


Abstract
The use of high-throughput array and sequencing technologies has produced unprecedented amounts of gene expression data in central public depositories, including the Gene Expression Omnibus (GEO). The immense amount of expression data in GEO provides both vast research opportunities and data analysis challenges. Co-expression analysis of high-dimensional expression data has proven effective for the study of gene functions, and several co-expression databases have been developed. Here, we present a new co-expression database, COEXPEDIA (www.coexpedia.org), which is distinctive from other co-expression databases in three aspects: (i) it contains only co-functional co-expressions that passed a rigorous statistical assessment for functional association, (ii) the co-expressions were inferred from individual studies, each of which was designed to investigate gene functions with respect to a particular biomedical context such as a disease and (iii) the co-expressions are associated with medical subject headings (MeSH) that provide biomedical information for anatomical, disease, and chemical relevance. COEXPEDIA currently contains approximately eight million co-expressions inferred from 384 and 248 GEO series for humans and mice, respectively. We describe how these MeSH-associated co-expressions enable the identification of diseases and drugs previously unknown to be related to a gene or a gene group of interest.
INTRODUCTION
Unprecedented amounts of gene expression data derived from high-throughput microarray and next-generation sequencing (NGS) technologies have accumulated in several public depositories such as the Gene Expression Omnibus (GEO) (1), ArrayExpress (2) and the Short Read Archive (SRA) (3). The cumulative size of the databases continues to grow at an increasing rate owing to the ever-decreasing cost for NGS. Therefore, these central depositories of gene expression data are considered important resources with huge potential for the study of gene functions. For example, as of July 2016, GEO contained over 1.8 million microarray or NGS samples, of which over 1.3 million samples were derived from either humans or laboratory mice. The majority of the samples are for gene expression profiling. This existing prohibitive amount of data becomes a major challenge when exploring functional hypotheses using the public data depository (4).
One of the popular approaches to study gene functions using high-dimensional expression data is co-expression analysis, which is based on the key observation that functionally associated genes tend to co-express across many different biological contexts (5). Aggregated co-expression relationships can be used to construct a functional gene network, in which a functional inference for each gene can be made using various network analysis algorithms (6). This network-based approach has proven useful in disease gene identifications and disease classifications (7,8). To increase the usability of the expression data in the central depositories, co-expression databases such as COXPRESdb (9) and GeneFriends (10) were developed through large-scale analysis efforts. These databases allow users to identify co-expressed genes and their associated biological concepts such as Gene Ontology (GO) terms (11), facilitating the functional characterization of a gene of interest.
Here, we present a new co-expression database, COEXPEDIA (www.coexpedia.org), which is distinctive from other co-expression databases in three aspects. First, we included only co-expressions in COEXPEDIA that passed a rigorous statistical test for co-functionality. We anticipated that a high correlation of expression across samples does not always indicate a functional association between genes. Therefore, we opted to measure the probability of functional coupling for the given co-expressed gene pairs and take gene pairs that were significantly co-expressed as well as highly likely to be co-functional. Second, we inferred co-expressions from individual studies rather than aggregating samples from multiple studies. With this study-centric co-expression analysis, we were able to focus more on context-associated co-expressions. We achieved this by leveraging co-expressions among samples for each GEO series (GSE), which generally corresponded to a published study that was designed and conducted to investigate gene functions with respect to a particular biomedical context such as a disease and drug treatment. Third, the co-expressions in COEXPEDIA are associated with medical subject headings (MeSH). We employed MeSH terms to systematically analyze the context-associated co-expressions. MeSH terminology was developed by the National Library of Medicine (NLM) as a controlled vocabulary thesaurus to index and catalog biomedical information in articles for PubMed (see https://www.nlm.nih.gov/mesh/ for more details). MeSH terms are hierarchically organized with 16 top-level categories. Because most GSEs are based on at least one PubMed article that has been indexed by MeSH terms, the co-expressions derived from each GSE are consequently associated with MeSH terms. We describe how the MeSH-associated co-expressions enable the identification of unknown gene-to-disease, gene-to-drug, disease-to-disease, and disease-to-drug associations.
INFERENCE OF CO-FUNCTIONAL CO-EXPRESSIONS
We analyzed human and mouse GSEs based on the major Affymatrix platforms (GPL96, GPL570, GPL571, GPL3921, GPL6244 for human; GPL339, GPL1261, GPL8321, GPL6246 for mouse) to infer co-expressions for biomedical research. GEO contains several hundred thousand human and mouse samples. We used the Pearson correlation coefficient (PCC) to measure the correlation of expression patterns between two genes. Using expression correlations across a large number of samples by concatenating multiple GSEs can identify statistically more robust co-expressions, yet co-expression signals for a specific context could be buried by the abundant noise spanning samples for irrelevant contexts. Therefore, we conducted a co-expression analysis for each GSE, as each is generally composed of samples from more coherent biological contexts. To manage the statistical robustness of the inferred co-expressions, we used only GSEs with at least 12 samples. Previous studies reported that the MAS5 normalization method was more effective than the RMA method in co-expression analysis (12,13). We therefore normalized all the raw array data using MAS5 except GPL6244 and GPL6246, the more recent platforms in which MAS5 is not applicable.
To ensure that the inferred co-expressed genes are also likely to be functionally coupled, we measured the likelihood of functional association for co-expressed gene pairs using the Bayesian statistics framework (14). For this benchmarking analysis, we generated a set of positive gold-standard co-functional gene pairs using pathway annotations with the GO biological process (GOBP) or MetaCyc (15) for both human and mouse data. We also defined a set of negative gold-standard data by pairing two annotated genes that do not share their pathway annotation at all. The log likelihood score (LLS) of co-expressions was calculated with the following equation:
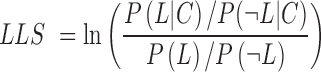 |
where 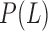 and
and 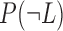 represent the probabilities of positive and negative gold-standard co-functional links, respectively.
represent the probabilities of positive and negative gold-standard co-functional links, respectively. 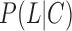 and
and 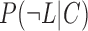 indicate the probabilities of positive and negative gold-standard co-functional links for the given strength of co-expression measured by the PCC, respectively. Gene pairs were sorted by decreasing order of the PCC, and then the LLS was calculated for each bin of 1000 gene pairs from the highest PCC. We then found a regression curve that best fit the data points between the PCC and LLS based on a sigmoid function. Using the best regression function, we assigned an LLS for all gene pairs with a PCC. To identify co-expressed gene pairs that are highly likely to be functionally coupled, we applied an LLS threshold of 1, which is equivalent to ∼2.7 times more likely than random chance (Figure 1A).
indicate the probabilities of positive and negative gold-standard co-functional links for the given strength of co-expression measured by the PCC, respectively. Gene pairs were sorted by decreasing order of the PCC, and then the LLS was calculated for each bin of 1000 gene pairs from the highest PCC. We then found a regression curve that best fit the data points between the PCC and LLS based on a sigmoid function. Using the best regression function, we assigned an LLS for all gene pairs with a PCC. To identify co-expressed gene pairs that are highly likely to be functionally coupled, we applied an LLS threshold of 1, which is equivalent to ∼2.7 times more likely than random chance (Figure 1A).
Figure 1.

Assessment of co-functional co-expression. (A) Co-functional co-expressions show a strong positive correlation between the strength of co-expression (measured by PCC) and likelihood of functional association (measured by LLS). For example, co-expressions across healthy and malignant human B-lymphocytes with or without B-cell receptor stimulation (GSE39411) show a strong correlation with the likelihood of co-functional associations. LLS scores are assigned for all co-expressed gene pairs based on sigmoid regression fitting of data points between PCC and LLS, and only those with at least an LLS of 1 (i.e. ∼2.7 more likely to be co-functional than random chance) are included in the COEXPEDIA database. (B) In many cases, the co-expressions inferred from a GSE do not implicate a co-functional relationship. For example, the co-expressions across time-course samples during the activation of human regulatory and effector T cells (GSE11292) show a poor correlation between PCC and LLS, indicating that these co-expressions are unlikely to be co-functional.
During our co-expression analysis for the GSEs, we frequently observed poor correlations between PCC and LLS values (see the example of GSE11292, Figure 1B), demonstrating that the co-expressions across the given samples do not necessarily indicate a co-functional relationship between the two genes. We analyzed a total of 2056 and 2468 GSEs but found meaningful correlations between PCC and LLS values and inferred co-functional co-expressions from only 384 and 248 GSEs for humans and mice, respectively (Supplementary Table S1). COEXPEDIA currently contains a total of 3 026 367 and 4 912 497 co-functional co-expressions for humans and mice, respectively.
OVERVIEW OF THE COEXPEDIA DATA STRUCTURE
All co-expressions in COEXPEDIA are based on an individual GSE, which generally corresponds to at least one published study. Given that each study was designed and conducted to investigate gene functions within a particular biomedical context, the co-expressions derived from the study are expected to be associated with the relevant biomedical context as well (Figure 2A). For example, the GSE6613 study was conducted to identify gene expression signatures of early Parkinson's disease. Thus, it is expected that co-expressions inferred from GSE6613 are associated with Parkinson's disease at the early stage. If we interpret co-expression networks in the context of Parkinson's disease, we may have a better chance of identifying causative genes or diagnostic biomarkers for Parkinson's disease. Therefore, the context information for the given co-expressions will enhance our ability to generate hypotheses that are more relevant to the given medical subjects. If the given GSE has been published, we can collect relevant biomedical information via the MeSH terms assigned for the article. We collected the list of PubMed articles for each GSE from Simple Omnibus Format in Text (SOFT) data files, and MeSH terms from XML-formatted summaries of each PubMed article. These cross references between GSEs and PubMed articles, and between MeSH terms and PubMed articles were then used to map associations between GSEs and MeSH terms. For example, GSE6613 can be associated with the MeSH terms ‘Parkinson's disease’, ‘biomarkers’, ‘diagnosis’ and ‘risk factors’ via the associated PubMed article (16). This biomedical information for co-expressions allows the exploration of novel biomedical hypotheses for a gene or a group of genes using enriched MeSH terms among co-expressions (Figure 2B).
Figure 2.

Overview of the COEXPEDIA data structure. (A) Co-expressions are inferred from individual GSEs, each of which is associated with at least one PubMed article. Each article has multiple MeSH terms indexed. Consequently, each co-expression can be associated with at least one MeSH term. (B) A real example of the data structure by CTLA4 co-expressions. The enrichment score of each MeSH term for the given co-expressions can be calculated by the summation of the sum of edge scores (i.e. LLS) derived from multiple GSEs (e.g. enrichment score for T-lymphocytes was calculated by summation of the sum of edge scores from GSE12195, GSE20711 and GSE14924). The given scoring scheme identified isotretinoin, T-lymphocytes, cyclophosphamide, epirubicin as top four MeSH terms enriched for CTLA4 co-expressions.
MeSH terms are composed of 16 categories, and we selected terms from only four categories that are thought to be more useful for generating biomedical hypotheses: Anatomy (A), Diseases (C), Chemicals and Drugs (D) and Psychiatry and Psychology (F). MeSH terms in category Anatomy (A) indicate particular organs, tissues, cell types, and other body structures. Considering that many diseases involve disorders of a particular organ, tissue, or cell-type, these MeSH terms are expected to be useful for generating disease-relevant hypotheses. We excluded MeSH terms under two subcategories of the Anatomy (A) category: ‘Cells, Cellular Structures’ (A11.284) and ‘Cells, Cultured’ (A11.251) owing to their low relevance to a specific disease. The Diseases (C) category has the largest number of MeSH terms. We excluded terms under three subcategories of this category: ‘Animal Diseases’ (C22), ‘Disease’ (C23.550.288), ‘Disease Attribute’ (C23.550.291) and ‘Chromosome Aberrations’ (C23.550.210), which are irrelevant to the specific diseases. For the Chemicals and Drugs (D) category, we used only terms under two subcategories: ‘Inorganic Chemicals’ (D01) and ‘Organic Chemicals’ (D02). MeSH terms in the Psychiatry and Psychology (F) category were used to supplement the disease terms for mental and behavior disorders.
HYPOTHESIS GENERATION VIA MESH-ASSOCIATED CO-EXPRESSIONS
With COEXPEDIA, we provide a companion web server that facilitates generating biomedical hypotheses for either a single gene or a group of genes queried by users. A single gene query starts with the identification of associated functions or diseases. When users submit a query gene, the web server first returns three types of search results: co-expressed partners sorted by the sum of LLSs from all supportive GSEs (i.e. sum of LLS) (Figure 3A), enriched GOBP terms (Figure 3B), and Disease Ontology (DO) (17) terms (Figure 3C) among co-expressed genes, sorted by the P-values from Fisher's exact test. These GOBP and DO terms suggest pathways and diseases associated with the query gene, respectively. The web server also provides a visual representation of the network of the co-expressions between the query gene and its co-expressed partners (Figure 3D). The publication-grade network images are also available for download.
Figure 3.

Screenshots of the BRCA1 query results. (A) A list of the co-expression partners of BRCA1 ranked by the sum of LLS scores. (B) A list of the enriched GOBP terms among BRCA1 co-expression partners ranked by the P-value from Fisher's exact test. (C) A list of the enriched DO terms among BRCA1 co-expression partners ranked by the P-value from Fisher's exact test. (D) A visualization of the gene network of BRCA1 and its co-expression partners. (E) A list of the enriched MeSH terms among the BRCA1 co-expression network. (F) A visualization of the gene network of BRCA1 and its co-expression partners only for the selected MeSH term ‘Heart’. (G) Information on the studies (GSEs and PubMed articles) that support the selected MeSH term ‘Heart’ for the co-expression network.
Next, the web server returns MeSH terms enriched among the co-expressions (Figure 3E). The enrichment of MeSH terms among the co-expressions is measured by the summation of LLSs from all supportive GSEs that are indexed by the MeSH terms. The top-ranked MeSH term can be interpreted as the most relevant biomedical context for the given co-expressions. We found that MeSH terms under Neoplasm (C04) are frequently indexed in PubMed articles, reflecting a study bias towards cancer in genomics. To facilitate the browsing of non-neoplasm diseases enriched among co-expressions, we opted to provide lists of the enriched MeSH terms that either include or exclude neoplasm MeSH terms in two separate tables (see Figure 3E). Users may conduct GOBP and DO enrichment analyses in the context of a particular MeSH term by clicking the ‘View’ link next to it. This context-specific functional enrichment analysis provides a different list of associated GOBP or DO terms. Then, the server provides a visualization of the network for the specific MeSH term (Figure 3F) and information on supportive GSEs and PubMed articles (Figure 3G).
A query using a gene group returns results similar to those from a single gene query. The enrichment analysis is not based on all the query genes but only on the query genes that are co-expressed with other query genes. The genome-wide association study (GWAS) and whole exome sequencing (WES) approaches have resulted in the reporting of many genes with disease implications. Co-expressions among a group of genes implicated for a disease can be enriched for some MeSH terms. The enriched disease- or chemical-associated MeSH terms for the co-expressions among the disease-associated genes may reveal novel disease-to-disease or disease-to-drug associations. The web server provides a table of gene sets precompiled from GWASdb (18) to enable easy queries of GWAS candidate genes.
COEXPEDIA predictions depend on not only the co-functionality between co-expressed genes but also the reliability of gene-to-MeSH relationships. We presumed that a high probability of a functional association between genes in COEXPEDIA could be achieved by using only co-functional co-expressions. Next, we assessed gene-to-MeSH relationships using literature-based gene-to-MeSH pairs from the Gene2MeSH database (http://gene2mesh.ncibi.org) as gold-standard data. We compiled a total of 39 657 gold-standard gene-to-MeSH pairs from the database. After excluding the prevalent Neoplasm (C04) terms, there were 38 224 gene-to-MeSH pairs left. We then counted the cumulative number of gold-standard gene-to-MeSH pairs in the top N ranked MeSH terms for each gene. We observed that the cumulative number of retrieved gold-standard gene-to-MeSH pairs increased as more MeSH term predictions were considered with either including or excluding Neoplasm terms (Figure 4). The number of gold-standard gene-to-MeSH pairs retrieved by COEXPEDIA is substantially higher than that by random predictions (by ∼25–50-fold), which indicates that relevant biomedical contexts for each gene can be reliably identified by utilizing MeSH-associated co-expressions.
Figure 4.

Assessment of gene-to-MeSH predictions. Literature-based gene-to-MeSH links compiled from the Gene2MeSH database are used as gold-standard data to evaluate predictions based on co-expressions in COEXPEDIA. The cumulative number of gold-standard gene-to-MeSH links is counted in the given top N ranked MeSH predictions for the query gene while excluding (A) or including (B) the prevalent neoplasm MeSH terms. The predictions by COEXPEDIA are compared with those by 1000 sets of randomly sampled gene-to-MeSH links, which are summarized as distributions for the same ranks.
CASE STUDIES
Predictions for a single gene: BRCA1 and CTLA4
The COEXPEDIA prediction for BRCA1, a tumor suppressor gene whose mutations substantially increase breast cancer susceptibility, returned GOBP terms related to DNA repair and cell cycle checkpoints as highly enriched pathways among BRCA1 co-expressed partners (see Figure 3B). Next, the DO enrichment analysis returned ‘cancer’ and ‘breast ovarian cancer’ terms as the diseases most associated with BRCA1. Notably, these associated diseases, which are already well known, were followed by ‘colorectal cancer’, whose association with BRCA1 is not widely known (see Figure 3C). Interestingly, recent studies have reported that the risk for colorectal cancer is increased in female carriers of BRCA1 mutations (19,20), which validates the gene-to-disease association predicted by COEXPEDIA.
Searching for non-neoplasm MeSH terms associated with BRCA1 returned ‘Heart’ as the second top associated term (see Figure 3E and F). Interestingly, BRCA1 was recently reported as an essential regulator of heart function and survival following myocardial infarction (21). Co-expressions that support the association between BRCA1 and heart function were derived from GSE8481 (22) (see Figure 3G), although this study was not designed explicitly to investigate the association between BRCA1 and heart function.
Next, we queried CTLA4, an immune checkpoint that is a major therapeutic target used in tumor immunotherapy (23). Immune-related GOBP terms were returned as the top enriched pathways, and autoimmune diseases such as lupus, multiple sclerosis, and arthritis were returned as the most associated DO terms, which are all consistent with the roles of CTLA4 in immune suppression. Interestingly, some chemicals used in cancer chemotherapy were suggested to be highly associated with CTLA4 via the enrichment analysis for non-neoplasm MeSH terms: isotretinoin (rank 1), cyclophosphamide (rank 3), epirubicin (rank 3) and paclitaxel (rank 3) (see Figure 2B). We found that the combination of low-dose cyclophosphamide and anti-CTLA4 blockade (ipilimumab) for melanoma is currently under clinical trial (https://clinicaltrials.gov/ ID: NCT01740401). Epirubicin was recently reported to inhibit regulatory T-cell activity (24), which also implicates the potential enhancement of anti-CTLA4 immunotherapy by combination therapy. Regarding paclitaxel, a preclinical investigation of the combination of anti-CTLA4 ipilimumab and paclitaxel showed a synergistic therapeutic effect in mouse tumor models (25). Excluding isotretinoin, we found evidence in the literature that supported the potential benefit of the combined use of cyclophosphamide, epirubicin, and paclitaxel in anti-CTLA4 cancer immunotherapy. All these chemical MeSH terms were supported by co-expressions derived from the GSE32646 data set composed of 115 breast cancer tumor samples with resistance to the chemotherapeutics (26). However, an association between the chemical drugs and CTLA-4 or tumor immunotherapy was not indicated in the study. These results suggest that MeSH-associated co-expressions can identify potential drugs for novel or enhanced treatments for diseases.
Predictions for a group of genes: GWAS candidates for Alzheimer's disease
COEXPEDIA predictions for a group of genes, particularly those associated with a genetic disorder, can be used to generate hypotheses about disease-to-disease associations (e.g., comorbidity) or novel drugs for disease treatment. When we submitted a group of 16 genes associated with Alzheimer's disease (AD) compiled from GWASdb, only 10 of them were found to be interconnected by co-expressions. The GOBP enrichment analysis for the 10 co-expressed AD candidate genes returned pathways involving beta-amyloid and neurofibrillary tangle assembly regulation as top predictions, which were all expected. Interestingly, the DO enrichment analysis for the same 10 genes returned ‘herpes simplex’ rather than AD as the most associated disease. Indeed, an international team of researchers has recently proposed that AD could be caused by herpes simplex virus type 1 (HSV1) infection, according to various lines of evidence (27). The results of the MeSH enrichment analysis for co-expressions seemed more intriguing. Among the top enriched non-neoplasm MeSH terms for the AD-associated co-expressions, we noticed ‘kidney’ (rank 3), which implicates an association of kidney function with AD. Indeed, multiple studies have reported chronic kidney disease as a risk factor for AD (28). The supportive co-expressions for the association of AD with kidney function were inferred from pancreatic tumor samples (GSE16515) (29) and clear cell carcinoma of the kidney (GSE14994) (30). These studies were neither designed nor conducted to investigate explicitly an association between AD and kidney disorders.
We also found some chemicals used in cancer treatment among the highly associated non-neoplasm MeSH terms: boronic acids (rank 4) and thalidomide (rank 5). Indeed, a recent study has reported that alkenylboronic acids have a neuroprotective function and can affect multiple biological targets involved in AD (31). This association between AD and boronic acids was inferred from co-expressions across 162 multiple myeloma tumor samples (GSE6477) (32) from a study that was not designed to study either AD or boronic acids. In addition, a therapeutic effect of thalidomide on AD has recently been suggested by a study involving its chronic administration in a mouse disease model (33). The association of thalidomide with AD was supported by co-expressions across 46 tumor samples from multiple myeloma patients who received an initial therapy of lenalidomide and dexamethasone (GSE31504) (34). Therefore, we concluded that MeSH-associated co-expressions can reveal potential drugs with therapeutic effects on a disease by querying a group of genes associated with the disease of interest.
CONCLUDING REMARKS
In this study, we present COEXPEDIA, a new database of co-expressions that are likely to be co-functional and associated with MeSH terms. By analyzing example queries using BRCA1, CTLA4, and AD-associated genes from GWAS, we demonstrate that MeSH-associated co-expressions enable the identification of unknown gene-to-disease, gene-to-drug, disease-to-disease, and disease-to-drug associations. To the best of our knowledge, COEXPEDIA is the first co-expression database to integrate MeSH terms, which are major sources of medical subject annotations. The incorporation of MeSH terms will greatly potentiate the use of co-expression information for generating biomedical hypotheses. As the size of gene expression data in GEO continues to grow at an ever-increasing rate and we constantly endeavor to index the literature using a controlled biomedical vocabulary system such as MeSH, the framework described in this study will become more useful for the biomedical research community.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea [2015R1A2A1A15055859, 2012M3A9B4028641, 2012M3A9C7050151 to I.L.]. Funding for the open access charge: National Research Foundation of Korea.
Conflict of interest statement. None declared.
REFERENCES
- 1.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolesnikov N., Hastings E., Keays M., Melnichuk O., Tang Y.A., Williams E., Dylag M., Kurbatova N., Brandizi M., Burdett T., et al. ArrayExpress update–simplifying data submissions. Nucleic Acids Res. 2015;43:D1113–D1116. doi: 10.1093/nar/gku1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kodama Y., Shumway M., Leinonen R., International Nucleotide Sequence Database, C. The sequence read archive: explosive growth of sequencing data. Nucleic Acids Res. 2012;40:D54–D56. doi: 10.1093/nar/gkr854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rung J., Brazma A. Reuse of public genome-wide gene expression data. Nature Rev. Genet. 2013;14:89–99. doi: 10.1038/nrg3394. [DOI] [PubMed] [Google Scholar]
- 5.Marcotte E.M., Pellegrini M., Thompson M.J., Yeates T.O., Eisenberg D. A combined algorithm for genome-wide prediction of protein function. Nature. 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- 6.Wang P.I., Marcotte E.M. It's the machine that matters: Predicting gene function and phenotype from protein networks. J. Proteomics. 2010;73:2277–2289. doi: 10.1016/j.jprot.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho D.Y., Kim Y.A., Przytycka T.M. Chapter 5: Network biology approach to complex diseases. PLoS Comput. Biol. 2012;8:e1002820. doi: 10.1371/journal.pcbi.1002820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shim J.E., Lee I. Network-assisted approaches for human disease research. Anim. Cells Syst. 2015;19:231–235. [Google Scholar]
- 9.Okamura Y., Aoki Y., Obayashi T., Tadaka S., Ito S., Narise T., Kinoshita K. COXPRESdb in 2015: coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems. Nucleic Acids Res. 2015;43:D82–D86. doi: 10.1093/nar/gku1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Dam S., Craig T., de Magalhaes J.P. GeneFriends: a human RNA-seq-based gene and transcript co-expression database. Nucleic Acids Res. 2015;43:D1124–D1132. doi: 10.1093/nar/gku1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gene Ontology, C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim W.K., Wang K., Lefebvre C., Califano A. Comparative analysis of microarray normalization procedures: effects on reverse engineering gene networks. Bioinformatics. 2007;23:i282–i288. doi: 10.1093/bioinformatics/btm201. [DOI] [PubMed] [Google Scholar]
- 13.Usadel B., Obayashi T., Mutwil M., Giorgi F.M., Bassel G.W., Tanimoto M., Chow A., Steinhauser D., Persson S., Provart N.J. Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ. 2009;32:1633–1651. doi: 10.1111/j.1365-3040.2009.02040.x. [DOI] [PubMed] [Google Scholar]
- 14.Lee I., Date S.V., Adai A.T., Marcotte E.M. A probabilistic functional network of yeast genes. Science. 2004;306:1555–1558. doi: 10.1126/science.1099511. [DOI] [PubMed] [Google Scholar]
- 15.Caspi R., Billington R., Ferrer L., Foerster H., Fulcher C.A., Keseler I.M., Kothari A., Krummenacker M., Latendresse M., Mueller L.A., et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44:D471–D480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scherzer C.R., Eklund A.C., Morse L.J., Liao Z., Locascio J.J., Fefer D., Schwarzschild M.A., Schlossmacher M.G., Hauser M.A., Vance J.M., et al. Molecular markers of early Parkinson's disease based on gene expression in blood. Proc. Natl. Acad. Sci. U.S.A. 2007;104:955–960. doi: 10.1073/pnas.0610204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kibbe W.A., Arze C., Felix V., Mitraka E., Bolton E., Fu G., Mungall C.J., Binder J.X., Malone J., Vasant D., et al. Disease Ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 2015;43:D1071–D1078. doi: 10.1093/nar/gku1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M.J., Liu Z., Wang P., Wong M.P., Nelson M.R., Kocher J.P., Yeager M., Sham P.C., Chanock S.J., Xia Z., et al. GWASdb v2: an update database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2016;44:D869–D876. doi: 10.1093/nar/gkv1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phelan C.M., Iqbal J., Lynch H.T., Lubinski J., Gronwald J., Moller P., Ghadirian P., Foulkes W.D., Armel S., Eisen A., et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: results from a follow-up study. Br. J. Cancer. 2014;110:530–534. doi: 10.1038/bjc.2013.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sopik V., Phelan C., Cybulski C., Narod S.A. BRCA1 and BRCA2 mutations and the risk for colorectal cancer. Clin. Genet. 2015;87:411–418. doi: 10.1111/cge.12497. [DOI] [PubMed] [Google Scholar]
- 21.Shukla P.C., Singh K.K., Quan A., Al-Omran M., Teoh H., Lovren F., Cao L., Rovira I.I., Pan Y., Brezden-Masley C., et al. BRCA1 is an essential regulator of heart function and survival following myocardial infarction. Nat. Commun. 2011;2:593. doi: 10.1038/ncomms1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kami D., Shiojima I., Makino H., Matsumoto K., Takahashi Y., Ishii R., Naito A.T., Toyoda M., Saito H., Watanabe M., et al. Gremlin enhances the determined path to cardiomyogenesis. PLoS One. 2008;3:e2407. doi: 10.1371/journal.pone.0002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egen J.G., Kuhns M.S., Allison J.P. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 2002;3:611–618. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 24.Kashima H., Momose F., Umehara H., Miyoshi N., Ogo N., Muraoka D., Shiku H., Harada N., Asai A. Epirubicin, identified using a novel luciferase reporter assay for Foxp3 inhibitors, inhibits regulatory T cell activity. PLoS One. 2016;11:e0156643. doi: 10.1371/journal.pone.0156643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jure-Kunkel M., Masters G., Girit E., Dito G., Lee F., Hunt J.T., Humphrey R. Synergy between chemotherapeutic agents and CTLA-4 blockade in preclinical tumor models. Cancer Immunol. Immun. 2013;62:1533–1545. doi: 10.1007/s00262-013-1451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyake T., Nakayama T., Naoi Y., Yamamoto N., Otani Y., Kim S.J., Shimazu K., Shimomura A., Maruyama N., Tamaki Y., et al. GSTP1 expression predicts poor pathological complete response to neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci. 2012;103:913–920. doi: 10.1111/j.1349-7006.2012.02231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itzhaki R.F., Lathe R., Balin B.J., Ball M.J., Bearer E.L., Braak H., Bullido M.J., Carter C., Clerici M., Cosby S.L., et al. Microbes and Alzheimer's Disease. J. Alzheimers Dis. 2016;51:979–984. doi: 10.3233/JAD-160152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Etgen T. Kidney disease as a determinant of cognitive decline and dementia. Alzheimers Res. Ther. 2015;7:29. doi: 10.1186/s13195-015-0115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pei H., Li L., Fridley B.L., Jenkins G.D., Kalari K.R., Lingle W., Petersen G., Lou Z., Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beroukhim R., Brunet J.P., Di Napoli A., Mertz K.D., Seeley A., Pires M.M., Linhart D., Worrell R.A., Moch H., Rubin M.A., et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009;69:4674–4681. doi: 10.1158/0008-5472.CAN-09-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jimenez-Aligaga K., Bermejo-Bescos P., Martin-Aragon S., Csaky A.G. Discovery of alkenylboronic acids as neuroprotective agents affecting multiple biological targets involved in Alzheimer's disease. Bioorg. Med. Chem. Lett. 2013;23:426–429. doi: 10.1016/j.bmcl.2012.11.068. [DOI] [PubMed] [Google Scholar]
- 32.Chng W.J., Kumar S., Vanwier S., Ahmann G., Price-Troska T., Henderson K., Chung T.H., Kim S., Mulligan G., Bryant B., et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007;67:2982–2989. doi: 10.1158/0008-5472.CAN-06-4046. [DOI] [PubMed] [Google Scholar]
- 33.He P., Cheng X., Staufenbiel M., Li R., Shen Y. Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of beta-secretase in a mouse model of Alzheimer's disease. PloS one. 2013;8:e55091. doi: 10.1371/journal.pone.0055091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar S.K., Uno H., Jacobus S.J., Van Wier S.A., Ahmann G.J., Henderson K.J., Callander N.S., Haug J.L., Siegel D.S., Greipp P.R., et al. Impact of gene expression profiling-based risk stratification in patients with myeloma receiving initial therapy with lenalidomide and dexamethasone. Blood. 2011;118:4359–4362. doi: 10.1182/blood-2011-03-342089. [DOI] [PMC free article] [PubMed] [Google Scholar]