


Abstract
DrugCentral (http://drugcentral.org) is an open-access online drug compendium. DrugCentral integrates structure, bioactivity, regulatory, pharmacologic actions and indications for active pharmaceutical ingredients approved by FDA and other regulatory agencies. Monitoring of regulatory agencies for new drugs approvals ensures the resource is up-to-date. DrugCentral integrates content for active ingredients with pharmaceutical formulations, indexing drugs and drug label annotations, complementing similar resources available online. Its complementarity with other online resources is facilitated by cross referencing to external resources. At the molecular level, DrugCentral bridges drug-target interactions with pharmacological action and indications. The integration with FDA drug labels enables text mining applications for drug adverse events and clinical trial information. Chemical structure overlap between DrugCentral and five online drug resources, and the overlap between DrugCentral FDA-approved drugs and their presence in four different chemical collections, are discussed. DrugCentral can be accessed via the web application or downloaded in relational database format.
INTRODUCTION
DrugCentral is a pharmaceutical information resource developed by combining information related to active pharmaceutical ingredients (APIs), regulatory information, bioactivity profiles, drug mechanism of action (MoA), pharmacological action, pharmaceutical products and indications. The majority of the data are collected and aggregated from online public resources, combined with manual curation of literature and drug label information. Two major entities are interlinked in DrugCentral: the APIs, referred to as ‘drugs’ by chemists, biologists and other basic scientists, and pharmaceutical products, referred to as ‘drugs’ by patients, pharmacists, nurses and physicians, as well as other clinician scientists. The conceptual links between associated entities in DrugCentral are illustrated in Figure 1. DrugCentral is based on a curated list of 4444 APIs, with an unambiguous list of over 20 617 drug synonyms and research codes that are continuously updated by automated monitoring of regulatory agencies approvals from U.S. Food and Drug Administration (FDA) (http://www.fda.gov), European Medicines Agency (EMA) (http://www.ema.europa.eu) and Japan Pharmaceutical and Medical Devices Agency (PMDA) (http://www.pmda.go.jp/english).
Figure 1.

DrugCentral main entities and relations diagram. Active pharmaceutical ingredients' is the central bridge entity that serves as a to link to the other database entities.
Mechanism of action annotations in DrugCentral are aggregated from several expert curated resources: ChEMBL database (1), Guide to Pharmacology (2), KEGG Drug (3), as well as manual curations extracted from drug labels and literature. Pharmacological actions are compiled from the Anatomical Therapeutic Chemical (ATC) classification system (http://www.whocc.no), the FDA Established Pharmacologic Class (EPC) (http://purl.access.gpo.gov/GPO/LPS118712), Medical Subject Headings (MeSH) (4) and Chemical Entities of Biological Interest (ChEBI) ontology (5), respectively. Indications in DrugCentral were collated from OMOP vocabularies (http://omop.org/Vocabularies) for those drugs approved before 2012; for drugs approved after 2012, indications were extracted from drug labels, and mapped to SNOMED-CT (6) concepts. Indication data from these two sources are currently being harmonized using the UMLS (7) application programming interface, as well as manual mapping.
DATABASE CONTENTS AND DATA SOURCES
DrugCentral entities and statistics, as of Jul 2016, are summarized in Table 1. Information on 4444 unique APIs is stored in DrugCentral as of June 2016 with new entries added constantly. A total of 1932 unique APIs from DrugCentral are mapped to Drugs@FDA (www.accessdata.fda.gov/scripts/cder/drugsatfda) that contains drug labels and approvals for drugs marketed in the United States. New molecular entities (NME) that are flagged in Drugs@FDA are the equivalent of DrugCentral APIs, and track the date of first approval by the FDA. Comprehensive NME approval dates are available going back to 1982 only, whereas earlier approval dates are incomplete. DrugCentral contains data for 1700 APIs with NME approval date as well as 232 APIs with missing NME approval date. The missing ones are typically for old drugs, e.g. for acetyl salicylic acid (aspirin). FDA NMEs approval dates provide the bulk of regulatory marketing data. These are supplemented by approval dates from EMA, and PMDA, particularly for APIs approved outside USA. The date of first approval, combined with MoA target annotations, also provides a reference date for first approval of specific targets, namely for first-in-class drugs as well as for specific indications. Websites and online databases of regulatory agencies are monitored to ensure up-to-date information concerning new APIs and new indications. New APIs/NMEs are registered whenever such information is published, complemented by literature surveys for accurate chemical structures, MoA targets, in vitro/in vivo potencies, cross references with external resources, approved indications, dosage and administration, and mapping to drug labels and pharmacological action classifications systems.
Table 1. DrugCentral database entities and statistics.
| Entities (Annotated APIs) | |
|---|---|
| Active pharmaceutical ingredients | 4444 |
| FDA drugs | 2021 |
| Drugs approved outside US | 2423 |
| Small molecules | 3799 |
| Salts and inorganic molecules | 112 |
| Biologics and peptides | 239 |
| Other drugs | 294 |
| Parent molecules | 199 (308) |
| Drug efficacy targets | 837 (1689) |
| Human protein targets | 600 (1387) |
| Infectious agents targets | 194 (221) |
| Metabolites & biopolymers | 43 (89) |
| Protein–drug crystal complex (PDB) | 48 (82) |
| Drug–protein crystal complex (PDB) | 1452 (283) |
| Bioactivity data points | 13 825 (1792) |
| Human proteins | 10 427 (1605) |
| Other species | 3398 (1002) |
| Pharmacological action | |
| WHO ATC code | 4195 (2941) |
| FDA Established Pharmacologic Class | 428 (1165) |
| MeSH pharmacological action | 424 (2529) |
| ChEBI ontology roles | 285 (1487) |
| Drug indications | 2224 (2247) |
| Drug contra-indications | 1458 (1376) |
| Drug off-label uses | 847 (646) |
| Pharmaceutical products | 67 064 (1660) |
| Rx pharmaceutical products | 29 665 (1561) |
| OTC pharmaceutical products | 37 399 (286) |
| External identifiers | 61 349 (4444) |
| CAS registry number | 6072 (4444) |
| PubChem Compound Id | 4175 (4175) |
| FDA Unique Ingredient Identifier (UNII) | 4304 (4304) |
| ChEMBL-db id | 5615 (4075) |
| WHO INN id | 3519 (3519) |
| SNOMED-CT | 4745 (2637) |
| KEGG DRUG | 3501 (3501) |
| NDFRT | 4171 (2406) |
| RxNorm RxCUI | 2897 (2897) |
| IUPHAR/BPS ligand id | 1345 (1345) |
| UMLS CUI | 2839 (2839) |
| CHEBI | 2557 (2557) |
| MeSH | 4063 (3846) |
| DrugBank | 2473 (2388) |
| Protein databank ligand id | 646 (618) |
API chemical structures and identifiers
DrugCentral places a special emphasis on curating chemical structures for APIs, to ensure accurate representation at the molecular level. DrugCentral stores APIs in pharmaceutical products with one important distinction: APIs formulated as ionic salts have counterions removed; we apply the same policy for hydrated/solvated formulations, where water/solvent has been removed. For example, the API atorvastatin, can be listed in FDA-approved drug labels as: atorvastatin calcium, atorvastatin calcium trihydrate or atorvastatin calcium propylene glycol solvate. DrugCentral, however, resolves all these forms into a single API, atorvastatin. Ester prodrug APIs that are covalently bound and are converted to their active form in the human body are stored as such. For example, olmesartan medoxomil, but not olmesartan, is the API stored in DrugCentral. Enalaprilat, on the other hand, is formulated both as free acid and as the maleate salt of enalapril the ethyl ester of enalaprilat, and therefore both enalapril and enalaprilat are stored as APIs in DrugCentral. A multistep manual curation and validation process was employed to collect, record and store correct chemical information for API molecular structures in the MDL (‘Molecular Design Limited’) MOL format (8). Chemical structure representation for small organic molecules and peptides were manually entered using depictions and information from the following sources listed in decreasing order of precedence: WHO INN (http://www.who.int/medicines/services/inn/en), USAN (http://www.ama-assn.org/ama/pub/physician-resources/medical-science/united-states-adopted-names-council.page), FDA SRS (http://fdasis.nlm.nih.gov), CAS (9) and FDA drug labels (http://www.fda.gov), respectively. Discrepancies were resolved by literature queries, priority being given to those studies that described chemical synthesis and structural analyses of new APIs. For example, the antimuscarinic drug difemerine has two different representations in the above listed data sources: 2-(dimethylamino)-1, 1-dimethylethyl benzilate (CAS) and 2-(dimethylamino)-2, 2-dimethylethyl benzilate (WHO INN, FDA SRS). Further investigations revealed that WHO INN corrected the initially published 2, 2-dimethylethyl structure, to resolve to 2-(dimethylamino)-1, 1-dimethylethyl benzilate (https://mednet-communities.net/inn/db/media/docs/r-innlist24.pdf), the amended structure confirmed by CAS is currently the version stored in DrugCentral. This example illustrates the challenge of manually curating API chemical structures, suggesting that structure-based matching with external resources is not error-proof. This forced us to adopt the name match as an alternative (see Table 1, external identifiers). To date, chemical structures for 4231 APIs have been manually entered and validated, with molecular weight (MW) ranging between 4 (Helium, a noble gas with medical use) and 22 kilodaltons (somatotropin, a 191-amino acid protein growth hormone), respectively. With respect to APIs, DrugCentral distinguishes between small molecules, biologics and peptides and other drugs, as shown in Table 1. Accurate representation of stereoisomers chemical structures is available for 1544 APIs, including 1383 APIs with tetrahedral chirality and 307 APIs with specified cis/trans isomerism. Racemic mixtures and APIs with relative stereoconfiguration (652 APIs) are stored using the enhanced stereochemical representation supported by MDL V3000 format (http://accelrys.com/products/collaborative-science/biovia-draw/ctfile-no-fee.html). DrugCentral allows for easy subsetting of chemical structures to create lists of molecules for specific applications, e.g. in drug repurposing applications, small molecules FDA approved drugs are of interest (see example scripts in Supplementary Material). DrugCentral contains 1608 unique small molecule FDA approved and discontinued drugs (FDA small molecules drugs subset).
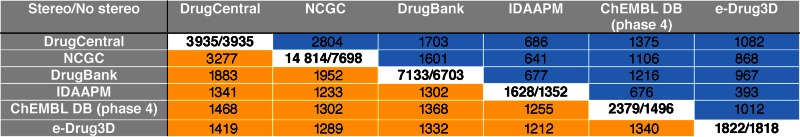
We performed chemical structure comparisons using InChI (10) strings generated from the MOL V2000 records, i.e. omitting the enhanced stereo option, with unique APIs from several drug-focused databases, after removing salt/solvent and invalid/duplicated structure records. Table 2 summarizes the variable degree of overlap between these resources and DrugCentral (MOL V2000 subset, 3935 unique APIs). The overlap matrix was computed for DrugCentral, in comparison with the following drug-focused resources: the NIH Center for Chemical Genomics (NCGC) pharmaceutical collection (11), DrugBank (12), the IDAAPM database (13), the phase 4 drugs subset from the ChEMBL 21 release (1) and the e-Drug3D database (14). Although the NCGC pharmaceutical collection is larger than DrugCentral, since it contains compounds undergoing clinical trials, its last published update was in 2012 and, as such, it does not include 240 FDA approved and 46 APIs approved elsewhere, after 2012. Also a larger collection, DrugBank has an update lag with the more recent approved APIs missing, a total of 269 FDA approved APIs absent from the DrugBank structures file; however, many of these are peptides which DrugCentral stores as chemical structures in MOL format whereas DrugBank provides sequence only, or have different structure record compared to DrugCentral. Most of the missing and discrepant API structures can be found by manual query online in DrugBank. An additional 65 non-FDA approved APIs have missing structures in the DrugBank structure file. The IDAAPM database, focused on FDA drugs only, does not cover APIs approved elsewhere. A total of 338 APIs are missing from IDAAPM, including some newer FDA-approved APIs, e.g. Flibanserin (approved in 2015), but also old over-the-counter drugs that are still marketed today, such as Salicylamide and Propylhexedrine. The overlap for the phase 4 subset of ChEMBL 21 (which designates approved drugs) reveals that 223 FDA-approved and 95 non-FDA approved APIs are not indexed in this particular subset. We note that most of these entries are represented in the ChEMBL database, just lack the phase 4 flag or may have missing structures. All overlap counts are summarized in Table 2, whereas overlap and non-overlapping lists for individual chemical libraries are provided as Supplementary Material.
Table 2. Structure overlap between 3935 DrugCentral structures (MOL V2000 subset) and other small molecule drug databases. The upper triangle (blue) summarizes the APIs overlap calculated using stereo chemistry information. The lower triangle (orange) contains overlaps determined with stereochemistry omitted. The Diagonal entries represent number of unique molecular structure records before and after removal of salt/solvent, or invalid/duplicate records.
|
Small organic API molecules are also annotated with the Lipinski rule of 5 (Ro5) criteria (15), as well as related properties such as the number of non-terminal rotatable bonds and the number of rings (16), which are calculated using definitions provided by Lipinski et al. for hydrogen bond donors/acceptors and the calculated 1-octanol/water partition coefficient, cLogP (17), to show Ro5 compliance according to the original Ro5 criteria. More than 83% of the 200 NMEs approved after 1997 with oral route of administration according to WHO ATC/DDD index (http://www.whocc.no/atc_ddd_index) are Ro5 compliant, a figure that is below the 90% cut-off proposed by Lipinski et al. (15). Summary statistics for Ro5 criteria and related properties for orally formulated APIs are presented in Table 3.
Table 3. Distribution of Ro5 criteria and related physicochemical properties for the orally formulated drugs subset of DrugCentral.
| Property | Min | 1st quantile | Median | 3rd quantile | Max |
|---|---|---|---|---|---|
| MW | 75.07 | 262.22 | 324.8 | 411.49 | 1526.74 |
| cLogP | −8.17 | 0.94 | 2.43 | 4.05 | 20.43 |
| HAC | 0 | 3 | 5 | 7 | 36 |
| HDO | 0 | 1 | 1 | 2 | 19 |
| Ro5 | 0 | 0 | 0 | 0 | 4 |
| ROTB | 0 | 3 | 4 | 7 | 31 |
| RGB | 0 | 12 | 17 | 21 | 74 |
| Rings | 0 | 2 | 3 | 4 | 10 |
| Rings Aro | 0 | 1 | 2 | 2 | 6 |
| Rings Aliph | 0 | 0 | 1 | 2 | 9 |
| TPSA | 0 | 40.73 | 67.2 | 98.78 | 573.91 |
MW - molecular weight; cLogP - estimated log octanol/water partition coefficient (CLOGP, BioByte); HAC - hydrogen bond acceptors; HDO - hydrogen bond donors; Ro5 - rule of 5 violations; ROTB - rotatable bonds; RGB - rigid bonds; Rings - smallest set of smallest rings (SSSR); Rings Aro - aromatic rings; Rings Aliph - aliphatic rings; TPSA - topological polar surface area (Å2).
Prodrug APIs chemical structures in DrugCentral match the representation in pharmaceutical formulations. For example, hydrocortisone (parent molecule) is the active form of eight different ester prodrugs (acetate, succinate, valerate, etc.) listed in pharmaceutical formulations. To avoid confusion and to support applications where different prodrugs with same parent molecule need to be merged, DrugCentral maintains a list of 199 parent molecules mapped to 308 prodrugs that is continuously updated as new information becomes available.
In the present version of DrugCentral, chemical structures cannot be searched online. However, they are available for download in the MDL SDF format, which can be used for cheminformatics-related activities such as virtual screening, computational drug repurposing (18), chemical library coverage comparison and similarity analyses, as well as other applications where accurate chemical structure information is required.
For example, an analysis of commercial chemical libraries to evaluate coverage of FDA approved APIs can assist those interested in determining the drug repurposing potential of various such libraries. The FDA drugs coverage for several commercially available libraries is summarized in Table 4. Since stereochemistry information is often omitted or incorrectly specified, the conservative estimates are between 42% and 82% for the FDA approved small molecule drugs are available from chemical vendors. While eMolecules provides the ‘best’ coverage (82%), this vendor does not sell these APIs in a ‘pre-packaged’ chemical library, but aggregates information on available chemicals from multiple vendors, which no doubt would makes it the pricier option.
Table 4. Commercial drug collections overlap with FDA small molecules drugs subset (1608 structures).
| Chemical catalog | Total number of chemicals | FDA small molecule drugs overlap with stereochemistry specified (%) | FDA small molecule drugs overlap with stereochemistry omitted (%) | URL |
|---|---|---|---|---|
| Prestwick Chemical Library | 1280 | 740 (46%) | 824 (51%) | http://www.prestwickchemical.com/prestwick-chemical-library.html |
| Selleckchem FDA-approved Drug Library | 978 | 595 (37%) | 670 (42%) | http://www.selleckchem.com/ |
| MicroSource Discovery US Drug Collection | 1385 | 687 (43%) | 929 (58%) | http://msdiscovery.com |
| eMolecules | 8 129 798 | 1208 (75%) | 1313 (82%) | https://www.emolecules.com/ |
The number of online resources dedicated to APIs and the amount of disjoint data in these resources poses significant challenges with respect to data integration and aggregation. Many of the online drug resources provide overlapping and complementary information on drugs.
One of the most prominent resources is DrugBank. Although similar in scope and content to DrugBank, there are significant differences in DrugCentral. Specifically, DrugCentral focuses on (i) drugs approved for human use, including currently marketed and discontinued APIs; (ii) regulatory approval information, provided whenever available; (iii) manually curated and validated chemical structures; (iv) drug indications, contra-indications and off-label indications mapped to terminologies from SNOMED-CT (6) concepts and UMLS (7) unique concepts identifiers; (v) API bioactivity profiles, providing numerical values where available. Furthermore, DrugCentral contains the complete text of all FDA drug labels. By contrast, DrugBank has better coverage with respect to Drug Metabolism, Health Economics and other areas that are complementary to DrugCentral contents.
Another online resource, SuperDrug (19), contains a subset of the APIs listed in the WHO ATC system, with focus on small organic molecules. Besides ATC codes and CAS registry numbers (also included in DrugCentral, together with 13 other external identifiers), SuperDrug contains 3D conformations of drugs, brand names and computed molecular properties. All the elements differentiating DrugCentral from DrugBank (listed above) remain valid with respect to SuperDrug as well. SuperTarget (20) contains biological activity profile annotations for drugs listed in SuperDrug and adverse events extracted from SIDER (21). While both DrugCentral and SuperTarget contain quantitative drug bioactivity profiles, DrugCentral pays specific importance to MoA drug targets, which are clearly annotated in bioactivity summary. Furthermore, DrugCentral separates bioactivity summaries for human and non-human targets (e.g. viral or bacterial targets), and maintains an up-to-date list of PDB identifiers for drug-target complexes. SuperTarget covers API side-effects, a feature that complements DrugCentral. However, as of June 2016 SuperTarget does not appear to cover many of the drugs approved after 2013.
The IDAAPM database (13) was recently described. The main differences between DrugCentral and IDAAPM are: (i) IDAAPM contains only FDA drugs; (ii) no drug indications and pharmacological action; (iii) chemical structures are not validated by a human curator (we found more than 70 APIs with incorrect molecular structure); (iv) target identifiers annotations are not normalized.
The e-Drug3D database (14) contains 3D conformations for Drugs@FDA entries, and provides information on commercially available drug fragments to facilitate computational drug repurposing and fragment based drug design. DrugCentral also covers biologics, peptides and inorganic drugs from Drug@FDA, whereas e-Drug3D focuses on small molecules having molecular weight below 2000 atomic mass units. Furthermore, DrugCentral covers APIs approved elsewhere, but not by FDA. While e-Drug3D links to FDA approved drug labels containing indications and dosage DrugCentral includes the information in the database and supports disease term queries. Most of the discrepancies observed between DrugCentral and e-Drug3D are related to different stereochemical configurations stored between two databases, and a small number of different (N = 23) structures having discrepancies at the connectivity matrix level.
To facilitate analyses and comparisons between DrugCentral and any other drug resource where no cross-mapping currently exists, we have cross-referenced APIs in DrugCentral with 15 external resources and mapped 61 258 identifiers to facilitate information use and exchange.
Drug mechanism of action and pharmacological action
Drug mechanism of action (MoA) target annotations provide mechanistic understanding of drug action at molecular level and relate protein targets to human disease and symptoms. We define the MoA or drug efficacy target as the molecule (e.g. protein, biopolymer, metabolite, metal atom, etc.) to which the API or its active metabolite binds directly, to exert the therapeutic drug action (22). DrugCentral MoA target annotations are collated from a carefully selected list of external resources combined with internal annotation efforts, for APIs that lack information in the current list of external resources. Our survey of resources indexing drug MoA targets led us to select only resources that are open access, employ human expert curators, are actively maintained and updated, and that provide supporting evidence for MoA, preferably as literature references or as drug labels. The following resources matched our above inclusion criteria: the ChEMBL database (1), the Guide to Pharmacology (2) and KEGG Drug (3). For APIs with missing MoA annotation in these resources, drug label and literature searches were used to determine consensus MoA targets. Pharmacological action provides useful grouping of APIs by modes of action and therapeutic effects. DrugCentral contains pharmacological action classification and annotations from: WHO ATC, FDA EPC, MeSH and ChEBI ontology (Table 1).
Bioactivity profiles
Quantitative bioactivity data are aggregated from multiple sources including: ChEMBLDb (1) (58.73% of the records); DrugMatrix (23) (15.95%); WOMBAT-PK (24) (13.9%); Guide to Pharmacology (2) (5.29%); PDSP (25) (3.41%); scientific literature (2.35%); and drug labels (0.36%), respectively. Over 14 000 numeric values are captured in DrugCentral, covering 2190 human and non-human targets for 1792 unique APIs. Only unique drug-target potency pairs are stored in the database. When multiple bioactivity values are available for the same drug–target pair, priority is given to binding (Kd) and inhibition (Ki) constants over other activity measurements types such as IC50.
Drug indications
Indications (10 707), contra-indications (27 851) and off-label indications (2496) were initially extracted from OMOP data model version 4.4 (http://omop.org/Vocabularies). Since the OMOP project transitioned to OHDSI (http://www.ohdsi.org), updated drug indication and contra-indication data are covered under a revised license agreement that in turn requires subscription licenses (i.e. it is no longer open-access). Therefore, indications for drugs approved after 2012 (322 pairs) were extracted from approved drug labels and mapped onto SNOMED-CT and UMLS concepts. Efforts are underway to map the remaining OMOP vocabulary terms to SNOMED-CT/UMLS disease concepts, with ∼67% of the concepts having already been mapped. Mapping to SNOMED-CT/UMLS disease concepts allows to extended mappings to other terminologies such as: International Classification of Diseases (www.who.int/classifications/icd/en), and Disease-Ontology (26) concepts.
Drug labels
FDA drug labels are downloaded from DailyMed (https://dailymed.nlm.nih.gov/dailymed) in the Structured Product Labeling (SPL) format, processed, imported and mapped to DrugCentral APIs entries using a customized pipeline. More than 67 000 drug labels containing information on over 84 000 pharmaceutical formulations are mapped to 1661 unique APIs. We note that, while the majority of the APIs (1562) are included in prescription (Rx) drug labels, only 286 unique APIs represent the ingredients for more than half of this formulary (55%), namely over the counter (OTC) products. This discrepancy is indicative not only of the increased competition on the OTC market, but also of the increased availability of Drug Safety and Patient Safety information, which allows manufacturers to combine OTC APIs. Furthermore, there is an overlap between Rx and OTC APIs (N = 187), these ingredients being present in both Rx and OTC drug labels. The most frequently re-formulated APIs (not counting sunscreen ingredients) as OTC products are paracetamol or acetaminophen (4530), phenylephrine (2660), dextromethorphan (2557), diphenhydramine (1436) and other decongestants/antihistamines indicated for management of cold symptoms and allergies.
Drug label data in SPL files are structured in sections annotated with Logical Observation Identifiers Names and Codes (LOINC) (27). DrugCentral stores drug label information for each section as a separate text blob, enabling section based text queries or the use of text mining tools to specific LOINC sections such as adverse events to text mining accuracy. For example, searching the LOINC sections related to Adverse events and Warnings with the keyword ‘Torsade de pointes’ returns 1727 Rx drug labels representing 79 unique APIs, suggesting that these drug labels contain information regarding this adverse event. To take full advantage of information stored in drug labels users are encouraged to download DrugCentral in the relational database format for processing with text mining tools.
WEB APPPLICATION AND DATA AVAILABILITY
DrugCentral can be accessed using a web browser at http://drugcentral.org from desktop or mobile devices. The web application was implemented using the Django Web framework (http://www.djangoproject.com) and PostgreSQL (http://www.postgresql.org) database as backend, with jQuery (http://jquery.com) and Bootstrap (http://getbootstrap.com) for a customized frontend. Web application search functionality supports multiple search term types as follows: (i) API name, synonym, identifier and brand name; (ii) target name, gene symbol and UniProt/SwissProt identifier; (iii) disease concept; (iv) pharmacological action; (v) drug label and description full text. Search results are sorted using a 4 level ranking system based on search term type match: highest rank (A) is assigned to API, MoA target and disease concepts listed in indication field hits; (B) non-MoA target, pharmacological action and disease concepts listed in drug contra-indications/off-label indications; (C) API description; with the lowest rank (D) is assigned to drug label full text search hits.
The web application is designed to render on both desktop and mobile devices (Figure 2). The jQuery framework detects the type of device used to access the web application and automatically adjusts the display correctly without user intervention. The entire database content is available for download under the Creative Commons Attribution-ShareAlike 4.0 license, and no user registration is required for access. Files for the Postgres relational database are available as database dump, and separate files containing APIs chemical structures are available as well in both MDL MOL V2000/V3000 and SMILES/InChI formats for user convenience.
Figure 2.

DrugCentral drug report page: (A) description, structure, properties and synonyms; (B) drug dosage, regulatory approvals and pharmacological action; (C) drug indications, contra-indications and off-label uses; (D) links to external resources; (E) bioactivity profile and mechanism of action target(s); (F) FDA approval pharmaceutical products containing API.
FUTURE DIRECTIONS
DrugCentral will be maintained to include new drug entries as soon as new regulatory approvals are published. Drugs withdrawn due to other than safety reasons will be flagged and annotated with indication/pharmacology/MoA to create a comprehensive list of drugs with potential for drug repurposing (28,29). Drug adverse events from FDA AERS database (https://open.fda.gov/) and SIDER (21) will be used to enhance the drug safety profile of existing drug reports. Target pathway and functional annotations will enable investigations of target upstream/downstream effects and provide new insight on drug action at molecular level. Web application search functionality will be extended to include chemical structure and similarity search. Categorical browsing based on pharmacological action, target and chemical structure type classifications will be added to enable users to view and download subsets of drug records for further analysis.
Acknowledgments
The authors would like to thank Anne Hersey (EMBL-EBI), Noel Southall (NIH-NCATS) and Aaron Pawlyk (NIH-NIDDK) for their suggestions on web application content and format.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [1U54CA189205-01 to O.U., J.H., C.B., J.Y., S.M., T.O.]. Funding for open access charge: National Institutes of Health [1U54CA189205-01].
Conflict of interest statement. None declared.
REFERENCES
- 1.Bento A.P., Gaulton A., Hersey A., Bellis L.J., Chambers J., Davies M., Krüger F.A., Light Y., Mak L., McGlinchey S., et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42:D1083–D1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Southan C., Sharman J.L., Benson H.E., Faccenda E., Pawson A.J., Alexander S.P.H., Buneman O.P., Davenport A.P., McGrath J.C., Peters J.A., et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res. 2016;44:D1054–D1068. doi: 10.1093/nar/gkv1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson S.J. Medical terminologies that work: the example of MeSH. 2009 10th International Symposium on Pervasive Systems, Algorithms, and Networks, Kaohsiung. 2009:380–384. [Google Scholar]
- 5.Hastings J., de Matos P., Dekker A., Ennis M., Harsha B., Kale N., Muthukrishnan V., Owen G., Turner S., Williams M., et al. The ChEBI reference database and ontology for biologically relevant chemistry: enhancements for 2013. Nucleic Acids Res. 2013;41:D456–D463. doi: 10.1093/nar/gks1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donnelly K. SNOMED-CT: the advanced terminology and coding system for eHealth. Stud. Health Technol. Inform. 2006;121:279–290. [PubMed] [Google Scholar]
- 7.Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004;32:D267–D270. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalby A., Nourse J.G., Hounshell W.D., Gushurst A.K.I., Grier D.L., Leland B.A., Laufer J. Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992;32:244–255. [Google Scholar]
- 9.CAS Registry System. CAS Registry System. J. Chem. Inf. Model. 1978;18:58–58. [Google Scholar]
- 10.Heller S., McNaught A., Stein S., Tchekhovskoi D., Pletnev I., Wiswesser W., Weininger D., Dalby A., Nourse J., Hounshell W., et al. InChI - the worldwide chemical structure identifier standard. J. Cheminform. 2013;5:7–16. doi: 10.1186/1758-2946-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang R., Southall N., Wang Y., Yasgar A., Shinn P., Jadhav A., Nguyen D.-T., Austin C.P. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 2011;3:80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y., Maciejewski A., Arndt D., Wilson M., Neveu V., et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Legehar A., Xhaard H., Ghemtio L., Bunnage M., Hay M., Thomas D., Craighead J., Economides C., Rosenthal J., Kola I., et al. IDAAPM: integrated database of ADMET and adverse effects of predictive modeling based on FDA approved drug data. J. Cheminform. 2016;8:33–44. doi: 10.1186/s13321-016-0141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pihan E., Colliandre L., Guichou J.-F., Douguet D. e-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. Bioinformatics. 2012;28:1540–1541. doi: 10.1093/bioinformatics/bts186. [DOI] [PubMed] [Google Scholar]
- 15.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 16.Oprea T.I. Property distribution of drug-related chemical databases. J. Comput. Aided. Mol. Des. 2000;14:251–264. doi: 10.1023/a:1008130001697. [DOI] [PubMed] [Google Scholar]
- 17.Leo A.J. Calculating log Poct from structures. Chem. Rev. 1993;93:1281–1306. [Google Scholar]
- 18.Oprea T.I., Kim Nielsen S., Ursu O., Yang J.J., Taboureau O., Mathias S.L., Kouskoumvekaki I., Sklar L.A., Bologa C.G. Associating drugs, targets and clinical outcomes into an integrated network affords a new platform for computer-aided drug repurposing. Mol. Inform. 2011;30:100–111. doi: 10.1002/minf.201100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goede A., Dunkel M., Mester N., Frommel C., Preissner R. SuperDrug: a conformational drug database. Bioinformatics. 2005;21:1751–1753. doi: 10.1093/bioinformatics/bti295. [DOI] [PubMed] [Google Scholar]
- 20.Hecker N., Ahmed J., von Eichborn J., Dunkel M., Macha K., Eckert A., Gilson M.K., Bourne P.E., Preissner R. SuperTarget goes quantitative: update on drug-target interactions. Nucleic Acids Res. 2012;40:D1113–D1117. doi: 10.1093/nar/gkr912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn M., Letunic I., Jensen L.J., Bork P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016;44:D1075–D1079. doi: 10.1093/nar/gkv1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santos R., Ursu O., Gaulton A, Bento A.P., Donadi R.S., Bologa C., Karlsson A., Al-Lazikani B., Hersey A., Oprea T.O., et al. A Comprehensive Map of Molecular Drug Targets. Nature Rev. Drug Discov. 2017 doi: 10.1038/nrd.2016.230. doi:10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganter B., Tugendreich S., Pearson C.I., Ayanoglu E., Baumhueter S., Bostian K.A., Brady L., Browne L.J., Calvin J.T., Day G.-J., et al. Development of a large-scale chemogenomics database to improve drug candidate selection and to understand mechanisms of chemical toxicity and action. J. Biotechnol. 2005;119:219–244. doi: 10.1016/j.jbiotec.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 24.Olah M., Rad R., Ostopovici L., Bora A., Hadaruga N., Hadaruga D., Moldovan R., Fulias A., Mractc M., Oprea T.I. Chemical Biology. Weinheim: Wiley-VCH Verlag GmbH; 2007. WOMBAT and WOMBAT-PK: bioactivity Databases for Lead and Drug Discovery; pp. 760–786. [Google Scholar]
- 25.Roth B.L., Lopez E., Patel S., Kroeze W.K. The multiplicity of serotonin receptors: uselessly diverse molecules or an embarrassment of riches. Neuroscientist. 2000;6:252–262. [Google Scholar]
- 26.Kibbe W.A., Arze C., Felix V., Mitraka E., Bolton E., Fu G., Mungall C.J., Binder J.X., Malone J., Vasant D., et al. Disease Ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 2015;43:D1071–D1078. doi: 10.1093/nar/gku1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huff S.M., Rocha R.A., McDonald C.J., De Moor G.J., Fiers T., Bidgood W.D., Forrey A.W., Francis W.G., Tracy W.R., Leavelle D., et al. Development of the Logical Observation Identifier Names and Codes (LOINC) vocabulary. J. Am. Med. Inform. Assoc. 5:276–292. doi: 10.1136/jamia.1998.0050276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oprea T.I., Bauman J.E., Bologa C.G., Buranda T., Chigaev A., Edwards B.S., Jarvik J.W., Gresham H.D., Haynes M.K., Hjelle B., et al. Drug repurposing from an academic perspective. Drug Discov. Today Ther. Strateg. 2011;8:61–69. doi: 10.1016/j.ddstr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oprea T.I., Overington J.P. Computational and practical aspects of drug repositioning. Assay Drug Dev. Technol. 2015;13:299–306. doi: 10.1089/adt.2015.29011.tiodrrr. [DOI] [PMC free article] [PubMed] [Google Scholar]