

Supplemental Digital Content is available in the text.
Keywords: diabetes mellitus, ischemia, metabolomics, proteomics, vascular remodeling
Abstract
Rationale:
The AMP-activated protein kinase (AMPK) is stimulated by hypoxia, and although the AMPKα1 catalytic subunit has been implicated in angiogenesis, little is known about the role played by the AMPKα2 subunit in vascular repair.
Objective:
To determine the role of the AMPKα2 subunit in vascular repair.
Methods and Results:
Recovery of blood flow after femoral artery ligation was impaired (>80%) in AMPKα2−/− versus wild-type mice, a phenotype reproduced in mice lacking AMPKα2 in myeloid cells (AMPKα2ΔMC). Three days after ligation, neutrophil infiltration into ischemic limbs of AMPKα2ΔMC mice was lower than that in wild-type mice despite being higher after 24 hours. Neutrophil survival in ischemic tissue is required to attract monocytes that contribute to the angiogenic response. Indeed, apoptosis was increased in hypoxic neutrophils from AMPKα2ΔMC mice, fewer monocytes were recruited, and gene array analysis revealed attenuated expression of proangiogenic proteins in ischemic AMPKα2ΔMC hindlimbs. Many angiogenic growth factors are regulated by hypoxia-inducible factor, and hypoxia-inducible factor-1α induction was attenuated in AMPKα2-deficient cells and accompanied by its enhanced hydroxylation. Also, fewer proteins were regulated by hypoxia in neutrophils from AMPKα2ΔMC mice. Mechanistically, isocitrate dehydrogenase expression and the production of α-ketoglutarate, which negatively regulate hypoxia-inducible factor-1α stability, were attenuated in neutrophils from wild-type mice but remained elevated in cells from AMPKα2ΔMC mice.
Conclusions:
AMPKα2 regulates α-ketoglutarate generation, hypoxia-inducible factor-1α stability, and neutrophil survival, which in turn determine further myeloid cell recruitment and repair potential. The activation of AMPKα2 in neutrophils is a decisive event in the initiation of vascular repair after ischemia.
AMP-activated protein kinase (AMPK) is a nutrient-sensitive kinase that can be regulated by Ca2+-elevating agents that activate upstream kinases and by various cellular stresses including hypoxia.1 In endothelial cells, the AMPK is required for an adequate angiogenic response to hypoxia2 and can induce the generation of vascular endothelial growth factor (VEGF).3 Angiogenesis and vascular repair after ischemia in vivo also rely on the recruitment and activation of circulating cells such as monocytes and neutrophils.4 Little is known about the role of the AMPK in such processes, but the kinase has been implicated in inflammatory signaling in myeloid cells,5 such as leukocyte homing and neutrophil activation,6 and monocyte differentiation.7–9
Editorial, see p 8
In This Issue, see p 2
The AMPK is a heterotrimeric kinase consisting of a catalytic α subunit and regulatory β and γ subunits. There are 2 different isoforms of the catalytic α subunit, that is, the α1 subunit that is mainly localized to the cytosol and the α2 subunit that can translocate to the nucleus, where it likely plays a more important role in transcription factor activation and gene regulation.1 Although many studies have reported a role for AMPK activation in angiogenesis in vitro and in vivo, they generally relied on unspecific pharmacological tools that cannot differentiate between the different catalytic α subunits. We reported previously that the AMPKα1 subunit in endothelial cells is a prerequisite for angiogenesis induced by cytokines and VEGF via a mechanism involving transforming growth factor-β–activated kinase 1 and superoxide dismutase 2 expression.10 The aim of the present study was to assess the role of AMPK, particularly the AMPKα2 subunit, in the regulation of vascular repair in vivo in a model where the outcome is largely dependent on local responses to hypoxia and the mobilization and recruitment of bone marrow–derived cells such as monocytes and neutrophils.
Methods
Animals
C57BL/6J mice were purchased from Charles River (Sulzfeld, Germany).
Genetically modified mice lacking either the AMPKα1 or the AMPKα2 subunits, their respective wild-type littermates, and floxed AMPKα2 mice were kindly provided by Benoit Viollet (INSERM, U1016, Paris, France) and bred at the Goethe University Hospital animal facility. Floxed AMPKα2 mice were crossed with animals expressing the Cre-deleter under the control of the Tie-2 promoter (B6.Cg-Tg(Tek-cre)12Flv/J; Jackson Laboratories, Bar Harbor, ME) to generate mice lacking the α2 subunit in endothelial cells and some myeloid cells (Tie2-α2), with VE-cadherin-Cre mice (B6-Tg(Cdh5-cre)/J; Polygene, Switzerland) to generate animals mice lacking the α2 subunit specifically in endothelial cells (AMPKα2ΔEC), or with LysM-Cre mice (B6.129P2-Lyz2tm1(cre)Ifo/J; Jackson Laboratories) to generate mice lacking the α2 subunit specifically in myeloid cells (AMPKα2ΔMC).
All animals were housed in conditions that conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23). Both the University Animal Care Committee and the Federal Authority for Animal Research at the Regierungspräsidium Darmstadt (Hessen, Germany) approved the study protocol (#F28/25). For the isolation of organs, mice were euthanized using 4% isoflurane in air and subsequent exsanguination or decapitation.
Hindlimb Ischemia
Arteriogenic and angiogenic capacity was investigated in a murine model of hindlimb ischemia using 6- to 8-week-old wild-type and transgenic animals as described.11
Statistical Analysis
Data are expressed as mean±SEM. Statistical evaluation was performed using Student t test for unpaired data, 1-way ANOVA followed by a Bonferroni t test, or ANOVA for repeated measures where appropriate. Values of P<0.05 were considered statistically significant.
Reagents and other detailed methods are described in the Online Data Supplement.
Results
Effects of AMPKα2 Deletion on Vascular Repair
Diabetes mellitus is not only a risk factor for the development of cardiovascular disease but is linked with impaired vascular repair after injury and ischemia.12,13 Given that the AMPKα2 subunit was reported to be a regulator of whole body insulin sensitivity,14 vascular repair after femoral artery ligation was assessed in wild-type and AMPKα2−/− mice. The recovery of blood flow in ischemic hindlimbs was markedly attenuated in AMPKα2−/− mice compared with their wild-type littermates over 14 days (Figure 1A). The restoration of hindlimb perfusion is dependent on arteriogenesis and angiogenesis, and micro computed tomography analyses revealed a clearly impaired arteriogenesis, defined as the development of remodeled collaterals with a corkscrew morphology, in the AMPKα2−/− mice (Figure 1B). The delayed functional recovery was also reflected in the decreased capillary density in the calf muscles 14 days after surgery (Figure 1C).
Figure 1.
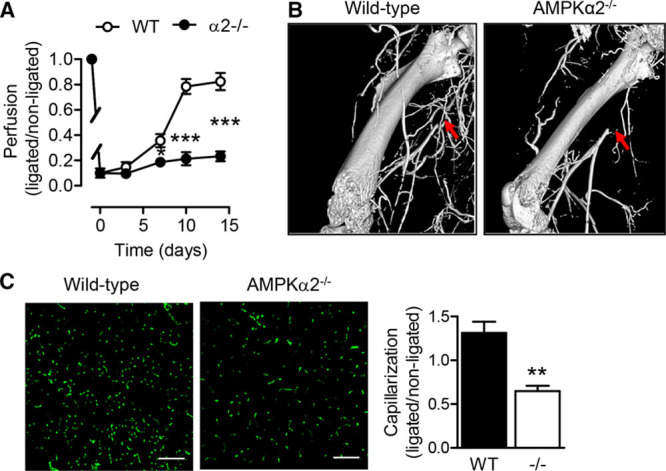
Recovery of blood flow and revascularization in ischemic hindlimbs after femoral artery ligation in diabetic and AMPKα2−/− mice. A, Reperfusion after femoral artery ligation as measured by laser Doppler analysis in wild-type (WT) and AMPKα2−/− littermates (n=9 per group). B, Micro computed tomography angiography of ligated limbs from WT and AMPKα2−/− mice 7 d post surgery. Arrows indicate site of ligation. Similar results were obtained in an additional 2 animals in each group. C, CD31+ capillaries in transverse sections of ischemic gastrocnemius muscles from WT and AMPKα2−/− mice (bar=100 µm; n=4 per group). *P<0.05, **P<0.01, ***P<0.001 vs control group. AMPK indicates AMP-activated protein kinase.
AMPKα2 in Endothelial Versus Myeloid Cells
Arteriogenesis and angiogenesis are determined by endothelial cells and circulating cells.4 To determine whether the reduced recovery after hindlimb ischemia was dependent on the deletion of AMPKα2 in endothelial cells or other cell types, experiments were repeated in AMPKα2flox/flox mice crossed with mice expressing the Cre-deleter under the control of the Tie-2 promoter (Tie2-α2 mice) and their wild-type littermates. Although there was delayed recovery of perfusion in the Tie2-α2 versus wild-type mice (Figure 2A), the effect was much less impressive than that in the global knockout animals. Although the Tie-2 promoter is frequently used to target endothelial cells, it is also expressed by a subset of myeloid cells.15,16 Therefore, endothelial cells were more specifically targeted by crossing AMPKα2flox/flox mice with mice expressing the Cre-deleter under the control of the VE-cadherin promoter. However, the endothelial cell–specific deletion of the AMPKα2 had no effect on the recovery from hindlimb ischemia (Figure 2B).
Figure 2.
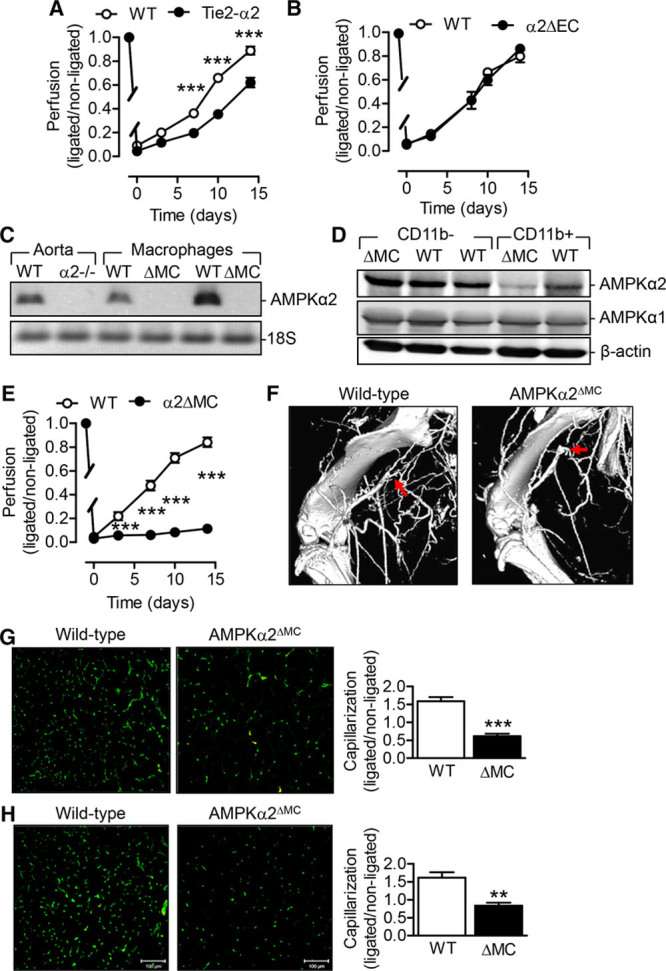
Comparison of the consequences of endothelial cell and myeloid cell AMP-activated protein kinase (AMPK) α2 deletion on vascular repair. A and B, Recovery of blood flow after femoral artery ligation in hindlimbs from (A) wild-type (WT), Tie2-α2 littermates (n=8 per group), and (B) WT and AMPKα2ΔEC littermates (n=4 per group). C, AMPKα2 mRNA expression in aorta and macrophages isolated from WT, AMPKα2−/− or AMPKα2ΔMC mice. Similar results were obtained in 2 additional experiments. D, AMPKα2 and AMPKα1 protein expression in CD11b− and CD11b+ cells isolated from spleens from WT and AMPKα2ΔMC littermates. Similar results were obtained in 2 additional experiments. E, Recovery of blood flow after femoral artery ligation in hindlimbs from WT and AMPKα2ΔMC mice (n=9 per group). F, Micro computed tomography angiography of ligated limbs from WT and AMPKα2ΔMC mice, 7 d post surgery. Arrows indicate site of ligation. G and H, Anti-CD31 immuno-stained capillaries (green) in the semimembranosus (G) and gastrocnemius (H) muscles of ligated limbs from WT and AMPKα2ΔMC mice 14 d after surgery. The quantification was performed relative to the nonligated limb (n=5 per group). *P<0.05, **P<0.01, ***P<0.001 vs WT.
As the results indicated that the AMPKα2 in mononuclear cells may explain the observations, the expression of the AMPKα2 subunit was assessed in myeloid cells. AMPKα2 mRNA was clearly expressed in the aorta and in peritoneal macrophages from wild-type mice (Figure 2C). Crossing AMPKα2flox/flox mice with LysM-Cre mice, to generate animals lacking the AMPKα2 in myeloid cells (AMPKα2ΔMC mice), resulted in the loss of AMPKα2 mRNA expression in macrophages (Figure 2C) and protein expression in CD11b+ (myeloid lineage) cells (Figure 2D). However, the CD11b− cell population from the same animals did express AMPKα2, confirming the specificity of the LysM-Cre–dependent deletion.
The myeloid cell–specific deletion of AMPKα2 resulted in a decrease in circulating neutrophils under basal conditions that could not be explained by altered hematopoietic stem cell differentiation or mobilization. The deletion of the AMPKα2 in myeloid cells did not alter the composition of hematopoietic stem cell populations or the more committed lineages in bone marrow from untreated mice or after treatment with granulocyte colony-stimulating factor (Online Figure IA). Neither the proliferation nor the differentiation of mobilized progenitors was altered 5 days after granulocyte colony-stimulating factor administration as assessed by using a colony-forming unit assay (Online Figure IB). These findings fit with the reported lack of effect of AMPK deletion on normal hematopoietic stem cell function.17
AMPKα2ΔMC mice reproduced the phenotype of the AMPKα2−/− mice in that only minimal recovery of hindlimb blood flow was observed over 14 days after the induction of ischemia (Figure 2E). Fitting with the delayed recovery of blood flow, arteriogenesis was less pronounced in the AMPKα2ΔMC mice compared with their wild-type littermates. The latter animals developed remodeled collaterals that largely failed to form in the AMPKα2ΔMC mice (Figure 2F). Angiogenesis, assessed as the number of capillaries (CD31+ cells) in the semimembranosus muscle (Figure 2G) and gastrocnemius muscle (Figure 2H), was also markedly decreased in ischemic hindlimbs from AMPKα2ΔMC mice. The AMPKα1 subunit is more highly expressed than the AMPKα2 isoform in myeloid cells. Despite this, the deletion of the AMPKα2 subunit attenuated neutrophil AMPK phosphorylation on the activating Thr172 site (the phospho-specific antibody used cannot differentiate between the AMPKα1 and α2 isoforms) under basal conditions and in cells exposed to hypoxia (Online Figure IIA). To confirm the importance of the AMPKα2 isoform in vascular repair, we also generated a myeloid cell–specific AMPKα1-deficient mouse. Although vascular repair after ischemia was attenuated in mice lacking the α1 subunit in myeloid cells, the effect was much less pronounced than that seen in AMPKα2ΔMC mice (compare Online Figure IIB and Figure 2E).
Altered Recruitment of Myeloid Cells to Ischemic Hindlimbs in AMPKα2ΔMC Mice
As the rapid recruitment of myeloid cells to the ischemic tissue has been implicated in arteriogenesis and angiogenesis, the recruitment of the different cell populations was studied 3 days after the induction of ischemia.
Using hematoxylin–eosin staining, distinctly fewer infiltrating cells were detected in ischemic muscles from AMPKα2ΔMC mice compared with their wild-type littermates (Figure 3A). In ischemic muscles from wild-type animals, leukocytes (CD45+ cells), neutrophils (Gr1+ cells), and macrophages (F4/80+ cells) were abundant but were significantly lower in the AMPKα2ΔMC group (Figure 3B). Infiltrating cells have a significant impact on the local environment in the ischemic skeletal muscle, and analysis of mRNA expression of a wide range of angiogenic markers and inflammatory chemokines and cytokines in the ischemic gastrocnemius muscle revealed pronounced differences (Figure 3C; Online Figure III). For example, levels of transforming growth factor (TGF)β, VEGFA and VEGFB, and the VEGF receptor 1 (VEGFR1) and VEGFR2 were lower in ischemic muscles from AMPKα2ΔMC mice compared with their wild-type littermates. On the contrary, the expression of several inflammatory mediators and chemokines were upregulated in ischemic muscles from AMPKα2ΔMC mice including tumor necrosis factor-α, interleukin (IL)-1β, and chemokine (C-C motif) ligand 2 (CCL2), also known as monocyte chemoattractant factor 1 (MCP-1) and its receptor C-C chemokine receptor type 2 (CCR2). The latter also coincided with endothelial cell activation as indicated by the upregulation of intercellular adhesion molecule-1, which was confirmed by immunohistochemistry (Figure 3D). This altered gene expression pattern was consistently observed in the gastrocnemius and in the semimembranosus muscles (Online Figure III).
Figure 3.
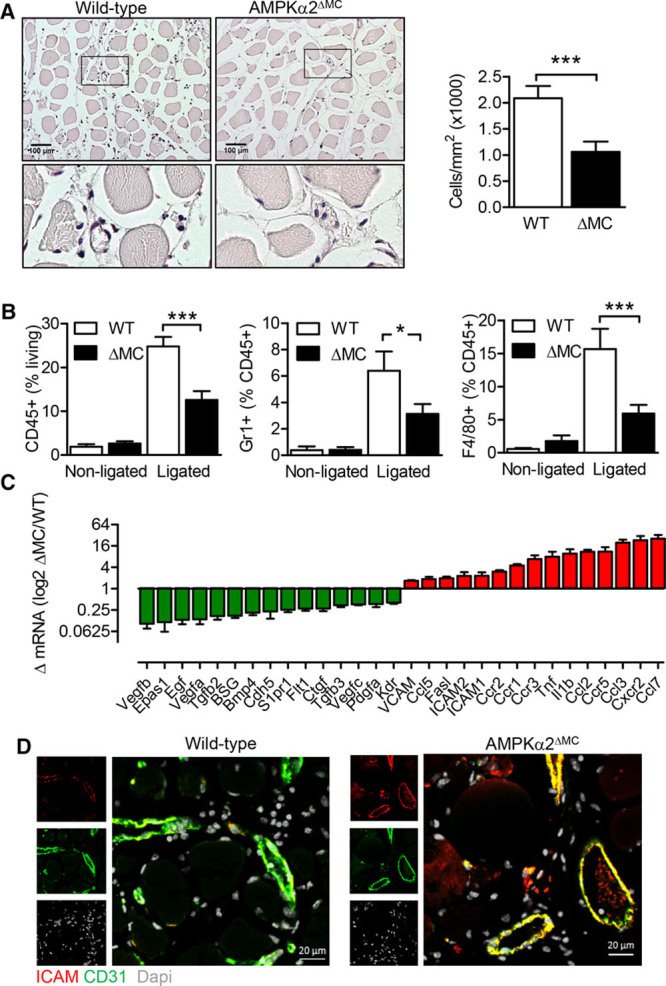
Myeloid cell infiltration into the ischemic hindlimb 72 h post surgery. A, Representative hematoxylin–eosin images and quantification of infiltrated cells into gastrocnemius muscle. Bar=100 µm. The graph summarizes data from 5 animals per group. B, CD45+, Gr1+, and F4/80+ cells isolated from the gastrocnemius muscle of the ligated and nonligated hindlimbs from wild-type (WT) and AMPKα2ΔMC (ΔMC) 3 d after ligation (n=5 per group). C, Chemokine and cytokine gene expression analysis in ischemic hindlimbs. Green bars show significantly (P<0.05) downregulated genes, and red bars indicate significantly upregulated genes in AMPKα2ΔMC compared with WT mice (n=4 animals in each group). D, Intercellular adhesion molecule (ICAM)-1 and CD31 immunostaining in the gastrocnemius muscle. Bar=20 µm. *P<0.05, **P<0.01, ***P<0.001. AMPK indicates AMP-activated protein kinase.
To determine whether myeloid cell recruitment to the ischemic muscle or myeloid cell survival in the hypoxic environment was altered by the deletion of the AMPKα2 subunit, experiments were repeated 1 day after surgery. Within 24 hours of ischemia, leukocyte infiltration was similar in the ischemic hindlimbs from wild-type and AMPKα2ΔMC mice (Figure 4A). However, a significantly higher number of neutrophils were recruited to the gastrocnemius muscle of AMPKα2ΔMC mice than their wild-type littermates (Figure 4B). At this early time point, no macrophages were consistently detected in the ischemic muscles. In in vitro migration assays, neutrophils from AMPKα2ΔMC mice more effectively migrated through an endothelial cell monolayer (Figure 4C) even though directed migration toward a cytokine, the murine IL-8 homologue CXCL1, did not differ between the 2 strains (data not shown). Confocal microscopy confirmed the elevated neutrophil infiltration into ischemic muscles from AMPKα2ΔMC mice 24 hours after surgery (Figure 4D). In these samples, intra- and extravascular neutrophils were easier to detect in whole mounts from AMPKα2ΔMC mice versus their wild-type littermates, indicating an improved transendothelial migration also in vivo (Online Figure IV). Moreover, although circulating numbers of neutrophils were lower under basal conditions, they were comparable in wild-type and AMPKα2ΔMC littermates 24 hours after the induction of ischemia, indicating a lack of a defect in mobilization (Online Figure V). Taken together, these data point out that, compared with responses in wild-type littermates, the response to ischemia in hindlimbs from AMPKα2ΔMC mice is linked to an increase in early neutrophil infiltration, the upregulation of inflammatory cytokines, and adhesion molecule expression followed by a later (72 hours) decline in neutrophil numbers.
Figure 4.
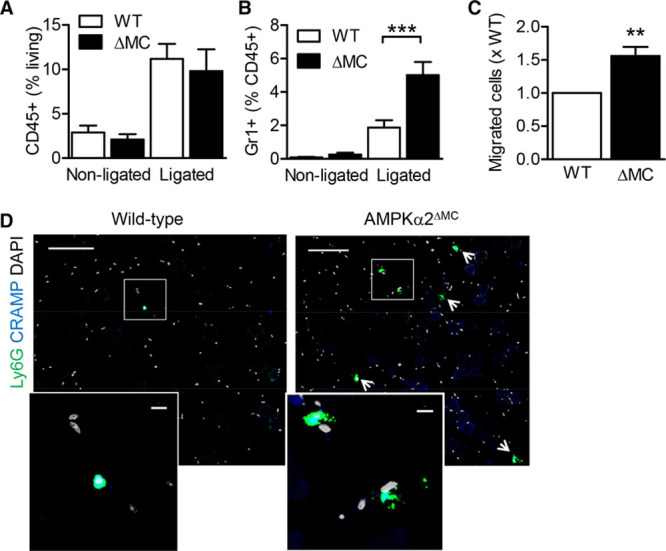
Myeloid cell infiltration into the ischemic hindlimb 24 h post surgery. A and B, CD45+ and Gr1+ cells isolated from the gastrocnemius muscle of the ligated and nonligated hindlimbs from wild-type (WT) and AMPKα2ΔMC (ΔMC) mice 24 h after ligation (n=6 per group). C, In vitro stromal cell–derived factor-1–induced transendothelial migration of bone marrow–derived neutrophils from WT and AMPKα2ΔMC mice (n=4–6 per group). D, Ly6G+ and CRAMP+ neutrophils in ischemic semimembranosus muscles from WT and ΔMC mice 1 d post surgery. The images are representative of data obtained in an additional 3 animals per group. Upper: bar=200 µm; lower: bar=10 µm. *P<0.05, **P<0.01, ***P<0.001 vs WT. AMPK indicates AMP-activated protein kinase.
AMPKα2 Regulates Neutrophil Survival Under Hypoxia
Circulating neutrophils are short-lived cells, but their life span can be prolonged under hypoxic conditions. The decreased recovery of neutrophils from ischemic hindlimbs 3 days after ligation hinted at an increased apoptosis of AMPKα2ΔMC neutrophils. To determine which AMPKα2-regulated proteins were altered by hypoxia, bone marrow–derived neutrophils were isolated from wild-type and AMPKα2ΔMC littermates and maintained under normoxic or hypoxic conditions for 16 hours. Thereafter, altered protein expression was determined by mass spectrometry. Online Table I summarizes all of the proteins identified, number of identified peptides, accession numbers, and sequence coverage of each sample. The quantification of the results is summarized in Online Table II.
Fitting with the in vivo observations, neutrophils from AMPKα2ΔMC mice expressed lower levels of the antiapoptotic proteins: translocase of inner mitochondrial membrane 50 and apoptosis inhibitor 5 (Figure 5A and 5B). These changes were accompanied by an increase in caspase-3 cleavage and Bax expression (Figure 5C) and an increase in caspase activity in neutrophils lacking AMPKα2 (Figure 5D). Thus, the expression of the AMPKα2 in neutrophils is required for protection against apoptosis in hypoxic conditions.
Figure 5.
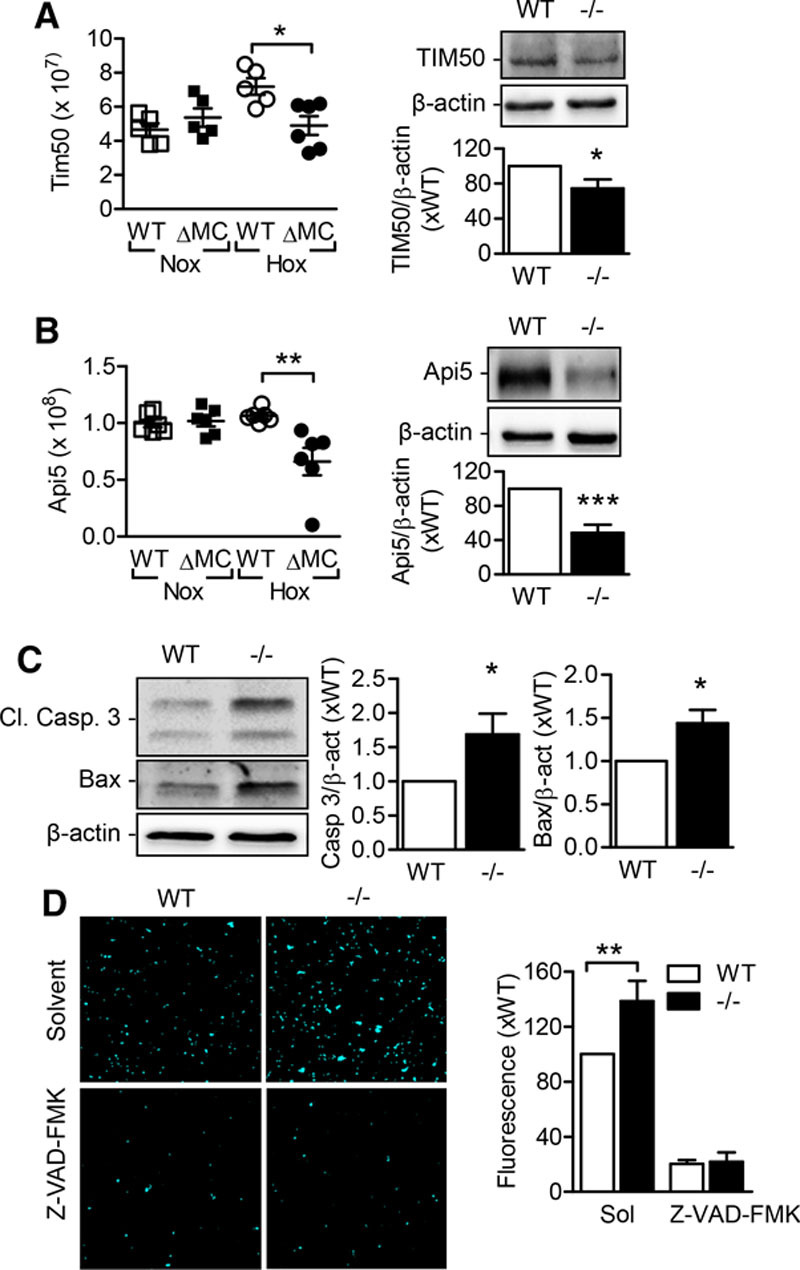
Effect of AMP-activated protein kinase (AMPK) α2 deletion on neutrophil apoptosis. Neutrophils from wild-type (WT) and AMPKα2ΔMC (ΔMC) littermates were incubated under normoxic (Nox) or hypoxic (Hox) conditions for 16 h (n=6 per group). A, Translocase of inner mitochondrial membrane 50 (TIM50) label-free quantification (LFQ) intensity (left) and Western blot of neutrophils maintained under hypoxic conditions (right). B, Api5 LFQ intensity (left) and Western blot of neutrophils maintained under hypoxic conditions (right). C, Expression of cleaved caspase-3 (Cl. Casp. 3) and Bax in bone marrow–derived neutrophils from wild-type (WT) and AMPKα2ΔMC (ΔMC) littermates (n=8 per group for Cl. Casp. 3 and n=5 per group for Bax expression). D, Caspase-3/7 activity measured as DEVD (7-amino-4-trifluoromethyl coumarin) fluorescence after cleavage from Asp-Glu-Val-Asp in neutrophils from WT and ΔMC littermates (n=5 per group). Cells were incubated with solvent or the caspase inhibitor (Z-VAD-FMK, 10 µmol/L; n=5 per group). *P<0.05, **P<0.01, ***P<0.001.
AMPKα2 Regulates Hypoxia-Inducible Factor-1α Hydroxylation and Stabilization and Hypoxia-Inducible Factor-1α–Dependent Responses in AMPKα2-Deficient Neutrophils
The most prominent difference in the proteome of neutrophils from the 2 strains was that the neutrophils lacking AMPKα2 demonstrated an impaired response to hypoxia (Figure 6A; Online Figure VI). This finding fits with reports that myeloid cell function is strongly dependent on and mediated by hypoxia-inducible factor (HIF)-1α,18 which is crucial for prolonging neutrophil survival under hypoxic conditions.19
Figure 6.
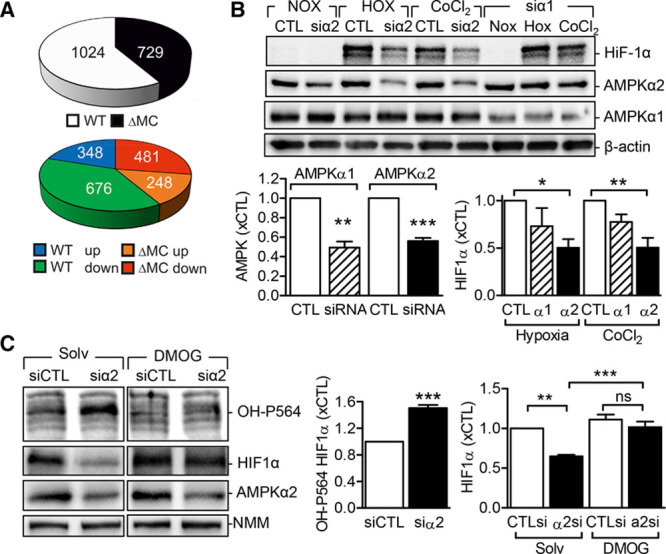
Link between AMP-activated protein kinase (AMPK) α2 and hypoxia-inducible factor-1α (HIF-1α). A, Pie charts depicting significantly regulated proteins in neutrophils from wild-type (WT) and AMPKα2ΔMC (ΔMC) mice after 16-h exposure to hypoxia. Upper: total number of significantly regulated proteins. Lower: number of significantly up- and downregulated proteins. B, HIF-1α levels in HEK293 cells transfected with a control siRNA (CTL) or siRNA directed against AMPKα1 (siα1) or AMPKα2 (siα2) and then either maintained under normoxic conditions, exposed to hypoxia (1% O2, 6 h) or stimulated with CoCl2 (150 µmol/L, 6 h). Graphs summarize data from 5 independent experiments. C, HEK293 cells transfected with a control siRNA (siCTL) or siRNA-directed against AMPKα2 (siα2) were exposed to hypoxia in the presence of solvent (Solv) or dimethyloxalylglycine (DMOG; 1 mmol/L) for 16 h. HIF-1α levels and its hydroxylation on proline 564 (OH-P564) were analyzed by Western blotting (n=9–12 per group). The break in the blots indicates that nonadjacent lanes in the same Western blot are presented. *P<0.05, **P<0.01, ***P<0.001.
HIF-1α protein levels can be modified by several kinases,20 and previous studies in a prostate cancer cell line indicated that global AMPK inhibition may attenuate hypoxia-induced responses such as HIF-1α target gene expression.21 Therefore, we determined whether or not AMPK can affect HIF-1α stability in HEK293 cells, which express both the AMPKα1 and α2 subunits. In cells maintained under normoxic conditions, HIF-1α levels were not detectable, but the protein was clearly induced after exposure of cells to hypoxia (1% O2, 6 and 16 hours) or cobalt chloride (150 µmol/L, 6 hours; Figure 6B). Although the downregulation of the AMPKα1 subunit had little effect on the induction of HIF-1α by either stimulus, HIF-1α levels were consistently lower in HEK293 cells treated with small interfering RNA directed against the AMPKα2 subunit. The serine phosphorylation of HIF-1α has been linked with its destabilization22,23; therefore, we determined the ability of AMPKα2 to phosphorylate a recombinant HIF-1α peptide (corresponding to amino acids 1–735) in vitro. We found that AMPKα2 was able to elicit the serine but not the threonine phosphorylation of HIF-1α (Online Figure VII); however, we were unable to confirm the AMPK-mediated phosphorylation of HIF-1α in isolated neutrophils.
HIF-1α protein levels are barely detectable in normoxic conditions largely because of the high activity of the prolyl hydroxylase domain (PHD)–containing enzymes, which hydroxylate HIF-1α (on Pro564) thus promoting its subsequent proteasome-mediated degradation.24 In response to hypoxia, PHD enzymes and the degradation of HIF-1α are inhibited. Interestingly, the downregulation of AMPKα2 enhanced the hydroxylation of HIF-1α on proline 564 in cells exposed to hypoxia, indicating that the effect of AMPKα2 on HIF-1α may be indirectly regulated via PHD activity. Indeed, treatment with the PHD inhibitor dimethyloxalylglycine restored HIF-1α levels in cells treated with AMPKα2 siRNA (Figure 6C). This implies that AMPKα2 regulates HIF-1α stabilization under hypoxia by interfering with its hydroxylation and thereby inhibiting its proteasomal degradation. Consistent with the observed effects on HIF-1α destabilization, the expression of the hypoxia-regulated genes, VEGF and stromal cell–derived factor-1, was decreased in lipopolysaccharide-stimulated neutrophils from AMPKα2ΔMC mice (Figure 7A). In the same cells, the expression of the proinflammatory markers, tumor necrosis factor-α and IL-1β, was increased (Figure 7B) and fit well with the altered cytokine and growth factor expression profile detected in the ischemic muscles (Figure 3C). Neutrophil recruitment to ischemic tissues involves several secreted factors including cathelicidin-related antimicrobial peptide (LL-37 in humans) and CD147 (basigin), both of which were increased in hypoxic neutrophils from wild-type mice but not in those lacking the α2 subunit (Figure 7C and 7D).
Figure 7.

Hypoxic response in neutrophils and mitochondrial metabolism. A and B, Bone marrow neutrophils were isolated from wild-type (WT) and AMPKα2ΔMC (ΔMC) littermates, and the expression of the angiogenic (VEGF and stromal cell–derived factor-1 [SDF-1]) and inflammatory markers (tumor necrosis factor [TNF]-α and interleukin [IL]-1β) were analyzed. The graphs summarize data from 8 to 9 individual animals in each group from 3 independent experiments. C and D, Neutrophils from WT and ΔMC littermates were incubated under normoxic (Nox) or hypoxic (Hox) conditions for 16 h (n=6 per group). C, cathelicidin-related antimicrobial peptide (CRAMP) protein expression (label-free quantification [LFQ] intensity). D, CD147 protein levels (LFQ intensity) and immunohistochemistry, CD147 (red), and phalloidin (green). Bar=10 µm. E, Matrix metalloproteinase (MMP)-9 mRNA expression (n=4 per group). F, Oxygen consumption rate (OCR) profile as an index of mitochondrial respiration in bone marrow–derived neutrophils from WT and ΔMC mice. G, DCHF (2′-7′-dichlorofluorescin) fluorescence in bone marrow–derived neutrophils from WT and ΔMC mice treated with solvent (Sol) or PMA (phorbol 12-myristate 13-acetate; 100 ng/mL) for 20 min (n=7–8 per group). *P<0.05, **P<0.01, ***P<0.001. AMPK indicates AMP-activated protein kinase.
AMPKα2 Regulates HIF-1α Hydroxylation by Modulating α-Ketoglutarate Levels
Because HIF-1α also regulates the cellular metabolic state, we analyzed metabolic parameters in isolated neutrophils. Under normoxic conditions, the deletion of AMPKα2 in neutrophils was associated with increased oxygen consumption, both in the absence of stimulation and when maximally stimulated with FCCP (Figure 7F; Online Figure VIIIA through VIIIE). The latter fits well with an increased phorbol ester–induced generation of reactive oxygen species (Figure 7G). There was no difference in glycolysis in neutrophils from the 2 strains under the conditions studied (Online Figure VIIIF). Hypoxia is a potent activator of AMPK, and because the Seahorse analyses could not be performed under hypoxic conditions, mass spectrometry was used to assess metabolic alterations in hypoxic neutrophils. Consistent with the extracellular acidification data, there were no detectable differences in glucose or lactate levels between the 2 strains of neutrophils maintained in normoxic or hypoxic conditions (Online Figure VIIIG through VIIIJ).
Proteomic analysis revealed a significant, hypoxia-induced decrease in 2 isoforms of the isocitrate dehydrogenase (IDH), that is, cytosolic IDH1 and mitochondrial IDH3b (Figure 8A and 8B). In the same cells, metabolomic analyses revealed that although the IDH product α-ketoglutarate decreased in wild-type neutrophils exposed to hypoxia, it failed to do so in hypoxic neutrophils from AMPKα2ΔMC mice (Figure 8C). α-Ketoglutarate is required as a cofactor for PHD enzymes and the subsequent hydroxylation of HIF-1α. The hypoxia-induced decrease in α-ketoglutarate production prevents the hydroxylation and degradation of HIF thus facilitating the induction of HIF-1α–regulated genes. Certainly, incubating human leukemic monocyte THP-1 cell line with octyl-α-ketoglutarate, a cell-permeable ester of α-ketoglutarate, was sufficient to abrogate HIF-1α expression in cells exposed to hypoxia (Figure 8D).
Figure 8.
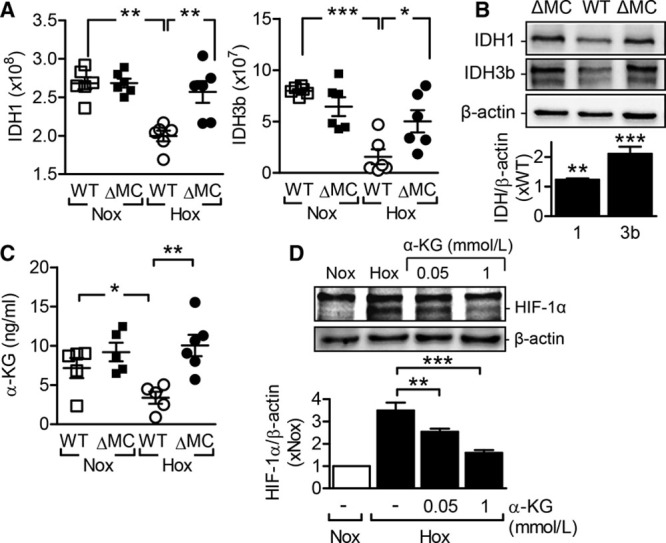
AMP-activated protein kinase (AMPK) regulates levels of isocitrate dehydrogenases and α-ketoglutarate. Neutrophils from wild-type (WT) and AMPKα2ΔMC (ΔMC) littermates were incubated under normoxic (Nox) or hypoxic (Hox) conditions for 16 h (n=6 per group). A, Isocitrate dehydrogenase (IDH) 1 and IDH3b expression (label-free quantification intensity). B, Western blot confirmation of altered IDH1 and IDH3b expression. C, α-Ketoglutarate (α-KG) levels in neutrophils from WT and ΔMC littermates incubated under Nox or Hox conditions for 16 h (n=5–6 per group). D, HIF-1α levels in THP-1 cells incubated in Hox conditions for 6 h with indicated concentrations of octyl-α-KG, a cell-permeable ester of α-KG (n=6 independent experiments). *P<0.05, **P<0.01, ***P<0.001.
Discussion
The results of the present study indicate that the AMPKα2 subunit is important in determining neutrophil survival and function in ischemic tissue. The mechanisms involved are related to the stabilization of HIF-1α protein by preventing its hydroxylation under hypoxia and the expression of a network of proteins—many of which affect mitochondrial function and cell survival. In the absence of these mechanisms, neutrophil survival is attenuated, and the recruitment of proangiogenic monocytes to ischemic areas is reduced, thus impairing the processes of arteriogenesis and angiogenesis (Online Figure IX).
Limb ischemia as a consequence of vascular injury initiates a series of events that involve an inflammatory phase, characterized by cell infiltration and the initiation of angiogenesis, followed by a resolution phase. The processes involved are complex, but many pharmaceutical tools, such as metformin, have been reported to improve vascular repair, at least in wild-type mice.25 One key signaling event that is altered in diabetes mellitus and targeted by metformin is the activation of the AMPK.26 To determine the importance of the AMPK in vascular repair, the recovery of blood flow after ischemia was assessed in wild-type and AMPKα-deficient mice. The focus of the studies was the AMPKα2 subunit, given its reported link to diabetes mellitus14 and its activation by hypoxia.21 We found that the recovery of blood flow was significantly impaired in ischemic hindlimbs from animals completely deficient in the AMPKα2 subunit. Interestingly, targeting AMPKα2 in endothelial cells did not impair vascular repair, whereas its deletion in myeloid cells reproduced the effects observed in the AMPKα2−/− mice. Certainly, the AMPKα2 isoform was clearly expressed in CD11b+ cells, and in animals lacking AMPKα2 in the myeloid linage (AMPKα2ΔMC mice), arteriogenesis and angiogenesis in the ischemic hindlimb were substantially reduced. The myeloid cell–specific deletion of the AMPKα1 subunit also delayed the recovery of blood flow after femoral artery ligation, but the defect was much less pronounced than that in the AMPKα2ΔMC mice. Although in our study the role of AMPKα2 deletion in neutrophils was more pronounced, it is possible that the AMPKα1 isoform plays a significant role in the regulation of monocyte function as described in an article published during the revision of this article.27
Consistent with a previous report that the deletion of the AMPK had no effect on normal hematopoietic stem cell differentiation or function,17 the myeloid cell–specific deletion of the AMPKα2 was without effect on hematopoietic stem cell differentiation. Interestingly, circulating levels of neutrophils were attenuated by the deletion of the AMPKα2 under basal conditions but were comparable in wild-type and AMPKα2ΔMC mice 24 hours after femoral artery ligation indicating normal neutrophil generation and mobilization.
Although neutrophils are early infiltrating cells that control critical steps during angiogenesis, collateral growth, and perfusion recovery, they have a short half-life within the ischemic tissue.28,29 Comparing the numbers of neutrophils that infiltrated ischemic skeletal muscle 1 and 3 days after femoral artery ligation revealed that despite increased neutrophil infiltration in ischemic AMPKα2ΔMC hindlimbs after 24 hours, the situation was reversed after 72 hours. These observations indicated that neutrophil survival was attenuated in the absence of the AMPKα2 subunit. Certainly, AMPK activation has previously been linked with neutrophil survival,30 and in this study, apoptosis was increased in AMPKα2-deficient neutrophils. Some of the proteins significantly altered in hypoxic AMPKα2ΔMC versus wild-type neutrophils were linked to apoptosis, in particular translocase of inner mitochondrial membrane 50, a component of the mitochondrial translocator,31 and apoptosis inhibitor 5, a negative regulator of E2F1-dependent cell death.32 Little is known about the role played by either protein in neutrophils, but the loss of translocase of inner mitochondrial membrane 50 has also been reported to increase apoptosis in breast cancer cells.33
Part of the arteriogenic/angiogenic response can be attributed to the liberation of neutrophil-derived VEGF, matrix metalloproteinases, and other proangiogenic growth factors,34 but these cells also release factors such as cathelicidins that promote the recruitment of monocytes,35,36 which then amplify the angiogenic and arteriogenic processes. Thus, the observed decrease in monocyte recruitment to the ischemic tissue 72 hours after ligation is likely to be linked to neutrophil loss. The same mechanism likely underlies the decreased expression of proangiogenic factors in hindlimbs from AMPKα2ΔMC mice. How can the increase in neutrophil infiltration at 24 hours be accounted for? This may be related a combination of enhanced neutrophil migratory activity and crosstalk between the neutrophils and the vasculature. Certainly, the expression of adhesion molecules such as intercellular adhesion molecule-1 was increased in hindlimbs from AMPKα2ΔMC mice, hinting that endothelial cell activation was enhanced. This is a neutrophil-dependent effect as endothelial cells from wild-type and AMPKα2ΔMC mice responded to IL-1β and lipopolysaccharide stimulation with similar increases in adhesion molecule expression (R. Abdel Malik et al, unpublished data, 2016).
The inhibition of neutrophil apoptosis in a hypoxic environment is dependent on the activation of HIF-1α,19 and interactions between HIF-1α and AMPK have been reported. Indeed, increased AMPKα expression and activity are paralleled by the upregulation of HIF-1α in cancer cells,37 and the inhibition of AMPK was found to impair the nuclear accumulation of HIF-1α under hypoxia or low glucose conditions.38 We found that the downregulation of the AMPKα2 but not the AMPKα1 subunit attenuated the increase in HIF-1α levels induced by either hypoxia or cobalt chloride. In agreement with our in vitro observations, the expression of the HIF-1α–regulated factors, stromal cell–derived factor-1 and VEGF, was attenuated in neutrophils from AMPKα2ΔMC mice. AMPK has also been proposed to regulate HIF-1α protein stability by phosphorylation.23 However, even though we could confirm these findings in an in vitro assay, we were unable to detect an AMPKα2-dependent phosphorylation of HIF-1α in intact cells. Instead, it was possible to demonstrate that the deletion of the AMPKα2 subunit enhanced the hydroxylation of HIF-1α on Pro564 in cells exposed to hypoxia. The latter response is indicative of increased PHD activity and proteasomal degradation. Indeed, treating AMPKα2-deficient neutrophils with a PHD inhibitor prevented the enhanced hydroxylation and rescued HIF-1α induction.
Unlike the AMPKα1 subunit, the AMPKα2 can translocate to the nucleus and affect gene and protein expression by several mechanisms.1 Mass spectrometry was used to ascertain which neutrophil proteins could be affected by AMPKα2 deletion and revealed that although wild-type neutrophils exposed to hypoxia increased expression of the granule protein cathelicidin-related antimicrobial peptide (LL-37 in humans) and CD147 (basigin or extracellular matrix metalloproteinase inducer), effects were blunted in AMPKα2ΔMC neutrophils. The latter proteins were of interest as both are regulated by HIF-1α39,40 and both are involved in neutrophil recruitment, invasion, and survival in ischemic tissues.35,41–43 Of interest for vascular repair is that CD147 is reportedly required for the increased synthesis of VEGF and matrix metalloproteinase-944,45; thus, the lower levels of matrix metalloproteinase-9 in the AMPKα2ΔMC neutrophils may be related to their failure to increase CD147 expression.
Prolyl hydroxylases require oxygen and the electron donor α-ketoglutarate to execute the hydroxylation and inactivation of HIF-1α.46 Neutrophil adaption to hypoxia and the stabilization of HIF-1α, therefore, require accompanying changes in metabolism and a decrease in α-ketoglutarate production. Isocitrate dehydrogenases catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate, and 2 IDH isoforms, that is, IDH1 and IDH3b, were differentially regulated by hypoxia in neutrophils from wild-type and AMPKα2ΔMC mice. Specifically, the decrease in IDH expression detected in wild-type neutrophils exposed to hypoxia failed to occur in AMPKα2-deficient neutrophils. The changes in IDH expression were reflected in α-ketoglutarate levels, which were decreased by hypoxia in wild-type neutrophils but remained elevated in neutrophils from AMPKα2ΔMC mice. It is interesting to note that higher IDH1 levels have been linked with increased HIF-1α degradation and the sensitization of melanoma cells to hypoxia-induced apoptosis,47 effects that parallel the fate of AMPKα2-deficient neutrophils. Moreover, inhibition of IDH by oxalomalic acid or IDH knockdown was reported to elevate HIF-1α levels.48 It, therefore, seems that the links between AMPKα2 and HIF-1α are complex and may be regulated at more levels than the direct phosphorylation of the transcription factor.
Taken together, these observations indicate that the activation of AMPKα2 in neutrophils is a decisive event in the initiation of vascular repair after ischemia. It is tempting to speculate that dysregulation of AMPK signaling, such as is the case in diabetes mellitus, may attenuate vascular repair by a combination of effects on HIF-1α stability mediated by alterations in phosphorylation and myeloid cell metabolism.
Acknowledgments
We are indebted to Isabel Winter, Mechtild Piepenbrock-Gyamfi, and Katharina Engel-Herbig for expert technical assistance.
Sources of Funding
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 834/A5 to B. Fisslthaler and I. Fleming, SFB 834/Z1 to M.A. Rieger, SFB 815/Z1 to I. Wittig, SFB 914/A2 to B. Walzog, “Exzellenzcluster 147 “Cardio-Pulmonary Systems” and Exzellenzcluster 115 “Macromolecular Complexes”). M.A. Rieger was also supported by the LOEWE Center for Cell and Gene Therapy (HMWK III L 4–518/17.004 [2013]).
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- AMPK
- AMP-activated protein kinase
- HIF
- hypoxia-inducible factor
- IDH
- isocitrate dehydrogenase
- IL
- interleukin
- PHD
- prolyl hydroxylase domain
- VEGF
- vascular endothelial growth factor
In September 2016, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12.73 days.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.116.309937/-/DC1.
Novelty and Significance
What Is Known?
Myeloid cells are rapidly recruited to ischemic tissue and are required for the initiation of vascular repair.
Neutrophils are the first cell population to infiltrate ischemic tissue and secrete factors that recruit a second wave of monocytes.
Neutrophils are short lived, but their half-life is prolonged in ischemic tissue by increased expression of the hypoxia-inducible factor (HIF)-1α.
HIF-1α protein levels are barely detectable in normoxic conditions because of the high activity of the prolyl hydroxylase domain–containing enzymes that hydroxylate HIF-1α and promote its degradation.
What New Information Does This Article Contribute?
Global deletion of the AMP-activated protein kinase (AMPKα2) subunit or its specific deletion in myeloid cells abrogate arteriogenesis and angiogenesis and prevents the recovery of hindlimb blood flow after femoral artery ligation.
Activation of the AMPKα2 subunit in neutrophils is essential for their survival in the ischemic hindlimb and subsequent monocyte recruitment.
In the absence of AMPKα2, the changes in neutrophil protein expression elicited by hypoxia are attenuated, a phenomenon attributed to the failure to stabilize HIF-1α protein expression.
In hypoxic conditions, AMPKα2 activation leads to a decrease in the expression of 2 isoforms of the isocitrate dehydrogenase and the generation of α-ketoglutarate, which is required as a cofactor for prolyl hydroxylase domain enzymes and the subsequent HIF-1α hydroxylation.
Limb ischemia, as a consequence of vascular injury, initiates a series of events that involve an inflammatory phase, characterized by cell infiltration and the initiation of angiogenesis, followed by a resolution phase. Neutrophils are early infiltrating cells that liberate several key factors that promote the subsequent recruitment of monocytes, which then amplify the arteriogenic and angiogenic processes. This study shows that the activation of AMPKα2 in neutrophils is required for arteriogenesis and angiogenesis in the ischemic hindlimb. In its absence, neutrophils fail to survive to recruit monocytes to the ischemic region, and as a consequence, the ischemic area is maintained in an inflammatory rather than an arteriogenic/angiogenic state. A combination of proteomic and metabolomic analyses show that AMPKα2 determines the response of neutrophils in an ischemic environment, partly by decreasing isocitrate dehydrogenase expression. The latter step is essential to decrease cellular levels of α-ketoglutarate and prevent the hydroxylation and degradation of HIF-1α. Diabetes mellitus is not only a risk factor for the development of cardiovascular disease but is linked with impaired vascular repair after injury and ischemia. Given that AMPK activity is dysregulated in diabetes mellitus, the rapid application of AMPK activators may promote vascular repair in diabetic individuals by a combination of effects on neutrophil metabolism and HIF-1α stability.
References
- 1.Hardie DG. AMPK–sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–952. doi: 10.1016/j.cmet.2014.09.013. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagata D, Mogi M, Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000–31006. doi: 10.1074/jbc.M300643200. doi: 10.1074/jbc.M300643200. [DOI] [PubMed] [Google Scholar]
- 3.Ouchi N, Shibata R, Walsh K. AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ Res. 2005;96:838–846. doi: 10.1161/01.RES.0000163633.10240.3b. doi: 10.1161/01.RES.0000163633.10240.3b. [DOI] [PubMed] [Google Scholar]
- 4.Jaipersad AS, Lip GY, Silverman S, Shantsila E. The role of monocytes in angiogenesis and atherosclerosis. J Am Coll Cardiol. 2014;63:1–11. doi: 10.1016/j.jacc.2013.09.019. doi: 10.1016/j.jacc.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 5.Carroll KC, Viollet B, Suttles J. AMPKα1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J Leukoc Biol. 2013;94:1113–1121. doi: 10.1189/jlb.0313157. doi: 10.1189/jlb.0313157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–L504. doi: 10.1152/ajplung.90210.2008. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wan X, Huo Y, Johns M, Piper E, Mason JC, Carling D, Haskard DO, Boyle JJ. 5’-AMP-activated protein kinase-activating transcription factor 1 cascade modulates human monocyte-derived macrophages to atheroprotective functions in response to heme or metformin. Arterioscler Thromb Vasc Biol. 2013;33:2470–2480. doi: 10.1161/ATVBAHA.113.300986. doi: 10.1161/ATVBAHA.113.300986. [DOI] [PubMed] [Google Scholar]
- 9.Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64:2028–2041. doi: 10.2337/db14-1225. doi: 10.2337/db14-1225. [DOI] [PubMed] [Google Scholar]
- 10.Zippel N, Malik RA, Frömel T, Popp R, Bess E, Strilic B, Wettschureck N, Fleming I, Fisslthaler B. Transforming growth factor-β-activated kinase 1 regulates angiogenesis via AMP-activated protein kinase-α1 and redox balance in endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:2792–2799. doi: 10.1161/ATVBAHA.113.301848. doi: 10.1161/ATVBAHA.113.301848. [DOI] [PubMed] [Google Scholar]
- 11.Frömel T, Jungblut B, Hu J, Trouvain C, Barbosa-Sicard E, Popp R, Liebner S, Dimmeler S, Hammock BD, Fleming I. Soluble epoxide hydrolase regulates hematopoietic progenitor cell function via generation of fatty acid diols. Proc Natl Acad Sci USA. 2012;109:9995–10000. doi: 10.1073/pnas.1206493109. doi: 10.1073/pnas.1206493109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howangyin KY, Silvestre JS. Diabetes mellitus and ischemic diseases: molecular mechanisms of vascular repair dysfunction. Arterioscler Thromb Vasc Biol. 2014;34:1126–1135. doi: 10.1161/ATVBAHA.114.303090. doi: 10.1161/ATVBAHA.114.303090. [DOI] [PubMed] [Google Scholar]
- 13.Yan J, Tie G, Park B, Yan Y, Nowicki PT, Messina LM. Recovery from hind limb ischemia is less effective in type 2 than in type 1 diabetic mice: roles of endothelial nitric oxide synthase and endothelial progenitor cells. J Vasc Surg. 2009;50:1412–1422. doi: 10.1016/j.jvs.2009.08.007. doi: 10.1016/j.jvs.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viollet B, Andreelli F, Jørgensen SB, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowak G, Karrar A, Holmén C, Nava S, Uzunel M, Hultenby K, Sumitran-Holgersson S. Expression of vascular endothelial growth factor receptor-2 or Tie-2 on peripheral blood cells defines functionally competent cell populations capable of reendothelialization. Circulation. 2004;110:3699–3707. doi: 10.1161/01.CIR.0000143626.16576.51. doi: 10.1161/01.CIR.0000143626.16576.51. [DOI] [PubMed] [Google Scholar]
- 16.De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201:105–115. doi: 10.1084/jem.20040624. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimova EY, Michiels C, Kietzmann T. Kinases as upstream regulators of the HIF system: their emerging potential as anti-cancer drug targets. Curr Pharm Des. 2009;15:3867–3877. doi: 10.2174/138161209789649358. [DOI] [PubMed] [Google Scholar]
- 21.Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003;278:39653–39661. doi: 10.1074/jbc.M306104200. doi: 10.1074/jbc.M306104200. [DOI] [PubMed] [Google Scholar]
- 22.Xu D, Wang Q, Jiang Y, Zhang Y, Vega-Saenzdemiera E, Osman I, Dai W. Roles of Polo-like kinase 3 in suppressing tumor angiogenesis. Exp Hematol Oncol. 2012;1:5. doi: 10.1186/2162-3619-1-5. doi: 10.1186/2162-3619-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A, Georgatsou E, Bonanou S, Simos G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem. 2006;281:33095–33106. doi: 10.1074/jbc.M605058200. doi: 10.1074/jbc.M605058200. [DOI] [PubMed] [Google Scholar]
- 24.Charpentier T, Hammami A, Stäger S. Hypoxia inducible factor 1a: a critical factor for the immune response to pathogens and Leishmania. Cell Immunol. 2016 doi: 10.1016/j.cellimm.2016.06.002. doi: 10.1016/j.cellimm.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi N, Shibata R, Ouchi N, Sugimoto M, Murohara T, Komori K. Metformin stimulates ischemia-induced revascularization through an eNOS dependent pathway in the ischemic hindlimb mice model. J Vasc Surg. 2015;61:489–496. doi: 10.1016/j.jvs.2013.09.061. doi: 10.1016/j.jvs.2013.09.061. [DOI] [PubMed] [Google Scholar]
- 26.Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 27.Zhu H, Zhang M, Liu Z, Xing J, Moriasi C, Dai X, Zou MH. AMP-activated protein kinase α1 in macrophages promotes collateral remodeling and arteriogenesis in mice in vivo. Arterioscler Thromb Vasc Biol. 2016;36:1868–1878. doi: 10.1161/ATVBAHA.116.307743. doi: 10.1161/ATVBAHA.116.307743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong Y, Koh DR. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2010;339:437–448. doi: 10.1007/s00441-009-0908-5. doi: 10.1007/s00441-009-0908-5. [DOI] [PubMed] [Google Scholar]
- 29.Massena S, Christoffersson G, Vågesjö E, et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood. 2015;126:2016–2026. doi: 10.1182/blood-2015-03-631572. doi: 10.1182/blood-2015-03-631572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossi A, Lord JM. Adiponectin inhibits neutrophil apoptosis via activation of AMP kinase, PKB and ERK 1/2 MAP kinase. Apoptosis. 2013;18:1469–1480. doi: 10.1007/s10495-013-0893-8. doi: 10.1007/s10495-013-0893-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo Y, Cheong N, Zhang Z, De Rose R, Deng Y, Farber SA, Fernandes-Alnemri T, Alnemri ES. Tim50, a component of the mitochondrial translocator, regulates mitochondrial integrity and cell death. J Biol Chem. 2004;279:24813–24825. doi: 10.1074/jbc.M402049200. doi: 10.1074/jbc.M402049200. [DOI] [PubMed] [Google Scholar]
- 32.Faye A, Poyet JL. Targeting AAC-11 in cancer therapy. Expert Opin Ther Targets. 2010;14:57–65. doi: 10.1517/14728220903431077. doi: 10.1517/14728220903431077. [DOI] [PubMed] [Google Scholar]
- 33.Gao SP, Sun HF, Jiang HL, Li LD, Hu X, Xu XE, Jin W. Loss of TIM50 suppresses proliferation and induces apoptosis in breast cancer. Tumour Biol. 2016;37:1279–1287. doi: 10.1007/s13277-015-3878-0. doi: 10.1007/s13277-015-3878-0. [DOI] [PubMed] [Google Scholar]
- 34.Ohki Y, Heissig B, Sato Y, Akiyama H, Zhu Z, Hicklin DJ, Shimada K, Ogawa H, Daida H, Hattori K, Ohsaka A. Granulocyte colony-stimulating factor promotes neovascularization by releasing vascular endothelial growth factor from neutrophils. FASEB J. 2005;19:2005–2007. doi: 10.1096/fj.04-3496fje. doi: 10.1096/fj.04-3496fje. [DOI] [PubMed] [Google Scholar]
- 35.Döring Y, Drechsler M, Wantha S, Kemmerich K, Lievens D, Vijayan S, Gallo RL, Weber C, Soehnlein O. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ Res. 2012;110:1052–1056. doi: 10.1161/CIRCRESAHA.112.265868. doi: 10.1161/CIRCRESAHA.112.265868. [DOI] [PubMed] [Google Scholar]
- 36.Wantha S, Alard JE, Megens RT, van der Does AM, Döring Y, Drechsler M, Pham CT, Wang MW, Wang JM, Gallo RL, von Hundelshausen P, Lindbom L, Hackeng T, Weber C, Soehnlein O. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ Res. 2013;112:792–801. doi: 10.1161/CIRCRESAHA.112.300666. doi: 10.1161/CIRCRESAHA.112.300666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L, Liu T, Zhang S, Zhou J, Wang Y, Di W. Succinate dehydrogenase subunit B inhibits the AMPK-HIF-1α pathway in human ovarian cancer in vitro. J Ovarian Res. 2014;7:115. doi: 10.1186/s13048-014-0115-1. doi: 10.1186/s13048-014-0115-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, Yin C, Lao T, Liang D, He D, Wang C, Sang N. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1α and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle. 2015;14:2520–2536. doi: 10.1080/15384101.2015.1055426. doi: 10.1080/15384101.2015.1055426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ke X, Fei F, Chen Y, Xu L, Zhang Z, Huang Q, Zhang H, Yang H, Chen Z, Xing J. Hypoxia upregulates CD147 through a combined effect of HIF-1α and Sp1 to promote glycolysis and tumor progression in epithelial solid tumors. Carcinogenesis. 2012;33:1598–1607. doi: 10.1093/carcin/bgs196. doi: 10.1093/carcin/bgs196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berger EA, McClellan SA, Vistisen KS, Hazlett LD. HIF-1α is essential for effective PMN bacterial killing, antimicrobial peptide production and apoptosis in Pseudomonas aeruginosa keratitis. PLoS Pathog. 2013;9:e1003457. doi: 10.1371/journal.ppat.1003457. doi: 10.1371/journal.ppat.1003457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang CH, Rong MY, Wang L, Ren Z, Chen LN, Jia JF, Li XY, Wu ZB, Chen ZN, Zhu P. CD147 up-regulates calcium-induced chemotaxis, adhesion ability and invasiveness of human neutrophils via a TRPM-7-mediated mechanism. Rheumatology (Oxford) 2014;53:2288–2296. doi: 10.1093/rheumatology/keu260. doi: 10.1093/rheumatology/keu260. [DOI] [PubMed] [Google Scholar]
- 42.Kosugi T, Maeda K, Sato W, Maruyama S, Kadomatsu K. CD147 (EMMPRIN/Basigin) in kidney diseases: from an inflammation and immune system viewpoint. Nephrol Dial Transplant. 2015;30:1097–1103. doi: 10.1093/ndt/gfu302. doi: 10.1093/ndt/gfu302. [DOI] [PubMed] [Google Scholar]
- 43.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 44.Tang Y, Nakada MT, Kesavan P, McCabe F, Millar H, Rafferty P, Bugelski P, Yan L. Extracellular matrix metalloproteinase inducer stimulates tumor angiogenesis by elevating vascular endothelial cell growth factor and matrix metalloproteinases. Cancer Res. 2005;65:3193–3199. doi: 10.1158/0008-5472.CAN-04-3605. doi: 10.1158/0008-5472.CAN-04-3605. [DOI] [PubMed] [Google Scholar]
- 45.Mauris J, Woodward AM, Cao Z, Panjwani N, Argüeso P. Molecular basis for MMP9 induction and disruption of epithelial cell-cell contacts by galectin-3. J Cell Sci. 2014;127:3141–3148. doi: 10.1242/jcs.148510. doi: 10.1242/jcs.148510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boulahbel H, Durán RV, Gottlieb E. Prolyl hydroxylases as regulators of cell metabolism. Biochem Soc Trans. 2009;37:291–294. doi: 10.1042/BST0370291. doi: 10.1042/BST0370291. [DOI] [PubMed] [Google Scholar]
- 47.Yang X, Du T, Wang X, Zhang Y, Hu W, Du X, Miao L, Han C. IDH1, a CHOP and C/EBPβ-responsive gene under ER stress, sensitizes human melanoma cells to hypoxia-induced apoptosis. Cancer Lett. 2015;365:201–210. doi: 10.1016/j.canlet.2015.05.027. doi: 10.1016/j.canlet.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 48.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]