options to retrieve evolutionarily conserved and unique RLFS-positive genes, searching across several mammalian and non-mammalian genomes in the R-loopDB. As more experimental R-loop data sets are generated, in the next update, R-loopDB should offer a more comprehensive collection of experimentally supported data and further development of the integrative and predictive tools.
Acknowledgments
The authors thank Tan Xuanru Mavis for the discussion and data processing. The collaboration between the co-authors would not have been possible without the travel grants from the School of Bioresources and Biotechnology, King Mongkut's University of Technology, Thonburi, Thailand.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Biomedical Research Council of the Agency for Science, Technology and Research (A*STAR), Singapore. Funding for open access charge: Bioinformatics Institute/A*STAR core budget.
Conflict of interest statement. None declared.
REFERENCES
- 1.Roy D., Yu K., Lieber M.R. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol. Cell. Biol. 2008;28:50–60. doi: 10.1128/MCB.01251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roy D., Lieber M.R. G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol. Cell. Biol. 2009;29:3124–3133. doi: 10.1128/MCB.00139-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Z.Z., Pannunzio N.R., Hsieh C.L., Yu K., Lieber M.R. The role of G-density in switch region repeats for immunoglobulin class switch recombination. Nucleic Acids Res. 2014;42:13186–13193. doi: 10.1093/nar/gku1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loomis E.W., Sanz L.A., Chedin F., Hagerman P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014;10:e1004294. doi: 10.1371/journal.pgen.1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguilera A., Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Maduike N.Z., Tehranchi A.K., Wang J.D., Kreuzer K.N. Replication of the Escherichia coli chromosome in RNase HI-deficient cells: multiple initiation regions and fork dynamics. Mol. Microbiol. 2014;91:39–56. doi: 10.1111/mmi.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos-Pereira J.M., Aguilera A. R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 2015;16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- 8.Richard P., Manley J.L. R loops and links to human disease. J. Mol. Biol. 2016 doi: 10.1016/j.jmb.2016.08.031. doi:10.1016/j.jmb.2016.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginno P.A., Lim Y.W., Lott P.L., Korf I., Chedin F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013;23:1590–1600. doi: 10.1101/gr.158436.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skourti-Stathaki K., Kamieniarz-Gdula K., Proudfoot N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 2014;516:436–439. doi: 10.1038/nature13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen P.B., Chen H.V., Acharya D., Rando O.J., Fazzio T.G. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol. 2015;22:999–1007. doi: 10.1038/nsmb.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanz L.A., Hartono S.R., Lim Y.W., Steyaert S., Rajpurkar A., Ginno P.A., Xu X., Chedin F. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell. 2016;63:167–178. doi: 10.1016/j.molcel.2016.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skourti-Stathaki K., Proudfoot N.J., Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 2011;42:794–805. doi: 10.1016/j.molcel.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim Y.W., Sanz L.A., Xu X., Hartono S.R., Chedin F. Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutieres syndrome. Elife. 2015;4:21–1. doi: 10.7554/eLife.08007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duquette M.L., Huber M.D., Maizels N. G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas. Cancer Res. 2007;67:2586–2594. doi: 10.1158/0008-5472.CAN-06-2419. [DOI] [PubMed] [Google Scholar]
- 16.Groh M., Lufino M.M., Wade-Martins R., Gromak N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014;10:e1004318. doi: 10.1371/journal.pgen.1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helmrich A., Ballarino M., Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell. 2011;44:966–977. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Wongsurawat T., Jenjaroenpun P., Kwoh C.K., Kuznetsov V. Quantitative model of R-loop forming structures reveals a novel level of RNA-DNA interactome complexity. Nucleic Acids Res. 2012;40:e16. doi: 10.1093/nar/gkr1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeo A.J., Becherel O.J., Luff J.E., Cullen J.K., Wongsurawat T., Jenjaroenpoon P., Kuznetsov V.A., McKinnon P.J., Lavin M.F. R-loops in proliferating cells but not in the brain: Implications for AOA2 and other autosomal recessive ataxias. PLoS One. 2014;9:e90219. doi: 10.1371/journal.pone.0090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenjaroenpun P., Wongsurawat T., Yenamandra S.P., Kuznetsov V.A. Quantitative Structural Model for Prediction and Analysis of R-loop Forming Sequences in the Genome. 2015. pp. 4–1. [Google Scholar]
- 21.Hartono S.R., Korf I.F., Chedin F. GC skew is a conserved property of unmethylated CpG island promoters across vertebrates. Nucleic Acids Res. 2015;43:9729–9741. doi: 10.1093/nar/gkv811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jenjaroenpun P., Wongsurawat T., Yenamandra S.P., Kuznetsov V.A. QmRLFS-finder: a model, web server and stand-alone tool for prediction and analysis of R-loop forming sequences. Nucleic Acids Res. 2015;43:W527–W534. doi: 10.1093/nar/gkv344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ginno P.A., Lott P.L., Christensen H.C., Korf I., Chedin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012;45:814–825. doi: 10.1016/j.molcel.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nadel J., Athanasiadou R., Lemetre C., Wijetunga N.A., OBroin P., Sato H., Zhang Z., Jeddeloh J., Montagna C., Golden A., et al. RNA:DNA hybrids in the human genome have distinctive nucleotide characteristics, chromatin composition, and transcriptional relationships. Epigenetics Chromatin. 2015;8:46–65. doi: 10.1186/s13072-015-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein L.D., Mungall C., Shu S., Caudy M., Mangone M., Day A., Nickerson E., Stajich J.E., Harris T.W., Arva A., et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Speir M.L., Zweig A.S., Rosenbloom K.R., Raney B.J., Paten B., Nejad P., Lee B.T., Learned K., Karolchik D., Hinrichs A.S., et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016;44:D717–D725. doi: 10.1093/nar/gkv1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanno J., Hutchin T., Kamada F., Narisawa A., Aoki Y., Matsubara Y., Kure S. Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia. J. Med. Genet. 2007;44:e69. doi: 10.1136/jmg.2006.043448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinaniemi M., Vuorenmaa T., Teppo S., Kaikkonen M.U., Bouvy-Liivrand M., Mehtonen J., Niskanen H., Zachariadis V., Laukkanen S., Liuksiala T., et al. Transcription-coupled genetic instability marks acute lymphoblastic leukemia structural variation hotspots. Elife. 2016;5:1–26. [Google Scholar]