

Abstract
The BrafV637E mutation is frequently reported in mouse hepatic tumors, depending on the mouse strain, and corresponds to the human BrafV600E mutation. In this study, we detected the BrafV637E mutation by whole‐exome analysis in 4/4 hepatic tumors induced by neonatal treatment with diethylnitrosamine (DEN) in male B6C3F1 mice. We also detected the BrafV637E mutation in 54/63 (85.7%) hepatic lesions, including microscopic foci and grossly visible tumors, by PCR‐direct sequencing. Although the mutation was detected in 5/7 (71.4%) hepatic tumors induced by neonatal DEN treatment followed by repeated CCl4 administration, it was not detected in 24 tumors induced by CCl4 treatment without DEN or in eight spontaneous lesions in B6C3F1 mice, suggesting that the mutation is induced by the genotoxic action of DEN. The DEN‐induced tumors exhibited hyperphosphorylation of ERK1 and Akt, suggesting that the BrafV637E mutation might activate the MAPK and Akt pathways. Moreover, the DEN‐induced tumors overexpressed mRNAs for the oncogene‐induced senescence (OIS) markers such as p15Ink4b and p19Arf as well as pro‐survival/pro‐proliferative cytokines/chemokines such as complement C5/C5a, ICAM‐1, IL‐1 receptor antagonist and CXCL9, suggesting that the BrafV637E mutation influences the expression of genes involved in either OIS or cellular growth/survival. Liver‐specific expression of mutated Braf under control of the albumin enhancer/promoter resulted in an enlarged liver that consisted entirely of small basophilic hepatocytes resembling DEN‐induced preneoplastic hepatocytes with ERK1/Akt hyperphosphorylation and C5/C5a overexpression. These results indicate that the BrafV637E mutation induces hepatocytic changes in DEN‐induced hepatic tumors. © 2016 The Authors. Molecular Carcinogenesis published by Wiley Periodicals, Inc.
Keywords: whole‐exome analysis, hepatic tumors, intracellular signaling, cytokines/chemokines, BrafV600E transgenic mice
Abbreviations
- DEN
diethylnitrosamine
- HCC
hepatocellular carcinoma
- OIS
oncogene‐induced senescence
- ICAM‐1
intercellular adhesion molecule‐1
- IL‐1ra
interleukin 1 receptor antagonist
INTRODUCTION
Hepatocellular carcinoma (HCC) is the sixth most common cancer and the third leading cause of cancer death worldwide 1. Many rodent hepatocarcinogenesis models have been developed to investigate the pathogenesis of HCC, to perform risk assessments of food/nutrition and to evaluate the effects of therapeutic agents. Diethylnitrosamine (DEN) is often used to induce hepatocarcinogenesis in rodents. Delivering a single low dose of DEN to neonatal mice results in the development of HCC without any further treatment 2. In this model, foci of cellular alterations are observed in the early stage of hepatocarcinogenesis, and in the late stages, adenomas and HCC develop.
The mutational changes associated with chemically induced hepatocarcinogenesis depend not only on the carcinogens used but also on the species, strain, age and gender of the animals 3. Buchmann et al. 4 have reported that in the hepatic tumors induced by neonatal treatment with DEN or 7, 12‐dimethylbenz(a)anthracene, the H‐ras codon 61 mutation is more prevalent in hepatocarcinogenesis‐susceptible strains such as C3H mice, whereas the BrafV637E mutation is more prevalent in resistant strains such as C57BL/6, indicating that either mutation may be selected depending on the genetic background.
The mouse BrafV637E mutation corresponds to the human BrafV600E mutation, which is frequently detected in certain human cancers, including melanoma and thyroid, ovarian and colon cancers 5. In this study, we determined that the BrafV637E mutation is very common in hepatic lesions induced by neonatal DEN treatment in male B6C3F1 mice. However, the frequency of the Braf mutation in hepatocarcinogenesis induced by agents other than DEN and the changes induced by the Braf mutation in hepatocytes, particularly with respect to intracellular signaling and gene expression, have not been well studied. We therefore investigated (i) whether this mutation is specific to the DEN‐induced model, (ii) how the BrafV637E mutation might provide a selective advantage to the mutated cells, and (iii) whether liver‐specific expression of mutated Braf using an albumin enhancer/promoter causes phenotypic changes resembling those of DEN‐induced hepatic tumors.
MATERIALS AND METHODS
Carcinogenic Treatment
Male B6C3F1 mice, which are commonly used for DEN‐induced hepatocarcinogenesis 2 and to assess the carcinogenicity of chemicals 6, were generated by mating female C57BL/6J and male C3H/HeJ mice (CLEA Japan, Tokyo, Japan). One group of mice was intraperitoneally injected with DEN at a dose of 5 μg/g body weight at 2 wk of age, and was fed a normal chow diet thereafter. The mice were sacrificed at 5, 8, and 13 months (Table 1). The second group was subcutaneously injected with CCl4 (1 ml/kg body weight) three times weekly beginning at 8 wk of age and was sacrificed at 6 months (Table 2). The third group was treated first with DEN (5 μg/g body weight) at 2 wk of age and then with intraperitoneal CCl4 injections twice weekly starting at 4 wk and was sacrificed at 6 months after the start of CCl4 treatment (Table 2). Spontaneous hepatic lesion samples isolated from 20‐month‐old male B6C3F1 mice were kindly provided by Dr. Takuji Tanaka (Tokai Cell Institute). Grossly visible tumors were isolated, frozen in liquid nitrogen and stored at −80°C until use. Microscopic foci were microdissected from 4‐μm‐thick paraffin sections using a 27‐gauge needle under microscopic observation. Livers of age‐matched untreated mice were used as controls. All experimental procedures performed on mice were approved by the Asahikawa Medical University animal experiment committee on the basis of the guidelines for the humane care and protection of animals.
Table 1.
Incidence of BrafV637E Mutation in DEN‐Induced Lesions
| Age (months) | No. mice | Foci | Tumors a |
|---|---|---|---|
| 5 | 7 | 16/18 (88.8%) | — |
| 8 | 3 | 22/28 (78.5%) | — |
| 13 | 3 | — | 16/17 (94.1%) |
Histological type, not determined.
Table 2.
Incidence of BrafV637E Mutation in Spontaneous, and CCl4 or DEN/CCl4‐Induced Lesions
| Histological types | |||||
|---|---|---|---|---|---|
| Lesions | Age (months) | No. mice | BrafV637E no. lesions | Foci | HCC |
| Spontaneous | 20 | 6 | 0/8 (0%) | 6 | 2 |
| CCl4 a | 6 | 16 | 0/24 (0%) | — | — |
| DEN/CCl4 a | 6 | 3 | 5/7 (71.4%) | — | — |
Histological type, not determined.
Generation of Alb‐Cre/BrafV600E Mice
Homozygous male BrafV600E mice (B6.129P2(Cg)‐Braftm1Mmcm>/J) reported by Dankort et al. 7 were purchased from Jackson Laboratories (Bar Harbor, ME). In these mice, a LoxP‐flanked cassette containing the human Braf exon 15–18 cDNA, mouse Braf polyadenylation sequences and neo gene is inserted into the mouse Braf intron 14 upstream of the human Braf exon 15 cDNA encoding the BrafV600E mutation. Alb‐Cre/BrafV600E mice were produced by in vitro fertilization using heterozygous female Alb‐Cre (Jackson Laboratories) and homozygous male BrafV600E mice. The hepatic expression of the transgene was investigated by RT‐PCR and direct sequencing. Twenty male and 20 female Alb‐Cre/BrafV600E mice were generated.
Whole‐Exome Analysis
DNA was extracted from the DEN‐induced hepatic tumors and age‐matched normal liver samples and then purified using a DNeasy Blood and Tissue Kit (Qiagen, Netherlands). For hepatic tumors, non‐parenchymal cells were removed before DNA extraction as follows: the livers containing the DEN‐induced tumors were perfused with collagenase solution, and the tumors (5–10 mm in diameter) were removed, minced with scissors, and passed through a 100‐μm cell strainer (BD Biosciences, San Jose, CA). The tumor cells were collected by low‐speed centrifugation (90g for 1 min) and washed twice with phosphate‐buffered saline (PBS). After a quality check of the DNA using a Qubit Fluorometer (Thermo Fisher Scientific, Waltham, MA), the exons were enriched by using an Agilent SureSelect Mouse All Exon kit (Agilent Technologies, Santa Clara, CA), and this was followed by sequencing on a HiSeq 2000 sequencer (Illumina, San Diego, CA). The mutations were analyzed within the exon regions with more than 100‐fold coverage.
PCR or RT‐PCR and Direct Sequencing
For grossly visible tumors, DNA was extracted from frozen materials with a DNeasy Blood and Tissue Kit. For microscopic foci, DNA was extracted from microdissected samples with a DEXPAT kit (Takara Bio, Japan). The nomenclature of microscopic foci followed the criteria of the International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice 8. The region that included the Braf codon 637 was amplified by PCR using appropriate primers (forward: 5′‐gacctcacggtaaaaataggtgac, reverse: 5′‐gcaattatgcctggcttacaa) and Mighty Amp DNA Polymerase (Takara Bio). Total RNA was extracted from frozen liver tissues of normal C57BL/6J and Alb‐Cre/BrafV600E mice with Sepasol (Nacalai Tesque, Japan) and was reverse‐transcribed with a cDNA Synthesis Kit (Roche, Basel, Switzerland). The region that included the Braf codon 637 was amplified from the cDNA samples by PCR using primers (forward: 5′‐acttacacgccaagtcaatc, reverse: 5′‐gcatacacgtctgactgaaag) corresponding to the mouse Braf exons 14 and 16, respectively. The PCR products were purified with ExoStar (GE Healthcare, United Kingdom) and sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) on an Applied Biosystems 3500 Genetic Analyzer (Thermo Fisher Scientific).
Quantitative Real‐Time PCR (qRT‐PCR)
To quantify the p15Ink4b and p19Arf mRNA, RNA was extracted from DEN‐induced hepatic tumors and age‐matched normal livers. cDNA synthesized as described above was used in qRT‐PCR reactions with incorporation of LightCycler RNA Master SYBR Green I (Roche) using the following primers: p15Ink4b, 5′‐ccaatccaggtcatgatgat (forward) and 5′‐cgtgcacaggtctggtaag (reverse); p19Arf, 5′‐gctctggctttcgtgaacat (forward) and 5′‐tgagcagaagagctgctacg (reverse). Samples were normalized to the levels of Gapdh mRNA quantified by qRT‐PCR using the following primers: 5′‐accacagtccatgccatcac (forward) and 5′‐tccaccaccctgttgctgta (reverse). To quantify the PCR products, a standard curve was created using the PCR products purified with a PCR purification kit (Qiagen), and the DNA concentration was measured with a NanoDrop 2000 spectrophotometer (Thermo Fischer Scientific).
Immunohistochemistry
Tissue sections were incubated with primary antibodies against complement C5/C5a (Abbiotec, San Diego, CA) or Ki‐67 (Novus Biological, Littleton, CO), and this was followed by detection of antibody binding using a Vector‐stain kit (Vector, Burlingame, CA). Tissue sections that were not incubated with the primary antibodies served as negative controls.
Immunoblot Analysis
DEN‐induced tumor samples were lysed in RIPA buffer, and equal amounts of protein were separated on 10% SDS‐polyacrylamide gels and transferred to nitrocellulose membranes. The blots were probed with antibodies against phospho‐ and total ERK1/2 (Cell Signaling, Danvers, MA) and Akt (Cell Signaling), BrafV600E (Ventana, Tucson, AZ), total Braf (Cell Signaling) and C5/C5a. As a loading control, α‐tubulin was detected using anti‐α‐tubulin antibody (Santa Cruz, Santa Cruz, CA). Band images were acquired using ECL Prime Western Blotting Detection Reagent (GH Healthcare Life Sciences, Pittsburgh, PA) and by exposure of the membranes to X‐ray films.
Cytokine/Chemokine Array
The expression of cytokines and chemokines was investigated in two DEN‐induced hepatic tumors (13 months) and two age‐matched normal livers with a Mouse Cytokine Antibody Array Panel A, with loading of 40 types of specific antibodies (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. The reactions were detected by chemiluminescence, and the density of the spots was quantified with ImageJ software and normalized to the positive and negative control spots on the membranes.
Statistical Analysis
Statistical analyses were performed using Student's t‐tests, the Mann–Whitney test or Fischer's exact test by using Prism software (GraphPad, La Jolla, CA), and probability values less than 0.05 were defined as statistically significant.
RESULTS
BrafV637E Mutation in DEN‐Induced Hepatic Tumors
Whole‐exome analysis detected 98 mutations in the four tumors isolated from three mice 13 months after neonatal DEN treatment (Supplementary Table S1). Of these, 96 were missense mutations, one was a six‐base deletion, and one was a one‐base insertion. Six mutations were detected in all four tumors, three mutations in 3/4 tumors, seven mutations in 2/4 tumors, and 82 mutations in 1/4 tumors (Supplementary Table S1). DEN induces G:C (C:G) to A:T (T:A) transitions via O6‐ethyl guanine and T:A (A:T) to C:G (G:C) transitions via O4‐ethyl thymine adducts 9. In the present study, 47 of 96 (48.9%) base change mutations were attributed to O6‐ethyl guanine or O4‐ethyl thymine adducts. Although not attributable to O6‐ethyl guanine or O4‐ethyl thymine adducts, 38/96 (39.5%) base change mutations, including the BrafV637E mutation (GTG to GAG), were T:A (A:T) to A:T (T:A) transversions, which have been reported to be one of three types of the H‐ras codon 61 mutations (wild type CAA to AAA, CGA or CTA) in mouse hepatic tumors induced by neonatal treatment with DEN 9.
The A:T to T:A transversion at the 2nd base of codon 637 in the Braf gene, which causes a valine to glutamic acid substitution (BrafV637E), was detected in all four tumors (Supplemental Table S1). Importantly, however, no mutations were detected in other known oncogenes or tumor suppressor genes. We then investigated the BrafV637E mutation in 17 hepatic tumors isolated from the three mice 13 months after DEN treatment. The BrafV637E mutation was detected in 16 (including the 4 samples used for the whole‐exome analysis above) of 17 tumors by PCR/direct sequencing (Table 1). Because the H‐ras codon 61 mutation is frequently observed in DEN‐induced mouse liver tumors 10, 11, we investigated the mutations in codons 12, 13, and 61 of the H‐ras, K‐ras and N‐ras genes, respectively, in the above 17 tumors. However, other than one K‐rasG13D mutation in one of the Braf mutated tumors, no mutations in these codons were detected in the samples (data not shown).
BrafV637E Mutation in Microscopic Lesions
We next addressed whether the BrafV637E mutation is also common in early DEN‐induced lesions. Foci of altered cells were microdissected from the tissue sections from the livers at 5 and 8 months, and the Braf mutation was investigated by the PCR direct sequencing method (Figure 1A and B). At 5 months, 16/18 (88.8%) of the foci contained the BrafV637E mutation, and at 8 months, 22/28 (78.5%) foci contained the mutation (Table 1); however, the BrafV637E mutation was not observed in the non‐tumor areas of the same sections (Figure 1B). Therefore, the BrafV637E mutation was equally frequent in the hepatic lesions at 5 and 8 months as at 13 months.
Figure 1.
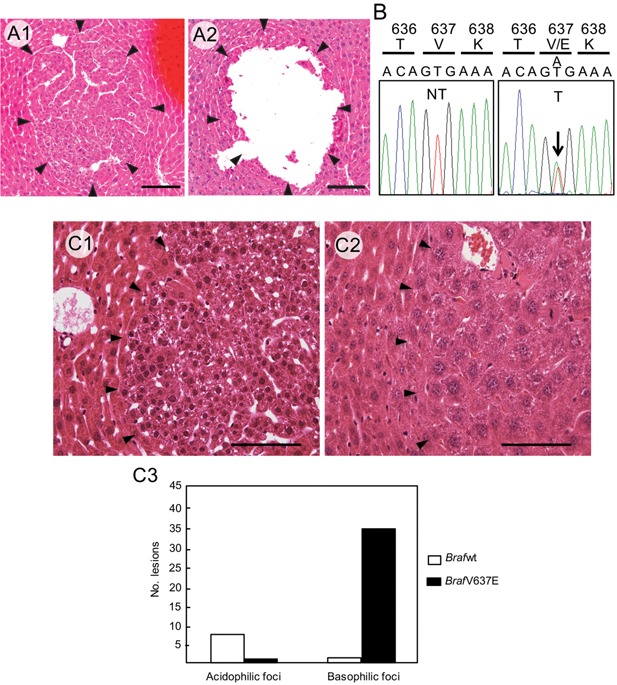
A: BrafV637E mutation in microscopic lesions. Focus of cellular alteration (indicated by arrowheads) before (A1) and after (right) microdissection (A2). Scale bars: 100 μm. B: Braf sequence from non‐tumor (NT) and tumor tissues (T). The A:T to T:A transversion at Braf codon 637 leads to the BrafV637E mutation in the focus (arrow). C: Focus cells with the BrafV637E mutation mainly exhibited basophilic cytoplasm with smaller nuclei (C1), whereas those with wild‐type Braf mainly displayed acidophilic cytoplasm with large nuclei (C2). Scale bars: 50 μm. Thirty‐six of 37 basophilic foci contained the BrafV637E mutation, whereas only 1/8 acidophilic foci contained the mutation (P < 0.0001, Fischer's exact test) (C3).
Foci of cellular alteration are classified into subtypes (basophilic, acidophilic, mixed cell, clear and amphophilic) 8, and we therefore investigated the types of lesions that contained the BrafV637E mutation. Of 45 foci investigated for the BrafV637E mutation, 37 were basophilic, and 8 were eosinophilic. Thirty‐six of 37 basophilic foci (97.3%) had the BrafV637E mutation, whereas only 1 of 8 (12.5%) eosinophilic foci contained the BrafV637E mutation (basophilic vs. eosinophilic foci: P < 0.0001) (Figure 1C3).
BrafV637E Mutation in Spontaneous Hepatic Tumors and in CCl4‐ or DEN/CCl4‐Induced Hepatic Tumors
We next explored whether the BrafV637E mutation is specific to DEN‐induced hepatic tumors. For this purpose, the mutation was investigated in spontaneous hepatic lesions and in CCl4‐ or DEN/CCl4‐induced hepatic tumors in male B6C3F1 mice. None of the 8 spontaneous hepatic lesions (6 foci and 2 HCCs) and none of the 24 CCl4‐induced hepatic tumors had the mutation, whereas 5/7 (71.4%) of the DEN/CCl4 tumors had the BrafV637E mutation (Table 2). The difference in the frequency of the BrafV637E mutation between DEN and spontaneous or CCl4 lesions was statistically significant (P < 0.0001), whereas the difference between DEN and DEN/CCl4 lesions were not (P = 0.3020).
Activation of Intracellular Signaling
We investigated whether the BrafV637E mutation might activate the downstream ERK1/2 pathway in DEN‐induced hepatic tumors. Weak phosphorylation of both ERK1/2 was detected in age‐matched normal liver tissue, whereas prominent phosphorylation of ERK1 was observed in DEN‐induced tumors isolated 13 months after DEN treatment (Figure 2). Because the PI3K/Akt pathway plays an important role in hepatocarcinogenesis 12, 13, 14 and because PI3K/Akt activation can reverse mutated Braf oncogene‐induced senescence (OIS) 15, we investigated Akt phosphorylation in the DEN‐induced hepatic tumors. The phosphorylation of Akt at S473 was increased in the hepatic tumors, whereas phosphorylation at T308 was weakly detected in normal livers but not in hepatic tumors (Figure 2).
Figure 2.
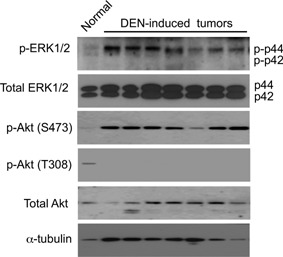
Phosphorylation status of ERK1/2 and Akt in normal liver and DEN‐induced tumors at 13 months after neonatal treatment with DEN. α‐tubulin: loading control.
Expression of Cell‐Cycle Regulators
The mutational activation of Braf leads to OIS, which is characterized by the overexpression of cell‐cycle regulators and may prevent cellular proliferation and induce a cellular senescence phenotype and/or apoptosis 16, 17, 18. We therefore addressed whether the DEN‐induced hepatic tumors exhibit OIS by investigating the expression of p15Ink4b and p19Arf, which play important roles in cell‐cycle arrest in mouse cells 19, 20. Quantitative RT‐PCR revealed that p15Ink4b and p19Arf mRNAs were increased in DEN‐induced hepatic tumors isolated at 13 months after DEN treatment, whereas no signals for p19Arf and p15Ink4b mRNAs were detected in the age‐matched normal livers after 35 cycles of PCR (Figure 3A and B).
Figure 3.
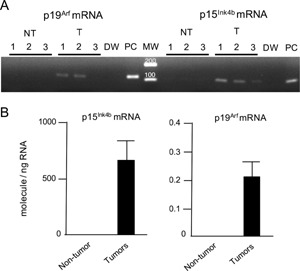
A: RT‐PCR for p15Ink4b and p19Arf mRNA in non‐tumor livers (NT), hepatic tumors (T), a negative control (distilled water: DW) and a positive control (PCR product used as template: PC). A total of 35 cycles of PCR were performed. MW: molecular weight marker. B: p15Ink4b and p19Arf mRNA levels were determined by qRT‐PCR of DEN‐induced hepatic tumors (n = 9) 13 months after neonatal DEN treatment and in age‐matched normal livers (n = 8).
Overexpression of Pro‐Proliferative/Pro‐Survival Cytokines/Chemokines in DEN‐Induced Hepatic Tumors
OIS may lead to the senescence‐associated secretory phenotype, which is characterized by the increased production of cytokines/chemokines that facilitate cell cycle arrest, apoptosis and immunosurveillance 21, 22. We, therefore, investigated cytokine/chemokine expression in two DEN‐induced hepatic tumors 13 months after DEN treatment and in two age‐matched normal livers by using a mouse array with antibodies specific to 40 types of cytokines/chemokines. As shown in Figure 4A and B, the expression of complement C5/C5a, intercellular adhesion molecule‐1 (ICAM‐1), interleukin 1 receptor antagonist (IL‐1ra) and chemokine CXCL9 was increased among the 40 factors specifically associated with hepatic tumors, whereas there were no significant differences in the expression levels of other factors between the tumors and age‐matched normal livers. The expression of ICAM‐1 23, IL‐1ra 24 and CXCL9 25 has previously been reported in DEN‐induced hepatic tumors; however, C5/C5a expression has not been reported. We therefore investigated C5/C5a expression in the hepatic lesions 5 and 8 months after DEN treatment by immunohistochemistry. Although the surrounding non‐tumor hepatocytes exhibited diffuse/weak cytoplasmic staining for C5/C5a, the cells in the foci displayed strong staining in the cytoplasm, presumably in the rough endoplasmic reticulum and Golgi apparatus compartments (Figure 4C and inset). No staining was observed when the primary antibody was omitted (data not shown).
Figure 4.
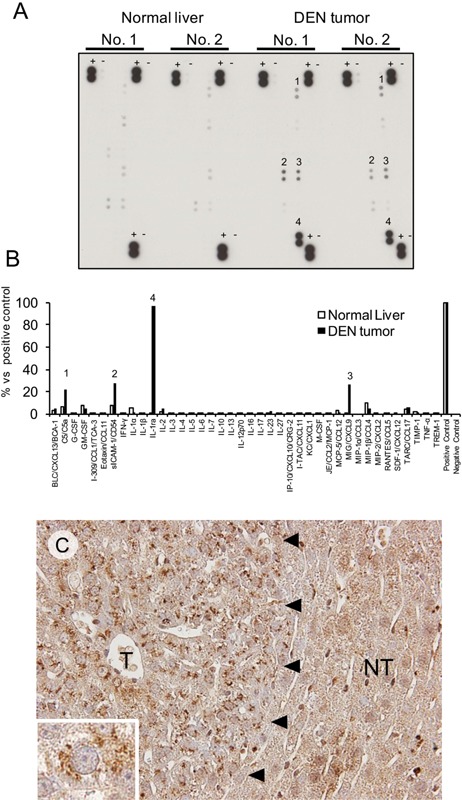
A: The cytokine/chemokine array analysis used a membrane to load the antibodies individually against 40 types of mouse cytokines/chemokines. Four factors (1: C5/C5a, 2: ICAM‐1, 3: CXCL9, 4: IL‐1ra) were increased in hepatic tumors compared with normal livers. +: positive control, −: negative control. B: Densitometric analysis of the above data. The data represent the average values for two normal livers and two DEN‐induced hepatic tumors. C: Immunohistochemical staining for C5/C5a in the focus (T) and in the surrounding non‐tumor hepatic tissue (NT) (left). Inset: high magnification of the focus cell.
Characteristics of the Liver of Alb‐Cre/BrafV600E Mice
Alb‐Cre/BrafV600E mice were normal at birth, but started to die 8 wk after birth. We sacrificed the surviving animals during the period between 8 and 12 wk after birth. The livers of Alb‐Cre/BrafV600E mice expressed mRNA derived both from normal mouse and mutated human Braf exon 15 (Figure 5A and B). The transgenic mice exhibited a 20% decrease in body weight compared with age‐matched normal mice, but showed a fivefold‐increase in the liver/body weight (Figure 5C, D, and E). Histological examination revealed that the liver consisted entirely of basophilic hepatocytes resembling basophilic foci at 8 wk of birth, and bile duct cells were further increased at 10–12 wk (Figure 5F1‐3). The Ki‐67 labeling index of hepatocytes in the Alb‐Cre/BrafV600E mice was 3 times higher than that in normal mice (Figure 5G). Immunoblot analysis revealed that the livers of Alb‐Cre/BrafV600E mice expressed the BrafV600E protein and showed hyperphosphorylation of ERK1 and AktS473 in a manner similar to that observed in DEN‐induced hepatic tumors (Figure 6). Furthermore, immunohistochemical staining detected increased C5/C5a expression in the hepatocytes of Alb‐Cre/BrafV600E mice (Figure 7A). In immunoblot analysis, although C5 levels were not different between age‐matched normal and Alb‐Cre/BrafV600E mice, a band with a smaller molecular weight probably corresponding to C5a was specifically detected in Alb‐Cre/BrafV600E livers (Figure 7B). There was no difference in hepatic phenotype between male and female Alb‐Cre/BrafV600E mice.
Figure 5.
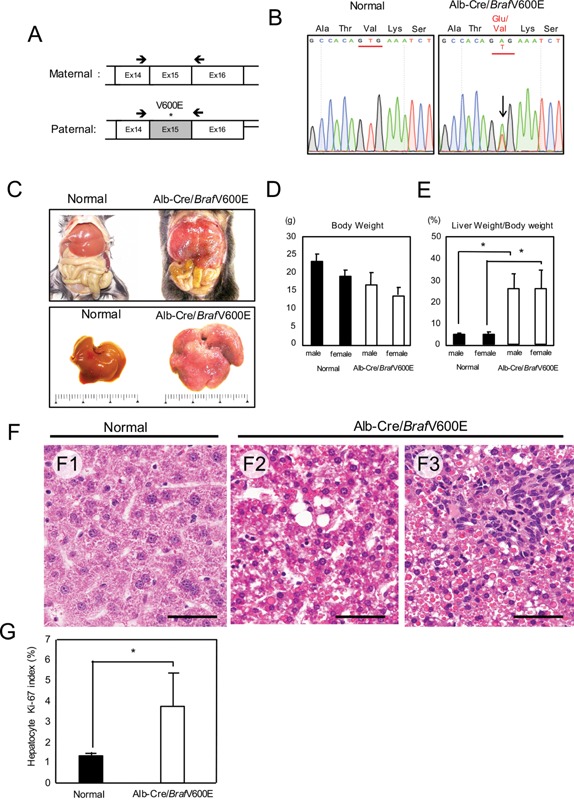
Alb‐Cre/BrafV600E mice. A: RT‐PCR primers (arrows) targeting mutated human and normal mouse Braf exon 15. B: The mutated (A) and normal bases (T) in Alb‐Cre/BrafV600E mice (arrow in the right panel). C: Livers of normal and Alb‐Cre/BrafV600E mice (C). D: Body weight of normal (n: 10 males, 10 females) and Alb‐Cre/BrafV600E mice (n: 20 males, 20 females). E: Liver weight relative to body weight in normal (n: 10 males, 10 females) and Alb‐Cre/BrafV600E mice (n: 20 males, 20 females). *P < 0.01. F: Histology of the liver in normal (F1) and Alb‐Cre/BrafV600E mice at 8 wk (F2) and 12 wk after birth (F3). Scale bars: 50 μm. G: The Ki‐67 labeling index of hepatocytes in normal (n = 5) and Alb‐Cre/BrafV600E mice (n = 6). *P < 0.05.
Figure 6.
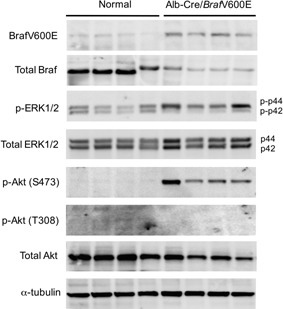
Expression of the BrafV600E protein and the phosphorylation status of ERK1/2 and Akt in the liver of normal and Alb‐Cre/BrafV600E mice.
Figure 7.
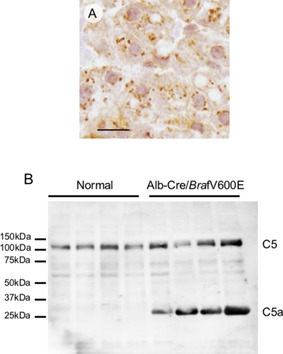
A: C5/C5a immunohistochemical staining in the livers of Alb‐Cre/BrafV600E mice. Scale bar: 10 μm. B: Immunoblot analysis of C5/C5a in the livers of normal and Alb‐Cre/BrafV600E mice.
DISCUSSION
Whole‐exome analysis of four DEN‐induced hepatic tumors at 13 months detected 98 mutations; however, with the exception of the BrafV637E mutation, no known oncogene or tumor suppressor gene mutations were observed. PCR‐direct sequencing of additional DEN‐induced lesions, including microscopic foci and grossly visible tumors, revealed that 54/63 (85.7%) lesions had the BrafV637E mutations. Although some mutations might be passenger mutations, other mutations, particularly those detected in two or more tumors, might function as oncogenic drivers. In the hepatocarcinogenesis model induced by neonatal DEN treatment, individual DEN‐induced tumors grow at different rates, and only a small fraction of preneoplastic lesions eventually progress to HCC 2. Therefore, it is possible that some mutations may contribute to tumor progression in cooperation with the BrafV637E mutation.
The BrafV637E mutation was not detected in the spontaneous and CCl4‐induced tumors but was detected in the tumors induced by neonatal DEN treatment followed by repeated CCl4 treatment, suggesting that the mutation was dependent on the genotoxic action of DEN. Further supporting this conclusion, 47/96 (48.9%) of the base‐change mutations detected by whole‐exome analysis were attributable to O6‐ethyl guanine and O4‐ethyl thymine, mutagenic adducts generated by DEN 9. O6‐ethyl guanine can form a G:T mismatch pair instead of a G:C pair, leading to G:C (C:G) to A:T (T:A) transition, whereas O4‐ethyl thymine can mismatch with guanine instead of adenine, leading to the T:A (A:T) to C:G (G:C) transition 9. Furthermore, although not attributable to O6‐ethyl guanine or O4‐ethyl thymine, 38/96 (39.5%) of the base‐change mutations, including the BrafV637E mutation (GTG to GAG), were T:A (A:T) to A:T (T:A) transversions. The A:T to T:A transversion has been detected as one of three types of the H‐ras codon 61 mutations (wild‐type CAA to CTA) in DEN‐induced mouse hepatic tumors, together with the CAA to AAA (not attributable to O6‐ethyl guanine or O4‐ethyl thymine) and CAA to CGA mutations (attributable to O4‐ethyl thymine) 9. Thus, the Braf V637E mutation (GTG to GAG) may be generated by an unknown mechanism similar to that resulting in the H‐ras codon 61 CAA to CTA mutation.
The BrafV637E mutation may lead to activation of the MAPK pathway. In the present study, although total ERK1/2 protein levels did not differ between normal livers and hepatic tumors, phosphorylation of ERK1 was increased in hepatic tumors, whereas both ERK1/2 were weakly phosphorylated in normal livers. Although the exact mechanism of hyperphosphorylation of ERK1 remains unclear, ERK1 may play a primary role in the activation of MAPK pathway. On the other hand the phosphorylation of Akt at S473 was increased in the hepatic tumors. Activation of the PI3K/Akt pathway has been reported to reverse OIS caused by the Braf mutation 15, and thus Akt activation may play a significant role in DEN‐induced hepatocarcinogenesis. Although the mechanism of Akt phosphorylation requires further investigation, an unknown signaling pathway linked to the BrafV637E mutation and/or the autocrine factors generated by the hepatic tumor cells may play a role, as discussed below.
The mRNAs of p15Ink4b and p19Arf were detected in the hepatic tumors, but not in normal livers, suggesting that the BrafV637E mutation activates the OIS‐signaling pathways along with pro‐proliferative/survival pathways. We have previously demonstrated that the cyclin‐dependent kinase inhibitor p27Kip1 is overexpressed in DEN‐induced hepatic tumors and that the BrdU labeling index is quite low in early tumors, indicating a slow rate of proliferation 12. Because grossly visible tumors are not usually observed until 6–8 months after treatment with DEN in our experimental model, the growth of BrafV637E‐mutated cells may be suppressed for a long period. Cyclin‐dependent kinase inhibitors, such as p15Ink4b, p19Arf, and p27Kip‐1, may counteract the BrafV637E‐induced pro‐proliferative signals, contributing to the dormancy of BrafV637E‐mutated cells.
Cytokines/chemokines that participate in sustained cell proliferation, insensitivity to apoptosis and escape from immunosurveillance in cancer, such as C5/C5a, ICAM‐1, IL‐1ra, and CXCL9, were increased in the DEN‐induced hepatic tumors, whereas factors that promote OIS, such as IL‐1α, β, IL‐6, INFγ, and TNFα 21, 22 were not increased. Kang et al. 26 have reported that introduction of the N‐rasG12V mutation into mouse hepatocytes in vivo leads to OIS, which in turn causes the OIS‐induced secretory phenotype. The chemokines and cytokines such as CTACK, IL‐1α, leptin, MCP1, and RANTES generated by N‐rasG12V expressing hepatocytes recruit inflammatory cells that contribute to clearance of the N‐rasG12V expressing hepatocytes. In the present study, however, we did not observe any inflammatory response against preneoplastic/neoplastic hepatocytes, and thus it is possible that the lesions that we investigated might have somehow escaped from the inflammatory attack in an early stage.
C5a, a C5 convertase‐cleaved product of C5, activates the PI3K‐Akt pathway via its receptor C5aR, which is involved in various tumor phenotypes, including cell proliferation, insensitivity to apoptosis, angiogenesis and escape from immunosurveillance 27, 28. Furthermore, C5‐deficient mice exhibit impaired liver regeneration after partial hepatectomy 29. ICAM‐1 is overexpressed in DEN‐induced rat hepatic tumor cells 23 and in human HCC cells 30, and levels of the secreted form sICAM‐1 are increased in the serum of HCC patients 31, 32. ICAM‐1 promotes metastasis via cell/cell and cell/extracellular matrix adhesion in cancer cells 33, and sICAM‐1 is thought to inhibit the interaction between tumor cells and immune cells, enabling escape from immunosurveillance 34. Our group has previously demonstrated overexpression of IL‐1ra, which antagonizes the functions of IL‐1α, β, in DEN‐induced hepatic tumors 24, and an increase in IL‐1ra over IL‐1α, β contributes to tumor growth 35, 36. CXCL9 is reportedly upregulated in livers containing DEN‐induced hepatic tumors in mice 25, and CXCL9 is excreted by tumor cells and subsequently recruits immune cells that express its receptor (CXCR3) towards the tumors. However, the CXCL9‐CXCR3 interaction can attenuate tumor immunity and produce pro‐tumorigenicity, depending on the tumor type 37. Furthermore, CXCL9 directly promotes invasion/infiltration via CXCR3 on HCC cells in vitro 38. In addition, our previous studies, together with those of other groups, have demonstrated that DEN‐induced hepatic tumor cells produce a number of growth factors, such as TGF‐α 39, insulin‐like growth factor II 40, VEGF 13 and NGF 41. Thus, the autocrine pro‐proliferative/survival factors produced by the hepatic tumor cells may provide a selective advantage by accumulating within the tumor microenvironment. Additional studies are needed to clarify the relevance of autocrine factors to the BrafV637E mutation.
The transgenic mice expressing human BrafV600E, which corresponds to mouse BrafV637E, in the liver under control of the albumin/enhancer/promoter exhibited a 5‐fold increase in liver/body weight ratio, and the liver consisted entirely of small basophilic hepatocytes mimicking those observed in basophilic foci induced by DEN. Furthermore, the liver exhibited hyperphosphorylation of ERK1 and AktS473 and increased expression of C5/C5a. Thus, the expression of BrafV637E protein is sufficient to induce the hepatocytic changes observed in DEN‐induced hepatic tumors. However, the Alb‐Cre/BrafV600E mice started to die 8 wk after birth, and most died by 12 wk. A similar phenomenon has previously been reported for the transgenic mice with liver‐specific H‐ras expression under control of the albumin enhancer/promoter 42. The possibility that aberrant activation of Braf, and presumably also H‐ras, in the liver causes fatal systemic disease(s) is now under investigation in our laboratory.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Table S1. List of mutated genes detected by whole exome analysis in four DEN‐induced hepatic tumors.
ACKNOWLEDGMENTS
This study was supported by grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology and grants from the Asahikawa Medical University Advanced Science Research Program. We are grateful to Dr. Takuji Tanaka of the Tokai Cell Institute for providing spontaneous mouse hepatic tumor samples.
[The copyright line for this article was changed on 17 June 2016 after original online publication.]
Conflict of interest: The authors declare no conflict of interest.
REFERENCES
- 1. Mittal S, Serag El. Epidemiology of hepatocellular carcinoma. J Clin Gastroenterol 2013; 47:S2–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vesselinovitch SD, Mihailovich N, Rao KV. Morphology and metastatic nature of induced hepatic nodular lesions in C57BL x C3H F1 mice. Cancer Res 1978; 38:2003–2010. [PubMed] [Google Scholar]
- 3. Maronpot RR, Fox T, Malarkey DE, Goldsworthy TL. Mutations in the ras proto‐oncogene: Clues to etiology and molecular pathogenesis of mouse liver tumors. Toxicology 1995; 101:125–156. [DOI] [PubMed] [Google Scholar]
- 4. Buchmann A, Karcier Z, Schmid B, Strathmann J, Schwarz M. Differential selection for B‐raf and Ha‐ras mutated liver tumors in mice with high and low susceptibility to hepatocarcinogenesis. Mutat Res 2008; 638:66–74. [DOI] [PubMed] [Google Scholar]
- 5. Pakneshan S, Salajegheh A, Smith RA, Lam AK‐Y. Clinicopathological relevance of BRAF mutations in human cancer. Pathology 2013; 45:346–356. [DOI] [PubMed] [Google Scholar]
- 6. Huff JE, McConnell EE, Haseman JK, et al. Carcinogenesis studies: Results of 398 experiments on 104 chemicals from the U.S. National Toxicology Program. Ann N Y Acad Sci 1988; 534:1–30. [DOI] [PubMed] [Google Scholar]
- 7. Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E‐induced lung tumors. Genes Dev 2007; 21:379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thoolen B, Maronpot RR, Harada T, et al. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol 2010; 38:5S–81S. [DOI] [PubMed] [Google Scholar]
- 9. Verna L, Whysner J, Williams GM. N‐nitrosodiethylamine mechanistic data and risk assessment: Bioactivaton, DNA‐adduct formation, mutagenicity, and initiation. Pharmacol Ther 1996; 71:57–81. [DOI] [PubMed] [Google Scholar]
- 10. Buchmann A, Bauer‐Hofmann R, Mahr J, Drinkwater NR, Luz A, Schwarz M. Mutational activation of the c‐Ha‐ras gene in liver tumors of different rodent strains: Correlation with susceptibility to hepatocarcinogenesis. Proc Natl Acad Sci USA 1991; 88:911–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dragani TA, Manenti G, Colombo BM, et al. Incidence of mutations at codon 61 of the Ha‐ras gene in liver tumors of mice genetically susceptible and resistant to hepatocarcinogenesis. Oncogene 1991; 6:333–338. [PubMed] [Google Scholar]
- 12. Yamamoto M, Tamakawa S, Yoshie M, Yaginuma Y, Ogawa K. Neoplastic hepatocyte growth associated with cyclin D1 redistribution from the cytoplasm to the nucleus in mouse hepatocarcinogenesis. Mol Carcinog 2006; 45:901–913. [DOI] [PubMed] [Google Scholar]
- 13. Tanaka H, Yamamoto M, Hashimoto N, et al. Hypoxia‐independent overexpression of hypoxia‐inducible factor 1 as an early change in mouse hepatocarcinogenesis. Cancer Res 2006; 66:11263–11270. [DOI] [PubMed] [Google Scholar]
- 14. Villanueva A, Chiang DY, Newell P, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008; 135:1972–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vredeveld LCW, Possik PA, Smit MA, et al. Abrogation of BRAFV600E‐induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012; 26:1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dhomen N, Reis‐Filho JS, Dias S, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 2009; 15:294–303. [DOI] [PubMed] [Google Scholar]
- 17. Michaloglou C, Vredeveld L, Soengas M, et al. BRAFE600‐associated senescence‐like cell cycle arrest of human naevi. Nature 2005; 436:720–724. [DOI] [PubMed] [Google Scholar]
- 18. Collado M, Serrano M. The power and the promise of oncogene‐induced senescence markers. Nat Rev Cancer 2006; 6:472–726. [DOI] [PubMed] [Google Scholar]
- 19. Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997; 91:649–659. [DOI] [PubMed] [Google Scholar]
- 20. Latres E, Malumbres M, Sotillo R, et al. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J 2000; 19:3496–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bartek J, Hodny Z, Lukas J. Cytokine loops driving senescence. Nat Cell Biol 2008; 10:1–3. [DOI] [PubMed] [Google Scholar]
- 22. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland J. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J Clin Invest 2013; 123:966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsuoka S, Matsumura H, Arakawa Y, et al. Expression of intercellular adhesion molecule‐1 in the livers of rats treated with diethylnitrosamine. J Clin Biochem Nutr 2009; 45:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamada Y, Karasaki H, Matsushima K, Lee GH, Ogawa K. Expression of an IL‐1 receptor antagonist during mouse hepatocarcinogenesis demonstrated by differential display analysis. Lab Invest 1999; 79:1059–1067. [PubMed] [Google Scholar]
- 25. Schneider C, Teufel A, Yevsa T, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine‐induced liver cancer. Gut 2012; 61:1733–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature 2011; 479:547–551. [DOI] [PubMed] [Google Scholar]
- 27. Cho MS, Vasquez HG, Rupaimoole R, et al. Autocrine effects of tumor‐derived complement. Cell Reports 2014; 6:1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol Cancer Res 2010; 8:1453–1465. [DOI] [PubMed] [Google Scholar]
- 29. Strey CW, Markiewski M, Mastellos D, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med 2003; 198:913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Momosaki S, Yano H, Ogasawara S, Higaki K, Hisaka T, Kojiro M. Expression of intercellular adhesion molecule 1 in human hepatocellular carcinoma. Hepatology 1995; 22:1708–1713. [PubMed] [Google Scholar]
- 31. Zöhrens G, Armbrust T, Pirzer U, Meyer zum Büschenfelde KH, Ramadori G. Intercellular adhesion molecule‐1 concentration in sera of patients with acute and chronic liver disease: Relationship to disease activity and cirrhosis. Hepatology 1993; 18:798–802. [DOI] [PubMed] [Google Scholar]
- 32. Shimizu Y, Minemura M, Tsukishiro T, et al. Serum concentration of intercellular adhesion molecule‐1 in patients with hepatocellular carcinoma is a marker of the disease progression and prognosis. Hepatology 1995; 22:525–531. [PubMed] [Google Scholar]
- 33. Hayes SH, Seigel GM. Immunoreactivity of ICAM‐1 in human tumors, metastases and normal tissues. Int J Clin Exp Pathol 2009; 2:553–560. [PMC free article] [PubMed] [Google Scholar]
- 34. Witkowska AM, Borawska MH. Soluble intercellular adhesion molecule‐1 (sICAM‐1): An overview. Eur Cytokine Netw 2004; 15:91–98. [PubMed] [Google Scholar]
- 35. La E, Rundhaug JE, Fischer SM. Role of intracellular interleukin‐1 receptor antagonist in skin carcinogenesis. Mol Carcinog 2001; 30:218–223. [DOI] [PubMed] [Google Scholar]
- 36. Arend WP. The balance between IL‐1 and IL‐1Ra in disease. Cytokine Growth Factor Rev 2002; 13:323–340. [DOI] [PubMed] [Google Scholar]
- 37. Billottet C, Quemener C, Bikfalvi A. CXCR3, a double‐edged sword in tumor progression and angiogenesis. Biochim Biophys Acta 2013; 1836:287–295. [DOI] [PubMed] [Google Scholar]
- 38. Lan X, Xiao F, Ding Q, et al. The effect of CXCL9 on the invasion ability of hepatocellular carcinoma through up‐regulation of PREX2. J Mol Histol 2014; 45:689–696. [DOI] [PubMed] [Google Scholar]
- 39. Tanno S, Ogawa K. Abundant TGFα precursor and EGF receptor expression as a possible mechanism for the preferential growth of carcinogen‐induced preneoplastic and neoplastic hepatocytes in rats. Carcinogenesis 1994; 15:1689–1694. [DOI] [PubMed] [Google Scholar]
- 40. Ishizaki T, Yoshie M, Yaginuma Y, Tanaka T, Ogawa K. Loss of Igf2 imprinting in monoclonal mouse hepatic tumor cells is not associated with abnormal methylation patterns for the H19, Igf2, and Kvlqt1 differentially methylated regions. J Biol Chem 2003; 278:6222–6228. [DOI] [PubMed] [Google Scholar]
- 41. Kishibe K, Yamada Y, Ogawa K. Production of nerve growth factor by mouse hepatocellular carcinoma cells and expression of TrkA in tumor‐associated arteries in mice. Gastroenterology 2002; 122:1978–1986. [DOI] [PubMed] [Google Scholar]
- 42. Sandgren EP, Quaife CJ, Pinkert CA, Palmiter RD, Brinster RL. Oncogene‐induced liver neoplasia in transgenic mice. Oncogene 1989; 4:715–724. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Table S1. List of mutated genes detected by whole exome analysis in four DEN‐induced hepatic tumors.