

Abstract
Mechanical complications of myocardial infarction (MI) are often fatal. Little is known about endogenous factors that predispose to myocardial rupture after MI. Ectonucleoside triphosphate diphosphohydrolase (CD39) could be a critical mediator of propensity to myocardial rupture after MI due to its role in modulating inflammation and thrombosis. Using a model of permanent coronary artery ligation, rupture was virtually abrogated in cd39–/– mice versus cd39+/+ controls, with elevated fibrin and collagen deposition and marked neutrophil and macrophage influx. Macrophages were found to display increased surface expression of CD301 and CD206, marking a reparative phenotype, driven by increased extracellular ATP and IL-4 in the infarcted myocardium of cd39–/– mice. A myeloid-specific CD39-knockout mouse also demonstrated protection from rupture, with an attenuated rupture phenotype, suggesting that complete ablation of CD39 provides the greatest degree of protection in this model. Absence of CD39, either globally or in a myeloid lineage–restricted fashion, skews the phenotype toward alternatively activated (reparative) macrophage infiltration following MI. These studies reveal a previously unrecognized and unexpected role of endogenous CD39 to skew macrophage phenotype and promote a propensity to myocardial rupture after MI.
Ectonucleoside tri(di)phosphohydrolase (CD39) skews macrophage phenotype and promotes a propensity to cardiac rupture after myocardial infarction.
Introduction
Myocardial infarction (MI) can be complicated by left ventricular (LV) free wall rupture, ventricular septal rupture, and papillary muscle or chordae tendinae rupture, all of which are associated with a high rate of mortality (1). LV free wall rupture almost always results in rapid demise. Extensive research is needed to understand the pathophysiology of mechanical complications of MI, given the high rate of death and limited options for treatment of this condition. Little is known about endogenous factors which predispose to this serious consequence of MI.
We hypothesized that the ectonucleotidase CD39 (ectonucleoside triphosphate diphosphohydrolase 1 or ENTPD1) could modulate the response to MI. CD39 is responsible for the hydrolysis of extracellular ATP and ADP to AMP (2). CD39 is expressed by endothelial cells, B cells, T cells, and monocytes/macrophages (3–5). Under homeostatic conditions, cardiomyocytes express negligible amounts of CD39 (5, 6). After tissue injury, injured and dying cells release ATP and ADP into the extracellular space, triggering inflammatory signaling, including IL-1β production, and thrombus formation, by binding purinergic receptors in the P2X and P2Y classes (7). The main functions of CD39 are antithrombotic (2), mediated by hydrolysis of ADP, and antiinflammatory (8), mediated by ATP catabolism and the production of adenosine following terminal phosphohydrolysis of AMP by the 5′ nucleotidase CD73 (3, 9). The role of CD39 in sustained ischemia resulting in MI has not been described. We hypothesized that lack of CD39 would result in accumulation of ATP and antagonize normal myocardial wound healing after infarction. To test this hypothesis, we used a permanent coronary artery ligation model on CD39-null and genomic control mice to assess the reparative response to MI.
Results
Survival rate in cd39–/– and cd39+/+ mice after MI.
To explore the long-term consequences of permanent coronary artery occlusion, survival was assessed in mice globally deficient for CD39 (cd39–/– mice) and wild-type control mice (cd39+/+ mice) (2). After 28 days, the survival rate of cd39–/– mice was significantly higher than that of cd39+/+ mice after permanent coronary artery occlusion (Figure 1A). Cardiac rupture occurred within 7 days after MI, most frequently at 4 days after MI (Figure 1B). Necropsy revealed that cardiac rupture was the cause of death for both cd39–/– and cd39+/+ cohorts, based on the presence of a visible slit-like site of myocardial rupture and a blood-filled chest cavity (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/jci.insight.89504DS1). Infarct size was evaluated after MI to ascertain whether this differed between cohorts. Initial infarct size was similar 24 hours after MI in cd39–/– and cd39+/+ mice (Figure 2A and Supplemental Figure 2). This suggested that the initial ischemic burden was the same between the two groups. To understand how the gross structure of the infarcted myocardium evolved between the two groups, infarct expansion, as represented by the infarct expansion index ([LV cavity area/total heart area] × [uninfarcted septal thickness/infarcted LV free wall thickness]), was assessed 48 hours after MI (10). Infarct expansion was found to be greater in cd39+/+ mice relative to cd39–/– mice (Figure 2B). LV systolic function was assessed by echocardiography at baseline and in surviving mice on days 1, 7, and 28 after MI (Figure 2C). Postinfarction heart rate and blood pressure did not differ between cd39+/+ and cd39–/– mice; this suggested that infarct expansion was not caused by differences in hemodynamic stress (Supplemental Table 1). Plastination experiments performed to make a cast of the coronary arterial tree showed no anatomic differences in the major epicardial coronary arteries between genotypes to account for the observed difference in incidence of cardiac rupture (Supplemental Figure 3).
Figure 1. Survival of cd39+/+ and cd39–/– mice after myocardial infarction.
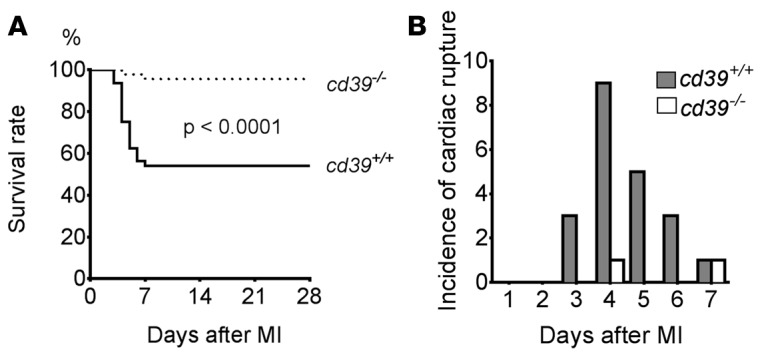
(A) Kaplan-Meier curve demonstrating survival of cd39+/+ and cd39–/– mice after permanent coronary occlusion (n = 46 for cd39–/– mice and n = 48 for cd39+/+ mice). Log-rank. (B) Incidence of cardiac rupture after myocardial infarction (MI). Genotype is indicated.
Figure 2. Myocardial infarct size and expansion and left ventricular systolic function.

(A) Infarct size was evaluated by TTC staining of 1-mm left ventricular (LV) cross sections 24 hours after permanent coronary occlusion. There was no difference in infarct size between cd39+/+ and cd39–/– mice. n = 4 per group. (B) Greater infarct expansion was seen in cd39+/+ mice in comparison with cd39–/– mice. Expansion index = (LV cavity area/total heart area) × (uninfarcted septal thickness/infarcted LV free wall thickness). n = 5–6 per group. Student’s t test. (C) Assessment of LV systolic function. Echocardiographic data for cd39+/+ and cd39–/– mice after myocardial infarction are shown. n = 6–7 per group, except for day 1, n = 3–4 per group. 1-way ANOVA, *P < 0.05 **P < 0.005, ***P < 0.001. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Differential response to MI.
In order to understand the unexpected finding of protection from cardiac rupture in mice lacking CD39, a number of possible drivers of myocardial wound healing were considered. PAI-1 activity, active MMP-2, and active MMP-9 were not found to differ following MI between cd39–/– and cd39+/+ mice (Supplemental Figures 4 and 5 ). Fibrin deposition was markedly increased in the infarct and peri-infarct tissue obtained from cd39–/– mice, compared with cd39+/+ mice (Figure 3). At baseline, there was no difference in fibrin deposition between cd39+/+ and cd39–/– mice (Figure 3).
Figure 3. Fibrin deposition after myocardial infarction.

Forty-eight hours after myocardial infarction (MI), the infarct and peri-infarct regions of myocardium were harvested, and fibrin deposition was evaluated by Western blot. Markedly increased fibrin deposition was noted in cd39–/– hearts compared with cd39+/+ hearts. Fibrin deposition did not differ in uninfarcted hearts of cd39+/+ and cd39–/– mice. n = 3–5 per group. 1-way ANOVA, **P < 0.005. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Role of CD39 in tissue fibrosis after MI.
To further evaluate the healing process after MI, the infarcted myocardium of cd39+/+ and cd39–/– mice was examined histologically 4 days after MI (Figure 4). The day 4 time point was chosen, as it was the most common time point at which cardiac rupture occurred during the initial 28-day observation period (Figure 1B). H&E staining was notable for a marked inflammatory cell infiltrate in the infarcted myocardium of cd39–/– mice, compared with cd39+/+ mice. Eosinophilic deposits consistent with fibrin were noted within the epicardial and subendocardial layers of the infarcted tissue of cd39–/– mice, which were less visible in cd39+/+ mice. Fibrin deposition, which was not appreciated in cd39+/+ mice, was also noted perivascularly in cd39–/– mice. Masson’s trichrome staining showed increased tissue fibrosis in the same distribution as fibrin deposition and the cellular infiltrate in the infarcted myocardium of cd39–/– mice compared with cd39+/+ mice (Figure 4A). The quantification of collagen by hydroxyproline assay demonstrated more collagen in the infarcted myocardium of cd39–/– mice compared with cd39+/+ mice (Figure 4B). Additionally, collagen I and fibronectin gene expression were higher in the infarcted myocardium of cd39–/– mice compared with cd39+/+ mice (Figure 4, C and D). These findings did not appear to be dependent upon TGF-β, as TGF-β expression did not differ between the two groups (Figure 4E). Taken together, these data support that an inflammatory, fibrin- and collagen-rich environment altered the phenotypic response to infarction in cd39–/– mice, compared with cd39+/+ mice.
Figure 4. Healing after myocardial infarction.

(A) Histological analysis of infarcted myocardium in cd39+/+ and cd39–/– mice; H&E (left) and Masson’s trichrome (right) staining 4 days after myocardial infarction (MI). Scale bar: 200 μm. (B) Hydroxyproline content in sham-operated myocardium and in the infarcted and uninfarcted regions of myocardium from mice with MI 4 days prior. n = 4–5 mice per group. 1-way ANOVA. (C) Collagen I and (D) fibronectin gene expression 4 days after MI. n = 3–4 per group. 1-way ANOVA. (E) TGF-β expression was evaluated in the infarct and peri-infarct regions 48 hours after MI in cd39+/+and cd39–/– mice using Western blot. TGF-β expression did not differ between the two groups. n = 3–5 per group. Student’s t test, **P < 0.001, ***P < 0.0001. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Levels of extracellular purinergic signaling molecules.
In order to understand whether protection from cardiac rupture could be a consequence of an altered extracellular milieu of ATP, ADP, AMP, and adenosine in the absence of CD39, circulating levels of these signaling factors were measured after MI. At baseline, circulating extracellular ATP and ADP levels do not differ between wild-type and cd39-/- mice (2). ATP was found to be elevated in the plasma of cd39–/– mice compared with cd39+/+ mice (Figure 5A). No differences were detected in circulating ADP, AMP, or adenosine (Figure 5). No compensatory changes in CD73 expression occurred in the absence of CD39 (Figure 5).
Figure 5. Circulating levels of ATP, ADP, AMP, and adenosine after myocardial infarction.

Circulating extracellular (A) ATP, (B) ADP, (C) AMP, and (D) adenosine concentrations were measured 48 hours after myocardial infarction using liquid chromatography–mass spectrometry. Metabolites were normalized to the amount hemolysis initially present in the sample. ATP was found to be elevated in the plasma of cd39–/– mice compared with cd39+/+ mice. There were no differences in circulating ADP, AMP, or adenosine levels between groups. (E) CD73 expression after myocardial infarction was evaluated using flow cytometry 4 days after myocardial infarction. There was no difference in (F) total or (G) leukocyte CD73 expression between cd39+/+ and cd39–/– mice. 10,000 events were captured per sample. n = 3 per group. Student’s t test, *P < 0.05. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Identification of leukocytes in the infarcted myocardium of cd39–/– and cd39+/+ mice.
As histologic examination had revealed greater numbers of leukocytes in cd39–/– mice in comparison with cd39+/+ mice after MI, we sought to elucidate the phenotype of these infiltrating cells and to understand their contribution to the observed protection from cardiac rupture in cd39–/– mice. Immunohistochemistry was performed on infarcted myocardial sections from cd39+/+ and cd39–/– mice. Greater numbers of neutrophils (detected by Ly/6G) and monocyte/macrophages (detected by CD68) were found in cd39–/– mice, when compared with cd39+/+ mice (Figure 6). This finding was confirmed and quantified by flow cytometry (Figure 6).
Figure 6. Identification of infiltrating leukocytes in infarcted myocardium.

Representative immunohistochemical staining of (A) CD68-positive monocytes/macrophages and (B) Ly-6G–positive neutrophils in the infarcted myocardium of cd39+/+ and cd39–/– mice 4 days after myocardial infarction (MI). Scale bar: 100 μm. (C) Quantification of CD68-positive monocyte/macrophage populations and (D) Ly-6G–positive neutrophils in the infarcted and sham-operated myocardium of cd39+/+ and cd39–/– mice, measured by flow cytometry. n = 3 mice per each group. 1-way ANOVA, ***P < 0.0001. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Effect of myeloid-specific deletion of CD39 on survival and wound healing after MI.
In order to determine the importance of cell-specific contribution of CD39 to promoting cardiac rupture after MI, we designed a myeloid-specific CD39-knockout mouse using the LoxP-Cre system (Figure 7A). LoxP sites were introduced into two poorly conserved regions flanking native CD39 and electroporated into a C57BL/6 stem cell line. Successful homologous recombination was confirmed in both the electroporated stem cell line and F1 generation of progeny (Figure 7B). Cd39flox/flox mice were crossed to a myeloid CRE (LYS-M) mouse to ablate CD39 in myeloid cells, which was later confirmed by FACS analysis of peritoneal macrophages (Figure 7C). At baseline, there were no differences in heart rate or blood pressure among cd39+/+, cd39–/–, cd39flox/flox, and cd39flox/floxLysMCre/– mice (Supplemental Table 2). Similarly, there were no differences in LV systolic or diastolic dimensions or volume, LV stroke volume, LV ejection fraction, cardiac output, aortic annulus dimension, interventricular septal dimension (diastole), or LV posterior wall dimension (diastole) among cd39+/+, cd39–/–, cd39flox/flox, and cd39flox/floxLysMCre/– mice, as assessed by echocardiography (Supplemental Table 3). A permanent coronary occlusion model of MI was used as before in cd39flox/flox and cd39flox/floxLysMCre/– mice. Infarct size was similar between cd39flox/flox and cd39flox/floxLysMCre/– mice (Figure 8). Mice with myeloid-specific ablation of CD39 had greater survival than control mice, although the phenotype was attenuated in comparison with the absolute difference observed between cd39+/+ and cd39–/– mice (Figure 8). In both groups, necropsies were performed, and they confirmed cardiac rupture as the primary cause of death.
Figure 7. Creation of myeloid CD39 conditional knockout.

(A) LoxP- and Frt-flanked neomycin sites were introduced to flank exon 1 of the native cd39 locus using homologous recombination. Neomycin was subsequently removed from all progeny using Flp recombinase. (B) Southern blot demonstrating successful transfer of the cd39flox allele in the F1 generation of progeny, the stem cell line identified with successful homologous recombination at the CD39 locus, and control C57BL/6 DNA. (C) CD39 expression by genotype. Flow cytometry demonstrating CD39 expression on myeloid cells (peritoneal macrophages) in cd39+/+, cd39–/–, cd39flox/flox, and cd39flox/floxLysMCre/– mice. Global CD39 knockouts were confirmed as having no CD39 expression, and myeloid-specific CD39 knockouts were confirmed as having no myeloid CD39 expression. n = 4 per group. 1-way ANOVA, ***P < 0.0001. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Figure 8. Role of myeloid CD39 in cardiac rupture.

(A) Kaplan-Meier survival curves for myeloid-specific CD39 knockout (cd39flox/floxLysMCre/–) and control (cd39flox/flox) mice after permanent coronary occlusion (n = 25 mice per group). Log-rank, P < 0.05. (B) Infarct size in cd39flox/flox and cd39flox/floxLysMCre/– mice after myocardial infarction (MI). Infarct size was evaluated using TTC staining 48 hours after permanent coronary occlusion. There was no difference in infarct size between the two groups. LV, left ventricle. n = 5–6 per group. Student’s t test. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Phenotype of tissue leukocytes.
The observation that cd39–/– mice were protected from cardiac rupture but demonstrated remarkably increased numbers of leukocytes in infarcted myocardium prompted further investigation into the postinfarction inflammatory milieu and its modulation by myeloid-specific CD39 ablation. Because cd39–/– mice were found to have a marked increase in macrophage infiltration compared with cd39+/+ controls and were protected from cardiac rupture, we hypothesized that persistently elevated extracellular ATP in cd39–/– mice could be modulating macrophage polarization toward a reparative phenotype. To test this hypothesis, we isolated bone marrow–derived macrophages (BMDMs) and polarized them with LPS (100 ng/ml) for 18 hours. Consistent with prior studies, LPS induced inflammatory genes, including TNF-α and IL-6 (Figure 9) (11, 12). To mimic the role of ATP in CD39 deficiency, we used the nonhydrolyzable ATP analog, γATP. Although macrophages are known to express P2X receptors, γATP (100 μM) treatment alone of BMDMs did not alter inflammatory gene expression (Figure 9). The concentration of 100 μM γATP likely approximates the local levels of ATP at the site of tissue damage, assuming an intracellular concentration of 10 mM ATP and an extracellular concentration of 10 nM ATP under homeostatic conditions (13). We next sought to determine if γATP altered inflammatory polarization of BMDMs in a setting in which there is inflammatory priming. For this purpose, we chose to use a priming mechanism, LPS, which mimics many of the features of hypoxia or ischemia. Concurrent treatment of BMDMs with LPS and γATP led to a decreased induction of IL-6 and TNF-α, compared with LPS alone (Figure 9). These results suggest that sustained levels of extracellular ATP, known to be released from infarcting myocardium, could be a major contributor to the observed protection from cardiac rupture after MI in mice lacking CD39.
Figure 9. Polarization of activated bone marrow–derived macrophages after γATP stimulation.
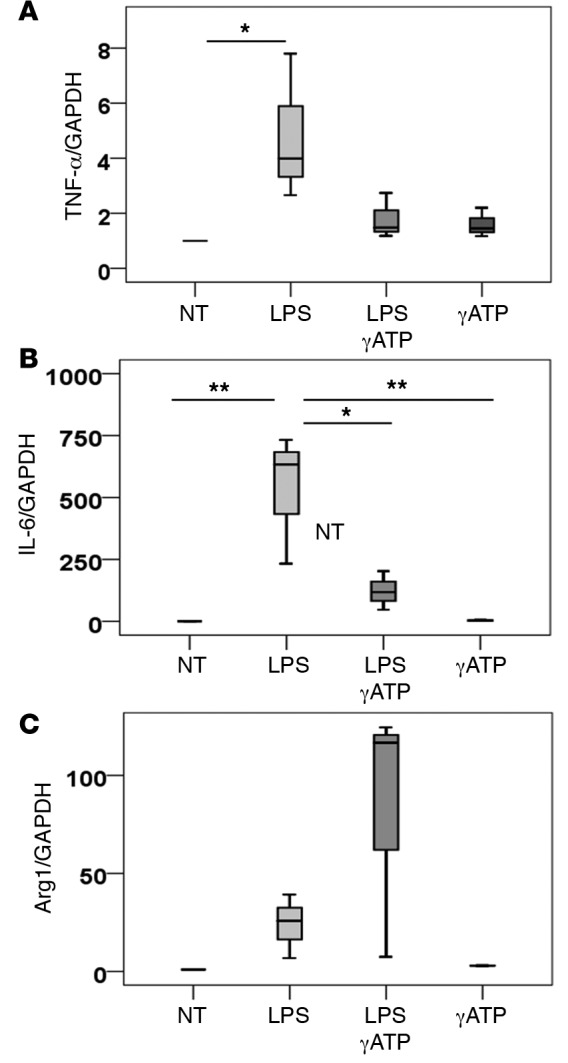
Bone marrow–derived macrophages were untreated (NT) or were stimulated with LPS (100 ng/ml), LPS with 100 μm γATP (nonhydrolyzable ATP), or γATP alone for 18 hours. Gene expression of (A) TNF-α, (B) IL-6, and (C) Arg1 was measured using real-time quantitative reverse transcriptase–PCR. LPS drove an inflammatory response as expected, and exposure with γATP as well as LPS reversed this response. n = 3. ANOVA, *P < 0.05, **P < 0.005. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Given the greatly increased numbers of macrophages present in the infarct and peri-infarct myocardium of cd39–/– mice, we proceeded to characterize and phenotype these cells further. CD68+ macrophages isolated from infarct tissue revealed differences in gene expression which overall favored a reparative (alternatively activated or M2) phenotype in mice lacking global and myeloid CD39, in comparison with controls (Figure 10). Il-10 gene expression was significantly increased in cd39–/– mice relative to cd39flox/flox mice. Mgl2 gene expression was elevated in cd39flox/floxLysMCre/– mice when compared with cd39+/+ and cd39flox/flox mice. In comparison to cd39+/+ mice, CD68+ macrophages infiltrating the infarct and peri-infarct tissue of cd39–/– mice were found to have increased surface expression of CD301 and CD206, indicating a skew toward a reparative (M2-type) macrophage phenotype in mice lacking CD39 (Figure 10). IL-4, a cytokine known to drive macrophage differentiation toward a reparative phenotype, was found to be elevated in the infarcted myocardium of cd39–/– mice, compared with cd39+/+ controls (Figure 11).
Figure 10. Phenotype of CD68+ macrophages infiltrating infarcted myocardium.

Three days after myocardial infarction, CD68+ macrophages infiltrating the infarcted myocardium of cd39+/+, cd39–/–, cd39flox/flox, and cd39flox/floxLysMCre/– mice were isolated, and gene expression of (A) TNF-α, (B) IL-1β, (C) Mrc1 (CD206), (D) Mgl2 (CD301), (E) IL-10, (F) Ym1, (G) Fizz1, and (H) IL-1Ra was measured using real-time quantitative reverse transcriptase–PCR. n = 3–4 per group. (I) Flow cytometric analysis of the M2 marker CD301 in cd39+/+ and cd39–/– mice at baseline and after myocardial infarction (MI). n = 3–4 per group. (J) Surface expression of the M2 marker CD206 in cd39–/– and cd39+/+ mice at baseline and after MI. n = 3 per control group and n = 10 per MI group. 1.7 × 106 events were captured per animal. 1-way ANOVA, *P < 0.05, **P < 0.005. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Figure 11. IL-4 expression in infarcted myocardium.

Western blot demonstrating increased IL-4 (a driver of M2 or alternatively activated macrophages) in cd39–/– mice compared with cd39+/+ mice in heart tissue harvested 2 days after myocardial infarction. n = 3–5 per group. Student’s t test, ***P < 0.0001. Box and whisker plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars).
Discussion
The key finding in this study is that endogenous CD39 promotes a propensity to cardiac rupture after MI and that lack of CD39 is protective, as myocardial rupture was nearly abolished in mice lacking CD39. Mice lacking global or myeloid CD39 were protected from myocardial rupture after MI, with an attenuated phenotype in mice lacking only myeloid CD39. Protection from rupture appears to have been driven by increased extracellular ATP, resulting in a reparative macrophage infiltrate, propelled by IL-4. The greatest degree of protection appeared to result from complete, rather than partial, absence of CD39.
Cardiac rupture represents a catastrophic event following MI, which, in most instances, leads to instantaneous demise (14). Myocardial rupture is difficult to study; by nature, it has an abrupt onset and high fatality rate. As such, further research is critically needed to understand how endogenous factors modulate the pathophysiologic response that leads to this serious consequence of MI. In this study, wild-type mice were found to have a rate of cardiac rupture of approximately 50% occurring within 7 days of MI, consistent with prior reports (15, 16). Despite similar sized initial infarcts, wild-type mice developed infarct expansion and ultimately had a higher rate of LV free wall rupture than cd39–/– mice (Becker type III) (17). Follow-up studies of cd39–/– and cd39+/+ mice that survived did not demonstrate any differences in echocardiographic parameters of LV systolic function. Survivorship bias could influence the echocardiographic results, given that approximately half of the wild-type control animals died prior to assessment. We deduce that once the subacute period after MI has passed, the mortality rates do not continue to diverge. It will be critical to examine the role of CD39 in long-term postinfarction myocardial remodeling. It is possible that, while the absence of CD39 protects from cardiac rupture in the short term, there could be deleterious effects on late remodeling and LV function due to lack of CD39.
We initially conjectured that CD39 deficiency would lead to local ATP accumulation and result in increased cardiac rupture. But contrariwise, our study demonstrated that, in the setting of MI, lack of CD39 promoted cardiac rupture. This was an unexpected finding, given that CD39 has been shown to be protective in the setting of myocardial ischemia/reperfusion injury, with absence of CD39 resulting in larger infarct size after ischemia and loss of cardioprotection afforded by ischemic preconditioning (5). Similarly, transgenic overexpression of CD39 was shown to result in decreased infarct size in a murine model of myocardial ischemic injury (18). Our results demonstrating a deleterious role for leukocyte CD39 following nonreperfused/necrotic MI are difficult to reconcile with prior reports demonstrating a protective role for CD39 and CD73 in other ischemia/reperfusion models (19, 20). In the permanent occlusion model of MI used in our studies, there are no preconditioning steps; therefore preconditioning protective mechanisms cannot contribute to myocardial protection. CD73 has been shown to suppress leukosequestration in ischemic tissue (21). We did not find any compensatory alterations in CD73 expression or circulating adenosine levels in this model. In summing these studies, deletion of CD39 may be beneficial after nonreperfused MI, by preventing cardiac rupture and accelerating wound healing. Both expression and deletion of CD39 could be therapeutic depending on the disease status and timing of the intervention (22). There is likely to be an optimum therapeutic window for augmenting or inhibiting CD39, as is seen in cerebral ischemia with tissue plasminogen activator therapy (23).
Consistent with prior studies, we found that the absence of CD39 after tissue injury was associated with increased tissue leukocyte infiltration (8, 24–26). Macrophages play an essential role in wound healing, removing necrotic tissue by secreting proteolytic enzymes and encouraging granulation tissue deposition to promote scar formation (27, 28). Macrophage depletion has been shown to result in significantly greater rates of cardiac rupture after MI, characterized by delayed necrotic tissue removal and reduced collagen deposition (29). It has been suggested that, in contrast to ischemia/reperfusion, unreperfused myocardium requires a greater amount of scar formation, a process that requires infiltration by monocytes (30). Mice null for CD39 have been shown to have greater vascular permeability after intestinal ischemia/reperfusion (31), which may facilitate transmigration. ATP has been shown to recruit phagocytes in vivo via the P2Y2 receptor (32). In addition to playing a role in leukocyte recruitment, elevated extracellular ATP has been shown to result in increased in vitro cardiac fibroblast expression of α-smooth muscle actin and collagen synthesis (33). This was shown to occur via P2Y2 receptors (34). This particular study emphasizes the dynamic nature of wound healing after MI, which results in a prevailing direction, and in this case, more myocardial wall integrity in the absence of CD39.
In this model of permanent coronary occlusion, elevated nucleotide levels likely played an important role both in the recruitment of leukocytes and in the modification of the macrophage phenotype at the site of cellular damage. We posit that the absolute increase in numbers of macrophages with a reparative phenotype contributed to the observed protection from cardiac rupture in CD39-null mice. These data complement and validate a recent report demonstrating the necessity of alternatively activated macrophages for repair of infarcted heart tissue and survival (35). In our study, we found that, in mice globally lacking CD39, circulating ATP was elevated and sustained, even days after MI. The addition of nonhydrolyzable ATP to activated BMDMs resulted in tempering of the inflammatory phenotype, suggesting that this process may not depend on the catabolism of ATP to adenosine, as has previously been suggested (36). Taken together, the data show that, via dissipation of extracellular ATP, CD39 modulates the quantity and characteristics of immune cells present in the myocardium after MI, which in turn likely critically affects wound healing.
Creation of the myeloid-specific CD39 knockout presented here allowed us to dissect out the importance and relative contributions of the origin of CD39 to the observed phenotype. We observed that mice lacking CD39 on myeloid cells were also protected from cardiac rupture, though the absolute difference from controls was attenuated in comparison to mice lacking global CD39 and the corresponding controls. This intermediate phenotype suggests a dose effect of CD39, such that the greatest degree of protection is drawn from complete lack of CD39. Mice globally lacking CD39 demonstrated elevation of collagen and fibrin deposition, which conferred protection from cardiac rupture. This is the first report to our knowledge to demonstrate that enhanced fibrin deposition in the absence of CD39 could play a protective role in wound healing, particularly in the setting of myocardial wound healing after a large MI (2, 4, 37).
In conclusion, CD39 promotes cardiac rupture in ischemic myocardium by limiting cellular infiltration with reparative macrophages, tempering fibrin deposition, and curbing the fibrogenic response to MI. These data contrast sharply with prior reports of an early protective role for CD39 in MI (5, 18). When considered together, understanding the timing and context of CD39 expression in MI can potentially lead to new therapeutic insights regarding expeditious myocardial wound healing.
Methods
Experimental animals.
cd39–/– mice were generated by replacement of the enzymatically active extracellular portion of the CD39 molecule, as previously described (2, 24). cd39+/+ mice of the same genetic background were purchased from Jackson Laboratory. cd39–/– mice were bred at the Animal Resource Facility at the University of Michigan under specific pathogen–free conditions. Male mice were used for experiments when they were between 9 and 12 weeks of age.
Generation of myeloid-specific cd39-deficient mice.
The vector pFlox-Flp-Neo was used as a backbone for generating a gene-targeting vector with LoxP sites flanking the first exon of CD39. BACPAC clone RP23-117D11 (derived from a C57BL/6 mouse) was used as source DNA for the insertion of BamHI sites at –490 and +950 of exon 1. This vector was introduced into C57BL/6-derived Bruce 4 embryonic stem cells and selected in G418. Embryonic stem cell clones with successful insertion of LoxP were identified by qPCR, confirmed by Southern blot analyses of BglI/SalI-digested DNA, and injected into C57BL/6 blastocysts. The resulting chimeras were bred with C57BL/6 mice to produce cd39+/flox heterozygous mice. Mice containing the CD39flox allele were crossed with mice expressing Cre recombinase under the control of the myeloid lineage-specific promotor, lysozome M (LysM), to generate mice in which the CD39 was deleted in monocytes/macrophages. The presence of the cd39flox/flox and LysM Cre transgene was determined by PCR analysis of genomic DNA from tail tips. Cardiac phenotype was assessed by measuring tail cuff blood pressure and heart rate and by evaluating for baseline differences in echocardiographic parameters. For heart rates and blood pressures, a minimum of 6 measurements per animal were obtained and averaged.
Study design.
To assess cardiac rupture and LV remodeling, we created MI in mice by permanently ligating the left anterior descending (LAD) coronary artery (38). Complete occlusion of blood flow was confirmed by the visible blanching of the heart and persistent ST segment elevation on the electrocardiogram. Sham operation included all procedures except for the ligation of the left coronary artery. Mice were observed for 28 days after MI, and the postoperative day and cause of death were noted. Echocardiograms were performed on surviving cd39+/+ and cd39–/– mice at 1, 7, and 28 days after MI. Heart rates and blood pressures were obtained by tail cuff on acclimatized animals 24 hours after MI using an average of 6 measurements.
Assessment of infarct size.
Infarcts were evaluated by harvesting hearts 24 hours and 48 hours after permanent LAD ligation. Hearts were trimmed as fresh tissue into 4 coronal sections using an acrylic rodent heart matrix (Kent Scientific). 1-mm sections were incubated at 37°C in 4% 2,3,5-triphenyltetrazolium chloride. Infarct area for each slice as a percentage of total LV area is reported. Infarct size is not reported in relation to area at risk, as the area at risk was essentially the same as the infarcted area. Infarct expansion index was calculated using the following formula: expansion index = (LV cavity area/total heart area) × (uninfarcted septal thickness/infarcted LV free wall thickness) (10). The TTC-stained sections were then fixed in 10% neutral buffered formalin for 24 hours. After 24 hours of fixation, the tissues were paraffin embedded on an automated histology processor (TissueTek). Sections were cut at 5 μm thickness on a rotary microtome and stained with hematoxylin and eosin on an automated stainer.
Evaluation of the coronary anatomy.
Coronary anatomy was evaluated by perfusing with heparinized saline via the inferior vena cava. Following this, a cast of the LV cavity, ascending aorta, and major epicardial coronary arteries was created using a plastination assay (Batson’s No. 17 Plastic Replica and Corrosion Kit, Polysciences) by injecting uncured plastic via the LV apex, followed by degradation of excess tissue with 30% potassium hydroxide.
Plasminogen activator inhibitor activity assay.
Permanent LAD occlusion was performed, and on day 3 after MI, the infarct and peri-infarct zones were harvested and protein was isolated. Uninfarcted heart tissue was isolated for comparison. 150 μg of protein from each sample was added to microwells in duplicate. Microwells were coated with urokinase, such that only functional PAI-1 present in the sample would bind and be detected by the assay (Molecular Innovations).
Zymography.
Permanent LAD occlusion was performed, and on day 3 after MI, the infarct and peri-infarct zones were harvested and protein was isolated, as for the plasminogen activator inhibitor (PAI) activity assay. Uninfarcted heart tissue was isolated for comparison. Protein samples were prepared with Novex Tris-Glycine SDS sample Buffer (Invitrogen), and 10 μg was loaded on a 10% zymogram gel (Invitrogen). After running the gel in Novex Tri-Glycine SDS Running Buffer (Invitrogen) for 90 minutes, the gel was incubated in a Zymogram Renaturing Buffer (Invitrogen) and a Zymogram Developing Buffer (Invitrogen). The gel was stained with the Coomassie Blue Staining Kit (Invitrogen) to detect protease activity.
Western blotting.
Protein was isolated from the infarct and peri-infarct zones on day 2 following MI. Protein was added to sample buffer under reducing conditions, heated at 90°C for 3 minutes, separated by 10% SDS-PAGE, and electrophoretically transferred onto PVDF membranes (Invitrogen). The membranes were incubated with anti-fibrin (provided by Charles Esmon, Oklahoma Medical Research Foundation, Oklahoma City, Oklahoma, USA) (39), IL-4 (ab11524, Abcam), or anti–TGF-β antibody (ab66043, Abcam) and autoradiographed using the enhanced chemiluminescence method (ECL detection system, GE Healthcare Bioscience).
Histological and immunohistochemical analysis.
Histological and immunohistochemical analyses were performed at 4 days after MI in cd39+/+ and cd39–/– mice. Sections were stained with hematoxylin and eosin; they were also stained with Masson’s trichrome. The following antibodies were used for immunohistochemistry: anti-CD68 (FA-11, Serotec) to detect monocytes/macrophages, anti-Ly-6G (RB6-8C5, Southern Biotechnology) to detect neutrophils, and Alexa Fluor 488–conjugated anti-rat IgG (Invitrogen) as a secondary antibody.
Hydroxyproline assay.
Four days after MI, tissues were freeze-dried, weighed, homogenized in 1 ml of 5 N HCl, and hydrolyzed. After the pH was adjusted to 7.0 with 0.1 N KOH, 0.1 M sodium borate buffer was added and mixed with 0.2 M chloramine-T solution. The sample solutions were centrifuged to separate aqueous and toluene layers. Ehrlich’s reagent was added to toluene layer. Absorbance at 560 nm was measured with a spectrophotometer.
Measurement of extracellular ATP and metabolites.
Permanent coronary occlusion was performed as above. Two days after MI, blood was drawn via direct cardiac puncture directly into a syringe prefilled with an inhibitor solution, with a final proportion of 1:1 inhibitor solution to whole blood. The inhibitor solution contained 118 mM NaCl, 5 mM KCl, 40 mM tricine buffer, 6 mM EDTA, 5 nM NBTI, 10 μM forskolin (Sigma-Aldrich), 100 μM IBMX (Calbiochem), 40 μM dipyridamole (Sigma-Aldrich), 10 μM EHNA (Sigma-Aldrich), and 10 μM ITU (Sigma-Aldrich) (40). Platelet-poor plasma was separated and assessed for free hemoglobin concentration, as an indicator of cellular hemolysis. Free hemoglobin concentration (mg/l) was calculated using the formula 10([16.72 × A415] – [8.36 × A380] – [8.36 × A450]), using a Nanodrop 2000 spectrophotometer (Thermo Scientific) (40). Samples from cd39+/+ and from cd39–/– mice with similar values of free hemoglobin (not exceeding 4.7 mg/l) were analyzed by mass spectrometry. A mixture of methanol, acetone, and acetonitrile (1:1:1) was used to extract metabolites from plasma samples. The extraction solvent was added to plasma samples (4:1 ratio). The supernatant was analyzed using LC-MS (Agilent 1200 chromatography, Luna NH2 hydrophilic interaction chromatography column, Agilent 6410 series triple quadrupole mass spectrometer with electrospray ionization source, operated in negative mode). A series of calibration standards was prepared along with samples to quantify metabolites. Data were processed by MassHunter workstation software, version B.04. Metabolites were normalized to the degree of hemolysis initially present in the sample.
Preparation of peritoneal macrophages.
To isolate peritoneal macrophages, mice were injected intraperitoneally with 3 ml of 4% thioglycollate. Peritoneal exudate cells were isolated from the peritoneal cavity 4 days after injection.
Flow cytometric quantification of leukocyte populations.
Four days after permanent coronary occlusion, hearts were perfused with cold normal saline and excised. Tissue was digested with 0.1% collagenase B (Roche) and homogenized. Erythrocyte contaminants were lysed using FACS Lyse (BD Biosciences). Cells were counted using a HEMAVET 950FS cell hemocytometer (DREW Scientific) and fixed with Cytofix fixation buffer (BD Biosciences). Nonspecific antibody interactions were inhibited using Fc Block (BD Biosciences). Leukocytes were initially identified as PE-conjugated CD45 (30-F11, BD Biosciences). CD45-positive cells were further identified as monocytes/macrophages or neutrophils based on FITC-conjugated anti-CD68 (Cl:A3-1, Serotec) or FITC-conjugated Ly-6G (1A8 [RUO], BD Biosciences) positivity. Flow cytometric analysis was performed on a FACS Calibur (BD Biosciences), using Cell Quest software (BD Biosciences). For flow cytometric analysis of macrophages, cells were isolated from the heart as described above but were not fixed. Cells were stained for Alexa Fluor 647–conjugated anti-CD301, which binds both macrophage galactose N-acetyl-galactosamine–specific lectin 1 and 2 (ER-MP23, Serotec), and Alexa Fluor 488–conjugated anti-CD206, which binds mannose receptor C type 1 (MRC1) (MR5D3, Serotec), markers of reparative type macrophages. Expression of CD73 was evaluated by isolating cells from the heart as described above, without fixing, and staining using APC-conjugated anti-CD73 (TY/11.8 Biolegend) and PE-conjugated anti-CD45 (30-F11 BD Biosciences). Flow cytometric analysis for macrophage phenotype and CD73 expression was performed on a FACS Canto II (BD Biosciences) and analyzed with FlowJo v.9.6.2 (Tree Star).
Real-time quantitative reverse transcription–PCR (qRT-PCR) assay.
For fibronectin and collagen gene expression, tissue was obtained from the infarct and peri-infarct regions 4 days after MI and mRNA was isolated (Qiagen). For macrophage gene expression analysis, tissue was obtained from the infarct and peri-infarct zones of the heart after permanent LAD occlusion. A single-cell suspension was prepared. Cells were sorted on a MACS (Miltenyi Biotec) based on CD68 positivity using PE-conjugated CD68 (FA-11 Biolegend) and anti-PE microbeads (Miltenyi Biotec). mRNA was then isolated from CD68+ cells via the RNAeasy Mini Kit (Qiagen). Isolated RNA was reverse transcribed into cDNA with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time PCR was performed by means of an ABI 7500 PCR system with TaqMan Universal PCR Master Mix and Assays-on-Demand gene expression probes. All primer and probe sets were purchased from Applied Biosystems (mouse Fn1, Col1a1, Tnf, Il1b, Mrc1, Mgl2, Il10, Chil3, Retnlb, and Il1rn). All qRT-PCRs were done in duplicate. mRNA expression was normalized to β-actin level by the 2 –ΔΔCt method.
Macrophage polarization experiments.
BMDMs were obtained as previously described (41). Briefly, bone marrow precursors from male C57BL/6 mice were isolated and matured for 7 days in RPMI 1640 media supplemented with penicillin (100 units/ml), streptomycin (100 mg/ml), and 10% heat-inactivated low endotoxin FBS with L-conditioned media as a source of M-CSF. After maturation, BMDMs were replated and then polarized with 100 ng/ml LPS with or without nonhydrolyzable ATP (100 μM γATP) for 18 hours. PCR was performed as above.
Statistics.
Data are presented as the mean ± standard deviation. Survival rates were analyzed using the log-rank method. Comparisons between paired groups were performed via Student’s paired 2-tailed t test. Comparisons between multiple groups were performed by 1-way ANOVA followed by the Student-Newman-Keuls test or Tukey HSD test. A P value of less than 0.05 was considered statistically significant.
Study approval.
Animal experiments were approved by and carried out in accordance with the guidelines of the University of Michigan Committee on Use and Care of Animals.
Author contributions
NRS, TH, and DJP designed the experiments. NRS and TH performed the experiments. MCH developed the cd39flox/floxLysMCre/– mouse. ACA, DPD, LB, and NW helped to design, execute, and analyze the flow cytometry experiments. AEB, KF, HL, SNG, YK, SHV, ELH, and JJR gave technical support and conceptual advice. NRS and DJP prepared and edited the manuscript.
Supplementary Material
Acknowledgments
We would like to thank Amgen for the use of cd39–/– mice. We would like to acknowledge Thom Saunders and the University of Michigan Transgenic Animal core for their assistance in the creation of the cd39flox/flox mouse. We would also like to thank Jim Shayman for the pFlox-Flp-Neo vector. We would like to thank Natasha Dolgacheva, Steven Whitesall, Kimber Converso-Baran, Ingrid Bergin, Kim Walacavage, Chari Cortez, and Chunhai Ruan for technical assistance. This work was supported in part by grants from the NIH (R01HL55397, R01HL085149, T32HL007853, DK097153, HL131993, HL119623, and HL123621), the J. Griswold and Margery Hopkins Ruth Professorship, and the A. Alfred Taubman Medical Research Institute of the University of Michigan.
Footnotes
TH’s present address is: Bayer Pharmaceuticals, Osaka, Japan. MCH’s present address is: University of Pennsylvania, Philadelphia, Pennsylvania, USA. AEB’s present address is: University of Illinois at Champaign-Urbana, Champaign-Urbana, Illinois, USA. KF’s present address is: Awaji Medical Center, Hyogo, Japan. ELH’s present address is: Memorial Sloan Kettering Cancer Center, New York, New York, USA. JJR’s present address is: University of Detroit Mercy School of Dentistry, Detroit, Michigan, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2017;2(1):e89504. doi:10.1172/jci.insight.89504.
Contributor Information
Takanori Hayasaki, Email: taka_djembe@mac.com.
Hui Liao, Email: huiliao@umich.edu.
Danica Petrovic-Djergovic, Email: danicap@med.umich.edu.
Linda Badri, Email: lbadri@umich.edu.
Natalie Walker, Email: nataliew@umich.edu.
Keigo Fukase, Email: keigofukase@gmail.com.
Yogendra Kanthi, Email: ykanthi@med.umich.edu.
David J. Pinsky, Email: dpinsky@umich.edu.
References
- 1.Honda S, et al. Trends in the clinical and pathological characteristics of cardiac rupture in patients with acute myocardial infarction over 35 years. J Am Heart Assoc. 2014;3(5):e000984. doi: 10.1161/JAHA.114.000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinsky DJ, et al. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest. 2002;109(8):1031–1040. doi: 10.1172/JCI10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6):1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enjyoji K, et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med. 1999;5(9):1010–1017. doi: 10.1038/12447. [DOI] [PubMed] [Google Scholar]
- 5.Köhler D, et al. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation. 2007;116(16):1784–1794. doi: 10.1161/CIRCULATIONAHA.107.690180. [DOI] [PubMed] [Google Scholar]
- 6.Bönner F, Borg N, Burghoff S, Schrader J. Resident cardiac immune cells and expression of the ectonucleotidase enzymes CD39 and CD73 after ischemic injury. PLoS One. 2012;7(4):e34730. doi: 10.1371/journal.pone.0034730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsukimoto M, Maehata M, Harada H, Ikari A, Takagi K, Degawa M. P2X7 receptor-dependent cell death is modulated during murine T cell maturation and mediated by dual signaling pathways. J Immunol. 2006;177(5):2842–2850. doi: 10.4049/jimmunol.177.5.2842. [DOI] [PubMed] [Google Scholar]
- 8.Friedman DJ, et al. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci USA. 2009;106(39):16788–16793. doi: 10.1073/pnas.0902869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanthi YM, Sutton NR, Pinsky DJ. CD39: Interface between vascular thrombosis and inflammation. Curr Atheroscler Rep. 2014;16(7):425. doi: 10.1007/s11883-014-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hochman JS, Choo H. Limitation of myocardial infarct expansion by reperfusion independent of myocardial salvage. Circulation. 1987;75(1):299–306. doi: 10.1161/01.CIR.75.1.299. [DOI] [PubMed] [Google Scholar]
- 11.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138(1):129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 13.Schwiebert EM, Zsembery A. Extracellular ATP as a signaling molecule for epithelial cells. Biochim Biophys Acta. 2003;1615(1-2):7–32. doi: 10.1016/s0005-2736(03)00210-4. [DOI] [PubMed] [Google Scholar]
- 14.Cheng TO. Cardiac rupture after myocardial infarction. JAMA. 1999;281(8):703. doi: 10.1001/jama.281.8.703. [DOI] [PubMed] [Google Scholar]
- 15.Sun M, et al. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation. 2004;110(20):3221–3228. doi: 10.1161/01.CIR.0000147233.10318.23. [DOI] [PubMed] [Google Scholar]
- 16.Nahrendorf M, et al. Factor XIII deficiency causes cardiac rupture, impairs wound healing, and aggravates cardiac remodeling in mice with myocardial infarction. Circulation. 2006;113(9):1196–1202. doi: 10.1161/CIRCULATIONAHA.105.602094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker AE, van Mantgem JP. Cardiac tamponade. A study of 50 hearts. Eur J Cardiol. 1975;3(4):349–358. [PubMed] [Google Scholar]
- 18.Cai M, et al. Transgenic over expression of ectonucleotide triphosphate diphosphohydrolase-1 protects against murine myocardial ischemic injury. J Mol Cell Cardiol. 2011;51(6):927–935. doi: 10.1016/j.yjmcc.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitakaze M, et al. Infarct size-limiting effect of ischemic preconditioning is blunted by inhibition of 5’-nucleotidase activity and attenuation of adenosine release. Circulation. 1994;89(3):1237–1246. doi: 10.1161/01.CIR.89.3.1237. [DOI] [PubMed] [Google Scholar]
- 20.Takano H, et al. A(1) or A(3) adenosine receptors induce late preconditioning against infarction in conscious rabbits by different mechanisms. Circ Res. 2001;88(5):520–528. doi: 10.1161/01.RES.88.5.520. [DOI] [PubMed] [Google Scholar]
- 21.Petrovic-Djergovic D, et al. Tissue-resident ecto-5’ nucleotidase (CD73) regulates leukocyte trafficking in the ischemic brain. J Immunol. 2012;188(5):2387–2398. doi: 10.4049/jimmunol.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanthi Y, et al. Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J Clin Invest. 2015;125(8):3027–3036. doi: 10.1172/JCI79514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lees KR, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375(9727):1695–1703. doi: 10.1016/S0140-6736(10)60491-6. [DOI] [PubMed] [Google Scholar]
- 24.Hyman MC, et al. Self-regulation of inflammatory cell trafficking in mice by the leukocyte surface apyrase CD39. J Clin Invest. 2009;119(5):1136–1149. doi: 10.1172/JCI36433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reutershan J, Vollmer I, Stark S, Wagner R, Ngamsri KC, Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 2009;23(2):473–482. doi: 10.1096/fj.08-119701. [DOI] [PubMed] [Google Scholar]
- 26.Mizumoto N, et al. CD39 is the dominant Langerhans cell-associated ecto-NTPDase: modulatory roles in inflammation and immune responsiveness. Nat Med. 2002;8(4):358–365. doi: 10.1038/nm0402-358. [DOI] [PubMed] [Google Scholar]
- 27.Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. 2009;175(6):2454–2462. doi: 10.2353/ajpath.2009.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulze PC, Lee RT. Macrophage-mediated cardiac fibrosis. Circ Res. 2004;95(6):552–553. doi: 10.1161/01.RES.0000143420.87587.9e. [DOI] [PubMed] [Google Scholar]
- 29.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170(3):818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121(22):2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guckelberger O, et al. Beneficial effects of CD39/ecto-nucleoside triphosphate diphosphohydrolase-1 in murine intestinal ischemia-reperfusion injury. Thromb Haemost. 2004;91(3):576–586. doi: 10.1160/TH03-06-0373. [DOI] [PubMed] [Google Scholar]
- 32.Elliott MR, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu D, Insel PA. Hydrolysis of extracellular ATP by ectonucleoside triphosphate diphosphohydrolase (ENTPD) establishes the set point for fibrotic activity of cardiac fibroblasts. J Biol Chem. 2013;288(26):19040–19049. doi: 10.1074/jbc.M113.466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu D, Soleymani S, Madakshire R, Insel PA. ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. FASEB J. 2012;26(6):2580–2591. doi: 10.1096/fj.12-204677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiraishi M, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126(6):2151–2166. doi: 10.1172/JCI85782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen HB, Briggs KT, Marino JP, Ravid K, Robson SC, Mosser DM. TLR stimulation initiates a CD39-based autoregulatory mechanism that limits macrophage inflammatory responses. Blood. 2013;122(11):1935–1945. doi: 10.1182/blood-2013-04-496216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res. 1999;44(2):232–241. doi: 10.1016/S0008-6363(99)00212-6. [DOI] [PubMed] [Google Scholar]
- 38.Salto-Tellez M, Yung Lim S, El-Oakley RM, Tang TP, ALmsherqi ZA, Lim SK. Myocardial infarction in the C57BL/6J mouse: a quantifiable and highly reproducible experimental model. Cardiovasc Pathol. 2004;13(2):91–97. doi: 10.1016/S1054-8807(03)00129-7. [DOI] [PubMed] [Google Scholar]
- 39.Weiler-Guettler H, et al. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J Clin Invest. 1998;101(9):1983–1991. doi: 10.1172/JCI2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorman MW, Feigl EO, Buffington CW. Human plasma ATP concentration. Clin Chem. 2007;53(2):318–325. doi: 10.1373/clinchem.2006.076364. [DOI] [PubMed] [Google Scholar]
- 41.Manzanero S. Generation of mouse bone marrow-derived macrophages. Methods Mol Biol. 2012;844:177–181. doi: 10.1007/978-1-61779-527-5_12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.