

Abstract
Background
Contrary to other areas of sequence analysis, a measure of statistical significance of a putative gene has not been devised to help in discriminating real genes from the masses of random Open Reading Frames (ORFs) in prokaryotic genomes. Therefore, many genomes have too many short ORFs annotated as genes.
Results
In this paper, we present a new automated gene-finding method, EasyGene, which estimates the statistical significance of a predicted gene. The gene finder is based on a hidden Markov model (HMM) that is automatically estimated for a new genome. Using extensions of similarities in Swiss-Prot, a high quality training set of genes is automatically extracted from the genome and used to estimate the HMM. Putative genes are then scored with the HMM, and based on score and length of an ORF, the statistical significance is calculated. The measure of statistical significance for an ORF is the expected number of ORFs in one megabase of random sequence at the same significance level or better, where the random sequence has the same statistics as the genome in the sense of a third order Markov chain.
Conclusions
The result is a flexible gene finder whose overall performance matches or exceeds other methods. The entire pipeline of computer processing from the raw input of a genome or set of contigs to a list of putative genes with significance is automated, making it easy to apply EasyGene to newly sequenced organisms. EasyGene with pre-trained models can be accessed at http://www.cbs.dtu.dk/services/EasyGene.
Keywords: computational gene finding, statistical significance, hidden Markov model, short open reading frames, automated genome annotation
Background
As of February 2003, 106 microbial genomes have been sequenced and made publicly available and the race is now on to mine genomes such as these for interesting and/or valuable genes and motifs. It has been estimated [1] that 60–80% of the genes in newly sequenced organisms have known homologues in other species. This percentage will grow as genomic annotations progress and perhaps there will be a time when virtually all genes can be found by homology matches to known proteins. That day, however, is not around the corner and even if it were, the occasional odd genes which would nevertheless escape detection by homology may very well be the truely novel and most wanted ones.
It is a common misconception that identification of genes in prokaryotes is almost trivial. Any random sequence, as well as non-coding regions in real genomes, contain a large number of open reading frames (ORFs). Most of these are too short to be possible protein coding genes, but in many genomes there are many 'random' ORFs longer than e.g. 100 amino acids, a cut-off that is often used for considering an ORF a real gene. The large number of short 'random' ORFs makes is difficult to discriminate real genes from random ORFs below a certain length, which depends on the genome and in particular its GC content. Therefore many genomes are over-annotated [2]. In one genome, that of A. pernix [3], all ORFs longer than 100 amino acids are annotated as genes, but probably only around half the annotated genes are real [2]. The most severe problem today is to disciminate between short genes and random ORFs, and here the meaning of 'short' is quite organism dependent. One of the most important contributions of this paper is a way to deal with this problem by introducing a statistical significance for an ORF being a gene.
Computational gene finding exploits the statistical differences in codon usage between coding and non-coding regions of DNA [4-6]. The search for a mathematical frame work to efficiently capture these differences in codon usage led to Markov chain models and the GeneMark algorithm [7]. In order to facilitate the combination of various Markov chain scores, the application of Hidden Markov Models (HMMs) to gene finding was introduced in a gene finder for E. coli, Ecoparse [8]. These methods relied on a set of known genes for estimating parameters. More recently methods have been developed which start from a raw genome and automatically extract data for estimation. One of these, Glimmer [9], employs interpolated Markov models in order to use the maximum Markov chain order which can reliably be estimated for every oligomer. Another one, Orpheus [1], appeared the same year and calculates coding potentials of ORFs based on codon frequency of similarity-derived genes. Most of these gene finders also extract Shine-Dalgarno sequences in order to improve prediction of start codons.
Due to their modular structure, HMMs are a suitable frame work for gene finding, and they are now used in GeneMark.hmm [10], GeneMarkS [11,12] and Frame-by-Frame [13]. GeneMarkS uses a mixture of Markov chains of order 0, 1 and 2 in combination with features of already annotated genomes to bootstrap an initial estimation of coding statistics, which is then iteratively improved by the GeneMark.hmm2.1 algorithm. Gibbs sampling is also used to detect Ribosome Binding Sites (RBS). The Frame-by-Frame method was conceived to improve the accuracy of GeneMark.hmm and it employs standard Viterbi parsing of all six reading frames independently and subsequently combines these into a global parse.
In this paper, we describe a fully automated method for making an organism specific gene finder. It starts from a raw genome and searches for protein matches to get a good training set. Then an HMM with states for coding regions as well as RBS is estimated from the data set. This HMM is used to score all the ORFs in the genome and finally the score is converted to a measure of significance – R – which is the expected number of ORFs that would be predicted in one megabase of random DNA. The main advantage of this significance measure is that it takes the length distribution of random ORFs properly into account. The method is shown to match or exceed other gene finders currently available.
Methods
Automatic extraction of training set
In order to fully automate the construction of the gene finder, a data set of reliable genes was obtained through the following procedure, which is essentially the same as that used in [1]. All ORFs with a significant match to a protein from a different organism are assumed to be real genes. A subset of those have only one possible start codon, because there is only one start between the most upstream protein match and the nearest upstream in-frame stop codon. The details of the method are:
1. Extract the maximal ORFs longer than 120 bases from the query-genome. For every stop codon, extract the region from the first downstream (in frame) start codon to the next downstream (in frame) stop codon.
2. Translate the ORFs to proteins and search for significant protein matches in Swiss-Prot [14] using BLASTP without gaps and a threshold of 10-5 [15]excluding proteins from the query-genome and proteins listed with one or more of the keywords putative, unknown, possible, hypothetical, probable, bacteriophage, transposon, insertion, reverse transcriptase.
3. For each ORF with at least one significant protein match, identify the most upstream position of these matches (to compensate for random matches, we actually remove 6 bases from the most upstream match before proceeding). If there is no alternative start codon between this position and the start of the ORF, put it in set A' of genes with certain starts. The remaining ORFs are put in set B' of certain genes with uncertain starts.
4. Reduce similarity in set A' by removing genes with similarity within the set. All pairs of genes are compared using BLASTN with a significance cut-off of 0.001. Two genes that match are called neighbours. Genes are removed starting with the one having the largest number of neighbours. This continues until no gene has neighbours left in the set [16]. The reduced set is henceforth referred to as A.
5. Unite set A' and B' into set T' and reduce similarity of T' to obtain set T .
6. Add 50 bases of upstream flank to genes of all sets and 10 bases downstream flank.
This procedure is a means to identify ORFs in a genome which are almost certain to be (protein coding) genes. The ORFs in set A and T make out reliable and balanced sets of positive examples which may be used to estimate the model parameters.
HMM architecture
We use a hidden Markov model (HMM) to model the gene structure. An HMM is a probabilistic model in which it is possible to model the various types of signals in a gene in one integrated model. For introductions to HMMs, see e.g. [17-19]. We model standard 'text book genes' with an unbroken open reading frame, i.e. genes with no programmed frame shifts, no sequencing errors nor any other special feature obstructing the reading frame.
The general architecture of the EasyGene HMM is shown in figure 1. There is a begin and end state marked B and E respectively. Then, at each side of the gene model, there are null models to model everything that is not part of a gene nor lies in the immediate vicinity of a gene. The next submodel is the RBS model which models the RBS as well as the nucleotides between the RBS and the start codon. After the start codon we model 3 bases explicitly since it appears that the codon usage immediately downstream of start codons differs from the rest of the gene [13]. Similarly we model the last codon before the stop codon explicitly and 6 bases after the stop in order to capture information present around the stop codon.
Figure 1.

The overall HMM architecture. Each box corresponds to a submodel with more than one state. The number above the boxes indicates the number of bases modelled by the submodel. An 'X' indicates a variable number.
We employ 3 looped codon submodels of the interior of the gene as depicted on the right hand side of figure 2. The reason for using several codon models is to embed a realistic length distribution in the HMM instead of the geometric distribution which would be implicit in having only one looped codon submodel [[17], Section 3.4]. We chose 3 because it results in a good fit to the empirical length distribution (see Results). The states in the codon model are of 4th order [20] in order to capture the inter-codon dependencies. The three looped codon models are identical, i.e. they have the same emission and transition probabilities (the states are tied).
Figure 2.

Enlargement of null model and internal looped codons. LEFT: The state structure of the NULL model. The background state is of third order and models the general composition of the genome. The three shadow states model coding regions on the complementary strand. There are transitions from the background state to the first RBS state and to the first state modelling the start codon. RIGHT: Details of model of internal codons. A codon is modelled by three states with a transition from the last state back to the first and one out of the codon model. By putting several codon models in series, the length distribution of coding regions can be captured. From the last state there is a transition to the first state of the 'BSTOP' model, which models the last codon before the stop codon.
The RBS model in figure 3 models the ribosome binding site as well as the nucleotides between the RBS and the start codon. It has 7 states to capture any ribosome binding patterns and 12 tied spacer states for modelling the region between the RBS and the start codon. From the first spacer state there are transitions to all but the last of the following states, so the length distribution (with a minimum of 3 and maximum of 12) can be modelled exactly without imposing Gaussian assumptions as is done in e.g. [21]. These spacer states are of order zero.
Figure 3.

The state structure of the RBS model. The RBS model consists of seven states for modelling the ribosome binding site followed by a set of tied states for the variable region between the RBS and the start codon. From the last state there is a transition to the first of the three states modelling the start codon.
The null model depicted on the left side of figure 2 has a third order state for capturing intergenic regions and a reverse codon model for modelling reverse genes ('shadows') with states of second order. Note that transitions are allowed directly from the null model to a start codon; this facilitates detection of genes inside operons which may not have a clear RBS.
We found that the inclusion of two more branches of internal codons improved performance. This is presumably because it allows the HMM to keep separate statistics for atypical genes, some of which may be horizontally transfered. Adding a fourth branch did not improve performance further, so we stopped at three lending some support to the hypothesis that there are essentially three classes of genes in prokaryotic genomes [22].
Model estimation
The HMM parameters (transition and emission probabilities) are estimated with the Baum-Welch algorithm, which is a maximum likelihood approach that finds the parameters maximizing the probability of the training set, see e.g. [17]. The estimation is done in these stages:
1. The emission probabilities of the background state are estimated from both strands of the complete genome.
2. The genes from start to stop codons are extracted from the training set and reverse complemented. The shadow model (consisting of three states) is estimated from these sequences. The parameters of this model are fixed.
3. The RBS, start and astart submodels (cf. figure 1) are estimated using set A exclusively since this set has reliable gene starts and therefore aligned upstream regions. Regions of 50 bases upstream from the start codon are extracted from all genes in set A. A null model (with fixed parameters) is included before the RBS model. The RBS model is initially primed with a high probability for the consensus GG (a dinucleotide common to most RBS), but the precise pattern is found by the estimation method. During estimation, a type of simulated annealing is used, where noise of a decreasing amount is added to the parameters in each iteration of the Baum-Welch algorithm [23]. After training on set A, the parameters of the RBS, start and astart models are fixed.
4. The null model, RBS, start and astart models are now combined with the internal codons, bstop, stop and astop models to make up the complete model. The non-fixed parameters of this model are then trained on the (larger) set T .
The whole procedure can be completely automated. Note that no experimentally mapped RBSs are used for estimating the RBS model, the RBS is discovered during the estimation procedure.
When estimating the complete model (stage 4 above) we use labelled estimation [17,20], where each base of the sequence is labelled as coding or non-coding. For the part of set T where the exact start is not known, we leave the part of the sequence from the most upstream start codon until the first significant protein match unlabelled. The weight with which each base in the unlabelled region contributes to the estimation of the parameters in the coding states is automatically determined during the iterative estimation procedure.
Decoding
By decoding we mean the process of finding an optimal parse of the DNA string into coding and non-coding regions.
The commonly used Viterbi decoding returns the most probable path of the sequence given the model [17], but this is not appropriate when the length is modelled by duplicating codon states, since this length modelling is realized only as a sum over many HMM paths. Therefore we use posterior decoding where one calculates, for each nucleotide i, the probability that it was emitted by a given state S. The calculation is done by adding the probabilities of all paths compatible with having state S emit nucleotide i [24,17]. We use this to calculate the posterior probability of the first state of the start codon model, and thus obtain the probability that a gene starts at any position in the sequence. Given our assumption of perfect 'textbook-genes' with no errors or frame shifts, there is exactly one stop codon for each start, and thus the probability of a gene start is equal to the probability of the whole gene. The independent scoring of start codons makes it trivial to report several possible start codons for a gene in cases where there is no clear "winner".
Note that the HMM architecture in figure 1 is non-looped – ie. it would find only one gene if we were using Viterbi decoding, which only gives the single most likely parse. This architecture however, is the correct one for scoring ORFs with posterior decoding. It has the further advantage that overlapping genes are easily handled since each gene start will be scored independently whether or not it overlaps other genes. In contrast, using a looped model and Viterbi decoding would not facilitate detection of overlapping genes unless the model contains explicit states for overlapping genes as described in e.g. [8].
The state posterior probability itself is not a useful score, because it is a probability of the whole sequence, not just a single gene, and it therefore depends on the length of the sequence it is part of. By dividing the posterior probability by the probability of the whole sequence (the genome) according to the null model, the contribution to the state posterior probability of the sequences flanking a gene will cancel and effectively make the ratio independent of the flanking sequences (except the parts very close to the gene), see the Appendix. The log of this ratio is called the log-odds score, and that is our basic score for a gene.
Significance
As mentioned above, it is important to take into account the chance that a random ORF of the same length scores as high as a given gene. This is implicitly taken into account by our HMM because it models the length distribution of genes, but it turns out that one can calculate a significance score, which works slightly better (se results) and has a more intuitive interpretation.
The probability of finding high-scoring ORFs in a random sequence is highly length dependent; the number of ORFs decays exponentially with the length so there will be a lot more short ORFs than long ones. For a random sequence of given length (e.g. 1 Mb) the expected number of ORFs of length l' can be written as
N(l') = exp (A - Bl'), (1)
where A and B are constants that can be found from linear regression of the log of the number of ORFs against the length. All lengths are measured in codons and we count both start and stop codon. For convenience we introduce the variable length l', which is the length of the ORF modelled by the looped codon submodels. A number of codons l0 are modelled explicitly in the beginning and end of the ORF (l0 = 4 in our model), so l' = l - l0.
If the log-odds score is denoted β, we show in the appendix that β' = β - (n - 1) log(l' - n/2) is approximately normally distributed with a mean αμ + γμ l' and variance ασ + γσ l', which are both linear in l' (the α's and γ's are constants). Here n is the number of looped codon submodels (3 in our model). The coefficients of the linear mean and variance are estimated by linear regression on ORFs from random sequences. Then we define the standard score
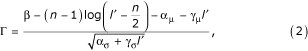
which is normally distributed with mean 0 and variance 1.
For any given length l' and standard score Γ, one can now estimate the expected number C(l', Γ) of ORFs of the same length scoring higher than Γ in a fixed (long) random sequence using equation (1),
C(l', Γ) = exp(A - Bl') [1 - Φ (Γ)], (3)
where Φ is the cumulative normal distribution. This number can be used directly to judge the significance of a gene predicted with length l and a standard score Γ. However, it would be preferable to know the total number of expected genes (of all lengths) predicted in a random sequence, rather than the expected number with a certain length. Therefore, instead of using C(l', Γ) to judge significance, we use the total number of expected predictions in a random sequence.
Suppose a gene of length l1 is predicted with a standard score Γ1. Then the expected number of genes of that length predicted in a random sequence is C = C(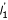 , Γ1). Now we want to calculate the total number of genes of all lengths predicted in a random sequence at the threshold C. For any length above a certain lC, the expected number of ORFs will fall below C due to the exponential decay of the length distribution. Therefore the total number of predicted genes (regardless of length) is roughly lC C + the sum of predicted genes above lC (a sum of a geometric series). We end up with the following expression for the total number of predicted genes in a random sequence:
, Γ1). Now we want to calculate the total number of genes of all lengths predicted in a random sequence at the threshold C. For any length above a certain lC, the expected number of ORFs will fall below C due to the exponential decay of the length distribution. Therefore the total number of predicted genes (regardless of length) is roughly lC C + the sum of predicted genes above lC (a sum of a geometric series). We end up with the following expression for the total number of predicted genes in a random sequence:
![]()
The number 16 arises from the fact that the minimum ORF length considered is 20 codons corresponding to a variable length of 20 - 4 = 16. See Appendix for details. This is the expected number of genes predicted in a random sequence for a given value of C, and that number is the one we quote for each predicted gene. The constants A and B characterize the random sequence.
R depends on the standard score Γ through C. This dependence is illustated in Figure 4. It clearly shows that a short ORF needs to have a much higher standard score than a long ORF in order to be significant (in the sense of having a low R-value).
Figure 4.

Relationship between R, Γ and variable length in codons l'. The numbers are taken from the E. coli runs described in Results and Discussion, but the qualitative behavior is independent of the genome
We have chosen to normalize R to a random sequence of 1 Mb rather than a random sequence the length of the genome, because then significance can be compared across genomes. The precise recipe we use for calculation of statistical significance is:
• For the genome in question, generate 40 Mb of random sequence using the 3rd order background model (estimated from the genome). Extract all ORFs and estimate the parameters A and B by linear regression of the log of the number of ORFs against the length and normalize to 1 Mb. To avoid distortion of regression lines due to noisy statistics of long ORFs, restrict the variable ORF length used to lie below 70 codons. This makes the range of variable length 16–70 codons, which corresponds to a total length range of 20–74 codons.
• Score all the ORFs in the random sequence with the model and calculate β' for all ORFs. For each length, calculate the average and variance and estimate the parameters αμ, γμ, ασ, and γσ by linear regression, again using ORFs in the range 20–74 codons.
• To calculate the significance of an ORF in the genome, first calculate the standard score Γ from equation 2, then C from equation 3, and finally the significance value R using equation 4.
There are of course other possible choices of significance measure, but we believe that this is a simple and intuitively clear one and we have prefered it to the more traditional significance measure. By reporting the number R of expected false positives in one megabase of random sequence, it is easy for the end-user to estimate the number of false positives in a random sequence the length of the entire query genome – one simply has to multiply R with the size of the genome measured in megabases.
Using other gene finders
In order to benchmark EasyGene we compare it with some of the existing gene finders.
GeneMark 2.4, Frame-by-Frame, GeneMark.hmm 2.1 and GeneMark.hmm/S all belong to the GeneMark suit of programs and are accessible via the web interfaces listed in the references.
For GeneMark.hmm2.1, GeneMark.hmm/S and Frame-by-Frame the output is a coordinate listing (start and stop positions) of all predicted genes.
GeneMark2.4 outputs a list of stop codons and corresponding high-scoring start codons. Each start/stop is listed with scores for coding potential and RBS. We collect all starts for a given stop and output the "Avg Prob" of the start with the highest RBS score. Whenever a threshold was needed for comparison purposes, we used 0.5 which is the default set on the web page.
Glimmer2.02 and Orpheus2 were installed locally. We changed the minimum ORF length predicted by these gene finders to 60 bp which seems to be the minimum used by the other gene finders. Orpheus and Glimmer provide two kinds of output: a verbose coordinate list of starts, stops and ORF scores and a simpler, post-processed list of coordinates for ORFs regarded as genes.
In order to test their ORF scoring we had to parse the scored output. We had some difficulties interpreting the scored Orpheus output since some ORFs were scored several times with identical results (several identical "Start chosen"). In cases where multiple copies were found, we simply chose one of them and used the corresponding "Coding potential" (with the recommended threshold of -1) for further analysis.
For Glimmer2.02 the scored output was parsed simply by selecting the Gene Score attributed to every scored ORF and using the recommended threshold of 90.
Finally, we used RBSfinder [25] for an alternative post-processing of Glimmer2.02 output. RBSfinder is designed to look for RBS sequences upstream of genes predicted by Glimmer2.02. If there are no RBS patterns in this region, RBSfinder searches for a start codon having a RBS pattern in the same reading frame upstream or downstream and relocates the start codon accordingly. The program may be iterated several times using revised predictions as new inputs. We found that running it twice was better than once but running it three times did not improve things further, so we chose to run it twice (with default options).
Results and Discussion
A number of tests were conducted in order to optimize the model architecture. We tested the number of codon models in series and found that three models yield a very good fit to the observed length distribution, see figure 5 for an example.
Figure 5.

Gene length distribution imposed by HMM architecture. The model length distribution given by a negative binomial (equation 8 with n = 3) compared to the length histogram of set A genes for H. pylori J99.
The results of our experiments with the number of branches in the coding model and the order of the coding states are shown in figure 6 and 7 respectively, which show (cross-validated) performance curves (ROC curves) for varying numbers of branches and various orders. The performance curves are made by plotting the average true positive rates for a range of average false positive rates (the fraction of false positives made on average by the 10 different cross-validation models in 1 Mb of random sequence).
Figure 6.

Assessing the optimal number of HMM coding branches. Performance curves for 1,2,3 and 4 Markov branches of looped codon submodels for E. coli. The performance curves are made by the following procedure: First we sort the positive R-values in ascending order for each of the 10 subsets of set T (test sets). Then for each ascending R-value we calculate the fraction of genes in set T scoring below R (true positive rate) and the fraction of ORFs (with lengths greater than or equal to 20 codons) in one megabase double-stranded sequence scoring below R (false positive rate). The resulting 10 files with true and false positive rates are concatenated and 30 false positive cutoffs are selected (from 0 to 0.15 with steps of 0.005). The false positive entries in the 10 files which fall between these cutoffs are found and the corresponding true positive entries are averaged. Hence for each average false positive rate (halfway between two consecutive false positive cutoffs) we associate an average true positive rate and these tuples are then plotted.
Figure 7.

Assessing the optimal order of looped codon states. Performance curves for 3rd, 4th and 5th order Markov states of looped codon submodels for E. coli. For explanation of the construction of performance curves please confer the caption of figure 6.
Figure 6 indicates that 3 branches is the best choice, while figure 7 suggests that the optimal order for these branches is 4. Note that the Y-axes of these figures have been zoomed in order to allow visual inspection of the performance differences. Note also that all architectures and orders in fact yield a relatively high true positive rate even at false positive rates below 0.02. Similar figures were also made for other organisms (not shown) and although the results were not always as clear as for E. coli, a choice of 3 branches of 4th order states as default models of the internal parts of genes works well for the organisms we tested.
Similarly, figure 8 shows performance curves to compare log-odds and significance scores and the significance scoring is seen to be slightly better in that it allows detection of more true positives for a given false positive rate. The significance scoring has the additional advantage of being genome independent and has an intuitively appealing interpretation.
Figure 8.

Comparing significance and log-odds. Performance curves comparing significance and log-odds scores for E. coli. For explanation of the construction of performance curves please confer the caption of figure 6.
Table 1 shows the average true and false positive rates for selected R-values in the case of E. coli models with three branches and fourth order branch states. This table gives an impression of the approximate R values corresponding to the graphs in figures 6, 7 and 8.
Table 1.
True and false positive rates for selected R-values.
| R-value | TP rate | FP rate |
| 0.1 | 0.971 | 0 |
| 2 | 0.980 | 2.3e-6 |
| 10 | 0.984 | 3.0e-5 |
| 50 | 0.987 | 3.1e-4 |
| 150 | 0.991 | 8.7e-4 |
| 500 | 0.995 | 2.8e-3 |
| 10000 | 0.999 | 0.059 |
True and false positive rates averaged over 10 cross-validations for selected R-values in the case of the three branches, fourth order E. coli model. The FP rate is measured as the average fraction of ORFs (with lengths greater than or equal to 20 codons) in one megabase of double-stranded random sequence scoring lower than the given R-value. The TP rate is measured as the average fraction of ORFs in data set T scoring lower than the given R-value
In order to test the validity of the approximations used in the derivation of the significance measure, we generated 1 Mb of random sequence from the Markov chain corresponding to the background state of the E. coli model. The plots in Figure 9 show the mean and variance of the length-corrected log-odds score, β', for each length. They are very close to being linear as we assumed. The length distribution of the ORFs in the same random sequence is also shown, which confirm the geometric length distribution. The predicted number of significant ORFs in 1 Mb random sequence is compared with the theoretical significance value R in Figure 10 and the agreement is seen to be rather good. We also compared the distribution of standard (3) scores for ORFs in the same random sequences to a normal distribution of unit variance and zero mean and the agreement also turned out to be good in this case as seen in Figure 11. We conclude that the assumptions and approximations used in the calculation of R are quite accurate.
Figure 9.

Statistical characteristics of random sequences. The top two panels show the mean and variance of log-odds scores versus variable ORF length in random sequences (E. coli model). Lowest subplot shows a logarithmic plot of the length distribution of random ORFs. The linear regression lines are shown in all three plots.
Figure 10.

Comparing predicted and found number of false positives. Empirical and theoretical number of false positives per Mb double-stranded random sequence according to the E. coli model.
Figure 11.

Probability density functions for the standard score. Empirical (dots) and theoretical (line) probability density functions for the standard scores (Γ) in random sequences (E. coli model). The lower plot is an enlargement of the distribution tail.
Table 2 shows the percentage of genes found for eight different gene finders and some sets of high-confidence genes from E. coli as well as the number of genes found in the whole genome and in random sequences. The eight gene finders are: EasyGene, Glimmer2.02, Glimmer2.02 with RBSfinder post-processing, Orpheus, GeneMark2.4, GeneMark.hmm2.1/GeneMarkS hybrid, "pure" GeneMark.hmm2.1 and Frame-by-Frame.
Table 2.
Specificity, sensitivity and precision estimates for different gene finders in E. coli.
| Data set | EasyGene | Glim | rbs-Glim | Orpheus | Gm24 | GmS | Gmhmm | Frame |
| A'-% found | 98.4 | 98.9/98.9 | 98.9 | 98.0/95.3 | 91.5 | 97.2 | 98.1 | 97.0 |
| A'-% exact | 93.8 | 98.9/95.3 | 84.1 | 95.1/92.4 | 41.6 | 88.0 | 85.7 | 93.2 |
| B'-% found | 98.4 | 98.5/98.6 | 98.6 | 95.9/96.5 | 90.2 | 96.6 | 97.2 | 96.4 |
| T-% found | 98.1(98.0) | 98.3/98.4 | 98.4 | 96.5/95.6 | 89.8 | 96.3 | 97.1 | 96.1 |
| Genome | 4145 | 6827/5756 | 5756 | 9333/7543 | 3552 | 4064 | 4230 | 4064 |
| zero order | 7 | 169/211 | 211 | 6761/5430 | 6 | 153 | 1459 | 0 |
| first order | 7 | 545/723 | 723 | 6836/4804 | 13 | 241 | 830 | 0 |
| third order | 1 | 2423/2694 | 2694 | 6582/4817 | 43 | 659 | 866 | 1 |
| shadows | 0 | 19/21 | 21 | 22/9 | 1 | 0 | 2 | 0 |
Upper part shows the percentage of genes found exactly (both 5' and 3' end) and partially (only 3' end exact) for different gene finders and sets of high confidence genes in E. coli. For Glimmer and Orpheus, the numbers before the "/" are based exclusively on their ORF scores and recommended threshold whereas the numbers after the "/" are based on their post-processing procedures. The number of genes predicted in the whole genome is also shown. This should be compared to the 4288 annotated genes in E. coli. The lower part of the table shows the number of false positives predicted in random sequences generated by Markov chains of order 0, 1 and 3 and the very last row shows the number of false predictions in the shadows of the high-confidence genes in data set A. All values listed for EasyGene are based on an R-value threshold of R = 2.
Sequence set A' consist of the 1136 genes with high-confidence starts extracted from the E. coli genome as described in Methods. The "% exact" row indicates the percentage of predictions where both start and stop codon are correctly predicted whereas the "% found" row indicates that only the stop codon is correct. Note that all genes in set A have the most upstream start (they are Longest Possible Open Reading Frames – LPORFs) by construction, and hence performance on this set favours gene finders which are biased towards LPORFs (such as Glimmer). Set B' consists of 1690 high-confidence genes with uncertain starts extracted as described in Methods. Due to the uncertainty of start codon placement, one cannot evaluate the exact start prediction performance for this set. The same is true for set T which is a similarity reduced union of A' and B' (2042 sequences).
The Genome row shows the number of genes predicted in the E. coli genome using default parameters and thresholds for the various gene finders. For Glimmer and Orpheus the minimum length of predicted genes was lowered to 60 in order to make their performance comparable to the others'. The next three rows show the number of false positives found in both strands of 1 Mb random sequences generated by zero, first and third order Markov chains estimated from the entire E. coli genome. The last row of the table shows the number of predictions wholly within the shadows of set A' – i.e. wholly within regions complementary to the genes in set A' where, ideally, no genes (or at least very few) should be predicted.
The test sets overlap with the training sets for EasyGene. Therefore, for set T the 10-fold cross validation sensitivity is shown in parenthesis for EasyGene and it is seen to be reassuringly close to the non-crossvalidated sensitivity suggesting that EasyGene employs an appropriate model complexity and steers free of overfitting. Note also that the other gene finders have also been estimated from sets that overlap (or even contain) set T .
For Orpheus and Glimmer we show two numbers N1/N2 for each entry corresponding to before and after post-processing. For Orpheus, N1 is the number of unique ORFs having a Coding Potential above the recommended minimum of -1 and N2 is the number of entries in the post-processed orfnuc file. The post-processing removes some overlaps but also appears to employ a less restrictive cutoff than the recommended -1. For Glimmer, N1 is the number of ORFs with Gene Scores greater than or equal to the recommended threshold of 90 and N2 is the number of entries in the post-processed list of putative genes. The post-processing elects ("votes") some ORFs as gene candidates despite a low score. On the other hand the post-procesing removes some same-strand overlaps in different reading frames so the N2 may be greater or less than N1 depending on the relative extent of overlaps and "voting".
It is always difficult to asses the specificity of a gene finder, because it is difficult to find genomic regions that are certain to contain no genes. We have therefore assessed specificity in three different ways. First, by counting the number of predicted genes in a genome. If this number is much higher than the number of annotated genes, it is likely that there are many false positive predictions, i.e. poor specificity. Our second test is based on random sequence. Clearly, a high number of predicted genes in a random sequence of bases indicates a poor specificity. However, it is probably not possible to find an exact quantitative correspondence between predictions in random sequences and real genomes. Also, it is not clear what sort of random sequence to use for such a test. By 0'th order we mean a sequence with bases generated randomly and independently with the base frequencies of the genome. Bases are quite correlated in DNA sequences, so we have also tested on sequences that are generated by Markov chains of orders 1 and 3. These Markov chains are estimated from the genome, so the sequences will have the same distribution of dinucleotides and 4-nucleotides, respectively, as the genome. Finally, the third test is the number of genes predicted on the opposite strand of genes (shadows); these shadow regions should contain very few genes if any.
All gene finders except EasyGene, Frame and GeneMark2.4 predict a rather large number of false positives in random sequences, but for GeneMark.hmm and GeneMarkS we do not see large over-prediction in the genome or in shadows. Evidently, Glimmer and Orpheus predict a lot more genes in the genome than the other gene finders, suggesting that these gene finders have very high false positive levels. This is supported by the high numbers of genes predicted in random sequences, and (to a much lesser extent) in shadows. Orpheus and Glimmer actually predict more genes pr. nucleotide in the third order random sequence than they do in the genome, suggesting that the coding potential calculated in these gene finders is far from optimal.
The HMM used by Frame assumes a minimum gene length of 69 bases which could make its false positive level seem somewhat better (lower) than it is, but there was no convenient way to lower the minimum length so we simply left it. It should also be noted that Frame relies on pre-existing annotations for training and is therefore not a self-training gene finder like Glimmer, Orpheus, GeneMarkS and Easygene.
The sensitivity of the gene finders is tested on sets of high-confidence genes. Glimmer has the highest sensitivity for all sets, but this is largely due to heavy over-prediction. One ought always to bear in mind that it is not difficult to achieve high sensitivity if high levels of false predictions are tolerated at the same time – sensitivity is 100% if all ORFs are predicted as genes! Although there are some very close competitors, EasyGene comes out as the second best in sensitivity for all sets.
The exact prediction of start codons is tested on set A' and on an experimentally ver-ified set. As mentioned above, set A is biased, because all genes of this set are LPORFs. Glimmer always predicts the most upstream start and consequently achieves a high performance on this set. When Glimmer's output is post-processed by RBSfinder the performance drops considerably.
The prediction of start codons was evaluated further on a set of 195 E. coli genes with experimentally verified starts [26] with results shown in table 3. Set LiC is the subset of 133 genes which coincide with the longest possible open reading frame (LPORF) while set LiD is the remaining 62 genes whose starts are downstream of the LPORF start. The goal is to find the starts of the challenging LiD set without loosing too many of the more trivial starts of set LiC. Table 3 shows that while most gene finders partially locate all genes in set LiC and LiD, there are large variations in their exact localization ability. Selecting for the highest combined performance on set LiC and LiD, one sees that EasyGene, GeneMarkS and Frame-by-Frame are best. Their performances also exceed that of [27] in which a cross-validated performance of 84.9% +/- 4% is reported on a subset of 184 genes out of the 195. In the low end we have Glimmer finding 0% of set LiD exact (1.6% with post-processing) and GeneMark2.4 finding 49.6% of set LiC exact. Using the RBSfinder post-processing [25] on the Glimmer predictions improves performance on set LiD to 75.8%, but at the cost of a substantial dip in set LiC performance to 88.7%.
Table 3.
Sensitivity and precision estimates for experimentally verified E. coli genes.
| Data set | EasyGene | Glim | rbs-Glim | Orpheus | Gm24 | GmS | Gmhmm | Frame |
| LiC-% found | 100 | 100/100 | 100 | 97.7/91.7 | 97.7 | 100 | 100 | 100 |
| LiC-% exact | 94.0 | 100/97.0 | 88.7 | 96.2/90.2 | 49.6 | 94.0 | 90.2 | 98.5 |
| LiD-% found | 100 | 100/100 | 100 | 96.8/98.4 | 100 | 100 | 100 | 100 |
| LiD-% exact | 96.8 | 0/1.6 | 75.8 | 51.5/51.6 | 67.7 | 95.2 | 80.6 | 87.0 |
Percentage of genes found exactly and partially in two subsets of the 195 experimentally verified genes published by [26]. All values listed for EasyGene are based on an R-value threshold of R = 2.
Many gene finders are first developed for E. coli and then later adapted to other organisms. It is therefore important also to test gene finders on other organisms. Based on table 2 and 3 we chose EasyGene, GeneMarkS and Frame-by-Frame as the gene finders with the best overall performances and then conducted further comparisons between these for M. tuberculosis [28], H. pylori [29] and B. subtilis [30]. M. tuberculosis presents a challenge due to GC richness, H. pylori due to small genome size and B. subtilis was chosen on account of its reputation of being well annotated [11]. The results are presented in table 4 with the same rows as table 2.
Table 4.
Specificity, sensitivity and precision estimates for M. tuberculosis, H. pylori J99 and B. subtilis.
| M. tuberculosis | |||
| Data set | EasyGene | GmS | Frame |
| A'-% found | 96.7 | 97.2 | 96.0 |
| A'-% exact | 89.1 | 80.9 | 87.9 |
| B'-% found | 96.8 | 97.1 | 96.3 |
| T-% found | 96.9(96.6) | 97.3 | 96.4 |
| Genome | 3749 | 3983 | 4341 |
| zero order | 0 | - | 8 |
| first order | 3 | - | 2 |
| third order | 2 | - | 12 |
| shadows | 1 | 0 | 1 |
| H. pylori J99 | |||
| Data set | EasyGene | GmS | Frame |
| A'-% found | 99.2 | 99.2 | 99.2 |
| A'-% exact | 97.5 | 95.7 | 96.7 |
| B'-% found | 100 | 99.6 | 98.9 |
| T-% found | 99.7(98.8) | 99.5 | 99.1 |
| Genome | 1491 | 1518 | 1479 |
| zero order | 2 | 1479 | 2 |
| first order | 1 | 336 | 2 |
| third order | 0 | 403 | 0 |
| shadows | 2 | 0 | 0 |
| B. subtilis | |||
| Data set | EasyGene | GmS | Frame |
| A'-% found | 99.3 | 98.1 | 98.8 |
| A'-% exacts | 94.8 | 94.1 | 93.3 |
| B'-% found | 99.2 | 99.0 | 98.2 |
| T-% found | 99.3(99.2) | 99.0 | 98.4 |
| Genome | 4083 | 4221 | 4006 |
| zero order | 1 | 675 | 0 |
| first order | 2 | 457 | 0 |
| third order | 1 | 813 | 2 |
| shadows | 0 | 0 | 0 |
Genes found exactly and partially for different gene finders and sets of high confidence genes in M. tuberculosis, H. pylori J99 and B. subtilis, where the number of annotated genes is 3918, 1491 and 4100 respectively. There are no pre-trained GeneMarkS models for M. tuberculosis, so it was not possible to obtain a false positive estimate for this organism. All values listed for EasyGene are based on an R-value threshold of R = 2.
For M. tuberculosis GeneMarkS and EasyGene are comparable, although GeneMarkS seems to over-predict slightly (assuming that the 3918 annotated genes are close to being correct), and Easygene might under-predict. We believe that Frame predicts too many genes in this organism and at the same time it has lower sensitivity than the two others, suggesting a worse performance overall. This indicates that Frame is not very robust with respect to high GC content.
The small dataset from H. pylori might give a slight over-fitting in EasyGene, where there is one percent difference between cross-validated results and non-crossvalidated. For this organism the three gene finders seem to have very similar performances. Finally, for B. subtilis EasyGene comes closest to the number of annotated genes and have higher sensitivity than the other methods.
Conclusions
The emerging overall picture is that the sensitivity of EasyGene tends to be comparable to GeneMarkS and higher than Frame. With regards to specificity, EasyGene and Frame tend to be comparable and both higher than GeneMarkS. Hence, EasyGene comes out with the best combined sensitivity/specificity performance. When it comes to exact starts, EasyGene also generally performs best.
Glimmer and Orpheus have good sensitivities but at the cost of very low specificities in this comparison. We have lowered the ORF length cutoff from their default values in these methods to make the results comparable. This may be unfair, but since the challenge is to find the short genes, we believe that any gene finder should be able to score them.
At present it is not possible to automatically find all genes in a prokaryotic genome. We believe the aim of a gene finding system is to help expert annotators as much as possible, and we consider the statistical significance of a gene an important help in classifying the predictions into almost certain genes and border-line genes needing more attention. Contrary to most other gene finders discussed here, it is up to the user which significance cut-off to use. EasyGene also predicts sub-optimal start codons if need be, so it will be easy to see if e.g. two alternative starts have almost the same score.
A shortcoming of the significance value as calculated here is that long ORFs score well simply on account of their length, since very long ORFs occur rarely in random sequences. For this reason, EasyGene also provides a log-odds score in the output which may be held up against the R-value for ORFs longer than say 500 bp. Genes of this length ought to have high log-odds values. If they do not – i.e. if they score below 0 – then they are probably not real genes despite their length (sometimes very long non-coding ORFs occur in regions of repetitive DNA).
If the amount of available genomic DNA is very small (as it may be in partially sequenced genomes) the 3 branches of 4th order coding models may have too many parameters to be reliably estimated. In such cases, one could reduce the parameter space simply by using fewer branches and/or lower orders. More generally, one could develop a method for automatic fine-tuning of HMM architecture for every new organism. Other lines of future research could focus on modelling of genes with errors and frame shifts.
Finally, it may be noted that a prototype of EasyGene has already been used in the annotation of S. typhi [31].
Appendix: The length dependent score distribution
The log-odds score distribution has several components. The probability of a sequence z containing and ORF given a model M may be written as
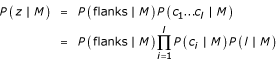
where c1, . . . , cl denotes the codons in the ORF and flanks denotes all the rest of the sequence. P (l|M) is the length distribution of the HMM. In logarithmic form we have
![]()
Similarly, the denominator of the log-odds score, log P (z|N ), may be rewritten in exactly the same way, so the entire log-odds score becomes
![]()
where
![]()
We will now look at the distribution of each of these terms.
The null model consists of a state with a loop and three reverse codon states with a loop. For a long sequence one of the loops will usually dominate the probability, so the length distribution is well approximated by a geometric distribution
P(l|N) 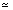 k1 exp (-k1l)
k1 exp (-k1l)
where k1 is a constant greater than zero.
The length distribution from the looped codon states is a negative binomial [17],
![]()
where p is loop probability and n is the number of looped codon submodels (in casu, n = 3). Some of the ORF (start and end) is modelled by non-looped states. The number of nucleotides modelled in the beginning and end of the ORF is denoted l0 and the variable length is denoted l', so the total ORF length is l = l0 + l'. Using that log 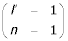 (n - 1) log(l' - n/2) - log(n-1)! for l' >>n, we obtain
(n - 1) log(l' - n/2) - log(n-1)! for l' >>n, we obtain
![]()
The next term, Q, in equation 6 is a sum of random variables since we are considering random sequences. The sum Q will therefore (according to the central limit theorem) converge to a normal distribution N (μl, σ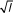 ), for large l.
), for large l.
The submodels flanking the gene model are identical to the null model. Therefore p(flanks|M) and p(flanks|N) will almost cancel in the last term of equation 6, except from the contribution from the RBS model and the states after the stop codon. This contribution will be independent of the ORF length. Since it is again a sum of random terms it is well approximated by a normal distribution.
Apart from the non-linear term from the negative binomial we have now shown that all terms in equation 6 are constant or scale linearly with l. Therefore
![]()
is normally distributed for fixed length with an average and variance that are linear functions of l:
average = αμ + γμl' (11)
![]()
The parameters of these linear functions can readily be estimated from random sequences by doing linear regression between the variable length and the mean and variance of log-odds scores as shown in the two upper plots of figure 9. (Note that one can switch from the variable length l' to the total length l in the formulas by changing the constants.)
Finally it is convenient to introduce the standard score Γ, defined in equation 2, which is normally distributed with average 0 and variation 1.
Since random ORF lengths are geometrically distributed (cf. equation 1), the expected number of ORFs of length l scoring more than Γ in a sequence is
C(l', Γ) = exp (A - Bl') [1 - Φ(Γ)], (13)
where the exponential term is the expected number of ORFs of variable length l' and Φ is the cumulative normal distribution. Thus, for a given score we can calculate the number of expected predictions in a random sequence.
For lengths l ≥ lC this expectation is always less than C (due to the exponential factor). So if we require that no more than C predictions be made at any length, the total number of predictions in a random genome is
![]()
where ls is the shortest ORF length considered (we use 20). lC is given by
![]()
Strictly speaking, we should take the smallest integer larger than 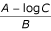 , but using the real expression introduces only an insignificant error. Inserting into equation 14 finally yields equation 4,
, but using the real expression introduces only an insignificant error. Inserting into equation 14 finally yields equation 4,
R = {(A - log C)/B - (ls - l0)}C + C/(1 - exp(-B)). (16)
Web sites used
Frame-by-Frame:
http://opal.biology.gatech.edu/GeneMark/fbf.cgi
GeneMark.hmm 2.1:
http://opal.biology.gatech.edu/GeneMark/gmhmm2_prok.cgi
GeneMark.hmm 2.1 using GeneMarkS models:
http://opal.biology.gatech.edu/GeneMark/gmhmm2_genemarks.cgi
GeneMark 2.4:
Acknowledgments
Acknowledgements
We wish to thank G. Kolesov for kindly providing the Orpheus package and the people behind http://www.tigr.org/software/glimmer for providing the Glimmer package. This work was supported by a grant from the Danish National Research Foundation.
Contributor Information
Thomas Schou Larsen, Email: thsl@novozymes.com.
Anders Krogh, Email: krogh@binf.ku.dk.
References
- Frishman D, Mironov A, Mewes HW, Gelfand M. Combining diverse evidence for gene recognition in completely sequenced bacterial genomes. Nucleic Acids Research. 1998;26:2941–2947. doi: 10.1093/nar/26.12.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovgaard M, Jensen LJ, Brunak S, Ussery D, Krogh A. On the total number of genes and their length distribution in complete microbial genomes. Trends in Genetics. 2001;17:425–428. doi: 10.1016/S0168-9525(01)02372-1. [DOI] [PubMed] [Google Scholar]
- Kawarabayasi Y, et al. Complete genome sequence of an aerobic hyperthermophilic crenarchaeon. Aeropyrum pernix K1 DNA Res. 1999;6:83–101. doi: 10.1093/dnares/6.2.83. [DOI] [PubMed] [Google Scholar]
- Fickett J. Recognition of protein coding regions in DNA sequences. Nucleic Acids Research. 1982;17:5303–5318. doi: 10.1093/nar/10.17.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribskov M, Devereux J, Burgess R. The codon preference plot: Graphic analysis of protein coding sequences and prediction of gene expression. Nucleic Acids Research. 1984;12:539–549. doi: 10.1093/nar/12.1part2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden R. Measurements of the effects that coding for a protein has on a dna sequence and their use in finding genes. Nucleic Acids Research. 1984;12:551–567. doi: 10.1093/nar/12.1part2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky M, McIninch J. GENMARK: Parallel gene recognition for both DNA strands. Computers and Chemistry. 1993;17:123–133. doi: 10.1016/0097-8485(93)85004-V. [DOI] [Google Scholar]
- Krogh A, Mian IS, Haussler D. A hidden Markov model that finds genes in E. coli DNA. Nucleic Acids Research. 1994;22:4768–4778. doi: 10.1093/nar/22.22.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Delcher AL, Kasif S, White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Research. 1998;26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Research. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. implications for finding sequence motifs in regulatory regions. Nucleic Acids Research. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. Heuristic approach to deriving models for gene finding. Nucleic Acids Research. 1999;27:3911–3920. doi: 10.1093/nar/27.19.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmatkov AM, Melikyan AA, Chernousko FL, Borodovsky M. Finding prokaryotic genes by the 'frame-by-frame' algorithm: targeting gene starts and overlapping genes. Bioinformatics. 1999;15:874–886. doi: 10.1093/bioinformatics/15.11.874. [DOI] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobohm U, Scharf M, Schneider R, Sander C. Selection of representative protein data sets. Protein Science. 1992;1:409–417. doi: 10.1002/pro.5560010313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin RM, Eddy SR, Krogh A, Mitchison G. Biological Sequence Analysis Cambridge University Press, Cambridge, UK. 1998.
- Krogh A. An introduction to hidden Markov models for biological sequences. In: Salzberg SL, Searls DB, Kasif S, editor. In Computational Methods in Molecular Biology. Vol. 4. Elsevier, Amsterdam; 1998. pp. 45–63. [Google Scholar]
- Rabiner LR. A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE. 1989;77:257–286. doi: 10.1109/5.18626. [DOI] [Google Scholar]
- Krogh A. Two methods for improving performance of a HMM and their application for gene finding. In: Gaasterland T, Karp P, Karplus K, Ouzounis C, Sander C, Valencia A, editor. In Proceedings of the Fifth International Conference on Intelligent Systems for Molecular Biology. Menlo Park, CA AAAI Press; 1997. pp. 179–186. [PubMed] [Google Scholar]
- Yada T, Totoki Y, Takagi T, Nakai K. A novel bacterial gene-finding system with improved accuracy in locating start codons. DNA Research. 2001;8:97–106. doi: 10.1093/dnares/8.3.97. [DOI] [PubMed] [Google Scholar]
- Medigue C, Rouxel T, Vigier P, Henaut A, Danchin A. Evidence for horizontal gene transfer in Escherichia coli speciation. Journal of Molecular Biology. 1991;222:851–856. doi: 10.1016/0022-2836(91)90575-q. [DOI] [PubMed] [Google Scholar]
- Hughey R, Krogh A. Hidden Markov models for sequence analysis: extension and analysis of the basic method. CABIOS. 1996;12:95–107. doi: 10.1093/bioinformatics/12.2.95. [DOI] [PubMed] [Google Scholar]
- Stormo GD, Haussler D. Optimally parsing a sequence into different classes based on multiple types of evidence. In Proc of Second Int Conf on Intelligent Systems for Molecular Biology. 1994. pp. 369–375. [PubMed]
- Suzek BE, Ermolaeva MD, Schreiber M, Salzberg SL. A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics. 2001;17:1123–1130. doi: 10.1093/bioinformatics/17.12.1123. [DOI] [PubMed] [Google Scholar]
- Link AJ, Robison K, Church GM. Comparing the predicted and observed properties of proteins encoded in the genome of escherichia coli k-12. Electrophoresis. 1997;18:1259–1313. doi: 10.1002/elps.1150180807. [DOI] [PubMed] [Google Scholar]
- Hannenhalli SS, Hayes WS, Hatzigeorgiou AG, Fickett JW. Bacterial start site prediction. Nucleic Acids Research. 1999;27:3577–3582. doi: 10.1093/nar/27.17.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- RA Alm, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- Kunst F, et al. The complete genome sequence of the gram-positive bacterium bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Parkhill J, et al. Complete genome sequence of a multiple drug resistant salmonella enterica serovar typhi ct18. Nature. 2001;413:848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]