

Summary
Although type 1 diabetes (T1D) is a T‐cell‐mediated disease in the effector stage, the mechanism behind the initial beta cell assault is less understood. Metabolomic differences, including elevated levels of glutamic acid, have been observed in patients with T1D before disease onset, as well as in pre‐diabetic non‐obese diabetic (NOD) mice. Increased levels of glutamic acid damage both neurons and beta cells, implying that this could contribute to the initial events of T1D pathogenesis. We investigated the underlying genetic factors and consequences of the increased levels of glutamic acid in NOD mice. Serum glutamic acid levels from a (NOD×B6)F2 cohort (n = 182) were measured. By genome‐wide and Idd region targeted microsatellite mapping, genetic association was detected for six regions including Idd2, Idd4 and Idd22. In silico analysis of potential enzymes and transporters located in and around the mapped regions that are involved in glutamic acid metabolism consisted of alanine aminotransferase, glutamic‐oxaloacetic transaminase, aldehyde dehydrogenase 18 family, alutamyl‐prolyl‐tRNA synthetase, glutamic acid transporters GLAST and EAAC1. Increased EAAC1 protein expression was observed in lysates from livers of NOD mice compared with B6 mice. Functional consequence of the elevated glutamic acid level in NOD mice was tested by culturing NOD. Rag2 −/− Langerhans’ islets with glutamic acid. Induction of apoptosis of the islets was detected upon glutamic acid challenge using TUNEL assay. Our results support the notion that a dysregulated metabolome could contribute to the initiation of T1D. We suggest that targeting of the increased glutamic acid in pre‐diabetic patients could be used as a potential therapy.
Keywords: beta cell apoptosis, genetics, glutamic acid, Idd, non‐obese diabetic mice
Abbreviations
- ALAT
alanine aminotransferase
- ASAT
glutamic‐oxaloacetic transaminase
- B6
C57BL/6
- EAAC1
excitatory amino acid carrier 1
- EAAT
excitatory amino acid transporter
- GLAST
glial glutamate and aspartate transporter
- GLT
glial glutamate transporter
- NOD
non‐obese diabetic
- SNP
single nucleotide polymorphism
- T1D
type 1 diabetes
Introduction
Type 1 Diabetes (T1D) is an autoimmune disease with an increasing incidence globally.1 A strong genetic contribution, including >50 loci, is an important factor underlying the disease,2 (www.t1dbase.org). However, a clear environmental contribution has also been shown, including viral infections, diet and sun exposure (reviewed in refs 3, 4, 5). In humans the first signs of pathogenesis of T1D has so far been limited to the appearance of autoantibodies in the sera,6 although this must be preceded by molecular and cellular events, including both the beta cells and the immune system. One example of a feature proceeding seropositivity has been demonstrated in children genetically predisposed to T1D,7 where levels of glutamic acid were elevated prior to the appearance of autoantibodies against insulin and glutamic acid decarboxylase. In addition, a correlation between age‐dependent appearance of autoantibodies and the metabolite methionine has been observed in pre‐diabetic children.8
The non‐obese diabetic (NOD) mouse is the most widely studied model for T1D.9 Not only does this strain develop spontaneous diabetes, the disease pathogenesis is similar to human diabetes.10 NOD mice show various immune deviations, including defective apoptosis in T cells,11 defective suppression by regulatory T cells,12 and enhanced and prolonged immune responses.13 In accordance with the human metabolic study,7 we have shown that NOD mice exhibit an altered metabolic profile before T1D onset,14 with elevated levels of glutamic acid in the sera. Similar results have been reported by others, i.e. a sustained elevated glutamic acid level was observed in a group of NOD T1D progressor mice.15
Although the autoimmune process has been well studied in NOD mice, the early events leading to full‐fledged autoimmunity are still to be completely elucidated. Apoptosis is considered important in development of autoimmune diseases16 as well as in triggering of T1D.17, 18, 19 Rodents, including NOD mice, exhibit beta cell mass remodelling and undergo waves of apoptosis during development.17, 20 Recently, it was shown that beta cells are specifically sensitive to extracellular glutamic acid and undergo apoptosis under physiological glutamic acid levels.21 This defines a novel mechanism of beta cell death and suggests that glutamic acid could contribute directly to pathogenesis in T1D.
Glutamate is the anionic/salt form of glutamic acid and is considered the most abundant neurotransmitter in the vertebrate brain. Glutamate is known to act on three families of ionotropic receptors: N‐methyl‐d‐aspartate, 2a‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid and kainite, reviewed in ref. 22. In addition, five glutamate transport enzymes, i.e. glial glutamate and aspartate transporter (GLAST), glial glutamate transporter (GLT), excitatory amino acid carrier 1 (EAAC1), excitatory amino acid transporter (EAAT) 4 and EAAT5 have been found in the mammalian nervous system.23 Although glutamate is an important neurotransmitter, it is also associated with neurotoxicity and chronic neurodegeneration.24, 25 In addition, glutamate seems to mediate beta cell apoptosis through oxidative stress,21 whereas pre‐treating them with ceftriaxone, which is known to increase GLT1 expression, rescues these cells by enhancing the expression of glutamate transporter GLT126.
In this study we analysed the genetic basis of the elevated glutamic acid levels in NOD mice – including possible single nucleotide polymorphisms (SNPs) in candidate genes and their regulatory regions and protein expression of selected glutamic acid transporters; and studied the effects of glutamic acid on Langerhans’ islets.
Materials and methods
Mice
All mice used in this study were bred and maintained in the general animal facility at Umeå University. Experimental procedures were performed in compliance with the relevant Swedish and Institutional laws and guidelines and approved by the Umeå research animal ethics committee (A44‐12;03/07/2012, A2‐15;15/1/2015). NODShilt/J, NOD.Rag2 −/− and C57BL/6J (B6) mice have been bred for more than 10 generation in our own animal facility. (NOD×B6)F2 mice were established by first crossing NOD female mice with B6 male mice, and then intercrossing the obtained (NOD×B6)F1 mice.
Sample collection and LC‐MS determination of glutamate levels
Blood was collected by retro‐orbital bleeding and left at room temperature for 30–60 min. After overnight clotting, serum was collected and frozen at −80° until analysis. All the extraction and LC‐MS procedures were performed by the Swedish Metabolomics Centre at Umeå universitet using standardized protocols. Samples were thawed, and an extraction mixture containing 80% methanol, and 3 ng/µl [15N]1‐glutamic acid (as internal standard; Cambridge Isotope Laboratories, Tewksbury, MA) was added to the serum. The mixture was shaken in a mixer mill for 3 min at 30 Hz and kept on ice for 30 min before being centrifuged for 10 min at 24,1000 × g RCF. Supernatants were transferred into LC‐MS vials and evaporated. For the derivatization, a Waters AccQ Tag™ derivatization kit (Waters, Milford, MA) was used as per the manufacturer's recommendations.
For the HPLC, an HP1290 ultra‐high‐performance liquid chromatography system from Agilent Technologies (Waldbronn, Germany) was used. An aliquot of the sample was injected into an HPLC column (Kinetex, Phenomenex) and held at 55° in a column oven. For gradient elution, two buffers were used: buffer A (H2O, 0·1% formic acid) and buffer B (acetonitrile, 0·1% formic acid).
The MS system used was an Agilent 6460 triple quadrupole mass spectrometer equipped with a jet‐stream electrospray source operating in positive ion mode. The LC‐MS analysis was controlled by masshunter™ software v 5.01 and the data were processed by masshunter™ Quantification software v 5.01 (Agilent Technologies).
Genotype analysis
Genomic DNA was extracted from tails according to standard techniques. Briefly, a 2‐mm tailpiece was digested with tail buffer I (25 mm NaOH, 0·2 mm EDTA) at 95° for 45 min and 4° for 45 min. The tail digest was neutralized by tail buffer II (40 mm Tris–HCl). The selected cohort of F2 mice (NOD×B6)F2 (n = 40) were genotyped using microsatellite markers and conventional PCR protocol (Sigma Aldrich, St. Louis, Missouri, USA). Selected markers covering the whole genome, including the Idd regions, were D1Mit430, D1Mit303, D1Mit181, D1Mit387, D1Mit498, D1Mit353, D1Mit117, D2Mit429, D2Mit367, D2Mit14a, D2Mit449, D2Mit504, D3Mit182, D3Mit244, D3Mit12, D3Mit106, D3Mit160, D4Mit235, D4Mit139, D4Mit166, D4Mit221, D4Mit251, D4Mit234, D4Mit343, D5Mit145, D5Mit387, D5Mit197, D5Mit275, D5Mit164, D6Mit86, D6Mit223, D6Mit263, D6Mit254, D6Mit14, D7Mit178, D7Mit191, D7Mit145, D7Mit235, D7Mit242, D8Mit294, D8Mit249, D8Mit80, D8Mit242, D8Mit113, D8Mit56, D9Mit323, D9Mit328, D9Mit105, D9Mit262, D9Mit182, D10Mit167, D10Mit257, D10Mit259, D10Mit233, D11Mit74, D11Mit140, D11Mit38, D11Mit168, D12Mit182, D12Mit136, D12Mit14, D12Mit101, D13Mit205, D13Mit61, D13Mit254, D13Mit228, D14Mit50, D14Mit212, D14Mit37, D14Mit197, D15Mit175, D15Mit206, D15Mit238, D15Mit149, D15Mit161, D16Mit182, D16Mit211, D16Mit43, D16Mit158, D17Mit164, D17Mit199, D17Mit28, D17Mit105, D17Mit218, D18Mit197, D18Mit149, D18Mit107, D18Mit4, D19Mit59, D19Mit117, D19Mit100 and D19Mit70. PCR products were run on 4% agarose gels using 2% low‐melting NuSieve GTG agarose (Cambrex, East Rutherford, NJ, USA) and 2% agarose (Sigma‐Aldrich) in TBE‐buffer, and visualized by GelRed (Biotium, San Francisco, CA). In general, the polymorphism between the NOD and the B6 microsatellite variants ranged between 6 and 200 bp.
Candidate gene analysis
Pathways involved in the metabolism of glutamic acid were collected from the KEGG pathway database for mice. Enzymes, receptors and transport proteins involved in glutamic acid metabolism were collected from a mouse genome informatics database (http://www.informatics.jax.org/). The SNPs in the genes were retrieved from the Mouse Phenome Database (http://phenome.jax.org/db/q?rtn=snp/home) using data for NOD and B6 mice. SNPs present in upstream or downstream regions were also collected.
The Ensembl Regulatory Build was used to identify the candidate gene specific regulatory regions and their associated regulatory proteins that were likely to be involved in gene regulation.27 Those SNPs that were localized within a 1‐kb region of promoter regions, within 500‐bp of the transcription factor binding/CTCF sites, enhancers and open chromatin regions were considered. The SNPs localized within a regulatory region as well as in the vicinity of another regulatory region were considered to be able to affect both, and so were counted twice.
Islet isolation, culture and apoptosis assay
Islets were isolated from 8‐ to 12–week‐old NOD.Rag2 −/− mice to exclude the risk of immune‐mediated beta cell death. Pancreata were cannulated with 5 ml of cold Hanks’ balanced salt solution containing 0·2 mg/ml collagenase P (Roche Diagnostics, Basel, Switzerland) through the common bile duct. Pancreata were collected and digested at 37°. Islets were purified on a Ficoll gradient and hand‐picked twice under a dissection microscope; 40–50 islets/well (96‐well flat bottom plate) were cultured in RPMI‐1640 medium containing 10% fetal bovine serum, antibiotics and different concentrations of glutamic acid as indicated in the Figure 4. Apoptosis was visualized by TUNEL assay according to the manufacturer's recommendations (Roche Diagnostics, Basel, Switzerland) with some modifications. For the washing steps with PBS, plates were centrifuged at 100 g for 30 seconds and the supernatant was removed manually under a microscope to avoid damage of the islets. In brief, post‐culture islets were washed with PBS and fixed with 2% paraformaldehyde for 1 hr at room temperature. After washing, the islets were permeabilized with freshly made 0·1% Triton‐X‐100 in 0·1% sodium citrate. After permeabilization, the islets were stained with reaction mixture. Stained islets were transferred to a clear‐bottom black‐sided plate (Greiner bio, Kremsmünster, Austria) and visualized with a Zeiss Axiovert 200 inverted live cell microscope.
Figure 4.
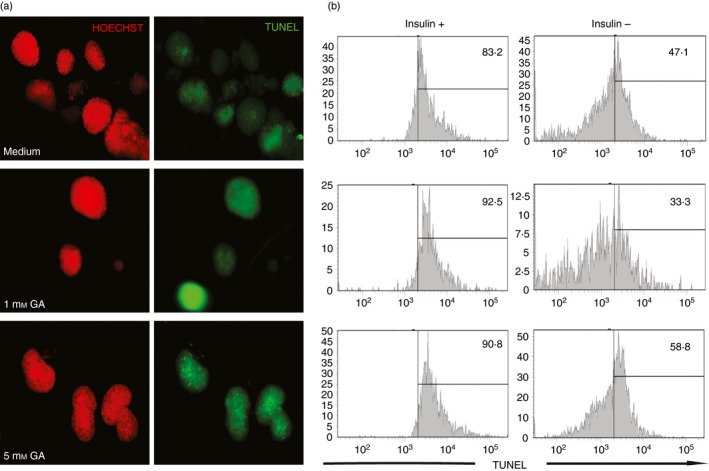
Glutamic acid induced apoptosis in beta cells. Islets from non‐obese diabetic (NOD) RAG females were isolated and cultured for 3 days in the absence of glutamic acid or 1 mm glutamic acid or 5 mm glutamic acid. (a) TUNEL (green) staining was used to detect islet cell apoptosis. Hoechst (red) was used to stain the DNA. (b) After microscopy, the same islets were dispersed to make a single‐cell suspension, stained with insulin and analysed by flow cytometry. [Colour figure can be viewed at wileyonlinelibrary.com]
Tissue collection and Western blotting
Liver and kidney from 4‐ to 8‐week‐old NOD and B6 mice were collected, snap frozen in liquid nitrogen and stored at −80° till analysis. For Western blot, frozen tissue samples were powdered in a mortar and weighed. Appropriate amounts of Proteojet mammalian cell lysis buffer (Fermentas, Waltham, MA, Canada) containing protease inhibitors (Roche Diagnostics) and 2% SDS were added to the tissue samples according to the manufacturer's recommendations. Protein lysates were vortexed for 20 min and incubated on ice for 15 min. DNA was sheared by passing the samples through a 27G needle. The samples were spun at 18 000 g for 15 min and supernatants were collected. Protein concentrations were measured by Bradford assay (Bio‐Rad, Hercules, CA, USA).
Protein amounts were normalized for all samples and 50 µg proteins were resolved on a 10% polyacrylamide gel under reducing conditions. As a positive control, lysates from rat cerebellum and EAAC1‐transfected cells were included (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The proteins were transferred to a nitrocellulose membrane (Bio‐Rad, Hercules, CA, USA) and non‐specific binding was blocked using 5% non‐fat milk in PBST. The membranes were probed with rabbit anti‐mouse EAAT1 (GLAST) antibody (Cell Signaling, Danvers, MA, USA; 1 : 1000, lot# 1), rabbit anti‐mouse EAAT3 (EAAC1) antibodies (Santa Cruz Biotechnology; 1 : 1000, lot# I2211) and rabbit anti‐mouse GAPDH (Proteintech, Chicago, IL, USA; 1 : 5000, lot# 18091) overnight at 4°. Proteins were visualized using a horseradish peroxidase‐conjugated goat anti‐rabbit IgG secondary antibody (Agrisera, Vännäs, Sweden; 1 : 10 000, lot# 1504) and SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA). Bands were detected using ImageQuant LAS4000 (GE Healthcare, Little Chalfont, UK) and intensity quantified using image analysis software from Licor (Lincoln, NE, USA). Expression levels in all the organs were normalized to the loading control, GAPDH. No significant difference in the expression levels of GAPDH between NOD and B6 liver and kidney samples was observed (data not shown).
Statistics
Phenotypic differences were analysed using two‐tailed, unpaired Student's t‐test or one‐way analysis of variance with Bonferroni correction as indicated. Phenotype–genotype associations were analysed by χ 2 analysis, with two degrees of freedom using ibm spss software (IBM, North Castle, NY, USA). In the mapping analysis a P‐value < 0·05 was considered significant. For protein expression comparisons, Mann–Whitney U‐test was applied using graphpad prism 6 (GraphPad, La Jolla, CA, USA).
Results
Increased glutamic acid levels in NOD
We have previously shown that glutamic acid levels are increased in NOD.14 To determine the genes influencing this phenotype, 4‐week‐old NOD, B6 and (NOD×B6)F1 mice were bled and their glutamic acid levels were measured by LC‐MS. Confirming our previous results, NOD mice displayed increased glutamic acid levels compared with B6 mice. Interestingly, the (B6×NOD)F1 mice displayed intermediate levels of glutamic acid, indicating a co‐dominant mode of action of the gene(s) to the phenotype (Fig. 1).
Figure 1.
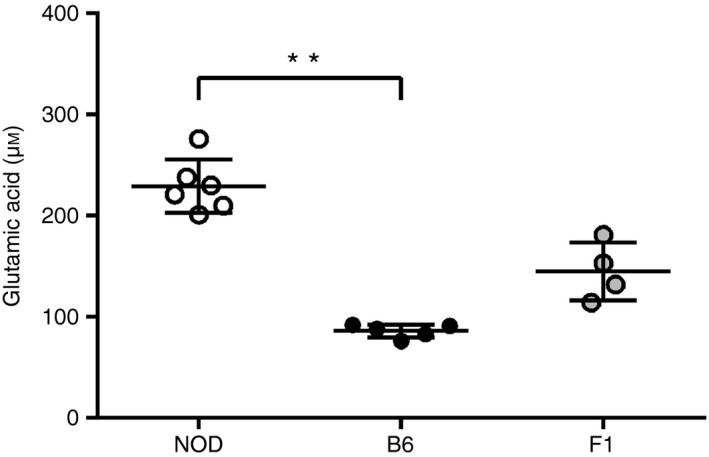
Increased glutamic acid levels in non‐obese diabetic (NOD) mice. Sera from NOD (n = 6), C57BL/6 (B6) (n = 5) and (B6×NOD)F1 (n = 4) mice were analysed by LC‐MS. NOD mice displayed the highest concentration (229 μm), F1 an intermediate level (145 μm), and B6 the lowest (86 μm). Bars depict the mean ± SD for each strain. **P = 0.005
Elevated glutamic acid levels are controlled by genes inside and outside Idd‐regions
To dissect the genetic component(s) responsible for the increase in glutamic acid, a cohort of 182 (NOD×B6)F2 mice were bred and serum was collected when the mice were 4 weeks old. The glutamic acid level in the serum of each individual was measured using LC‐MS. As shown in Figure 2, the levels varied in the F2 mice close to a normal distribution, indicating a multigenic control of the phenotype.
Figure 2.
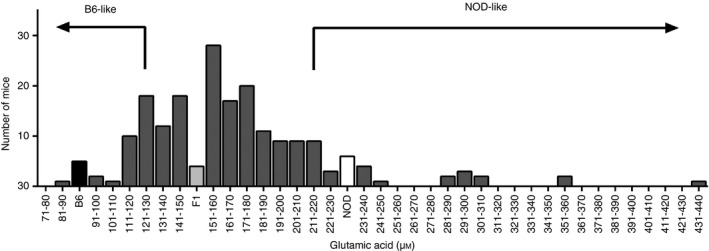
Phenotypic distribution of glutamic acid in a cohort of [C57BL/6 (B6) × non‐obese diabetic (NOD)]F2 mice. The glutamic acid level of the (B6×NOD)F2 mice (n = 182) was measured by LC‐MS. Based on parental strain glutamic acid levels, F2 mice were classified either as NOD‐like or B6‐like. These mice were used for mapping.
Selective genotyping of individuals with extreme phenotypes is known to impart power similar to that from genotyping the whole cohort28, 29, 30. Accordingly, 40 of the F2 mice were classified as either NOD‐like or B6‐like based on their serum glutamic acid levels. The 20 mice with the lowest glutamic acid levels were grouped as B6‐like and the 20 mice with the highest glutamic acid levels as NOD‐like. As this phenotype was controlled by several genes, this particular set of mice was included to achieve sufficient power for the study, with a risk of a potential loss of sensitivity.
To determine the responsible genetic region(s) for the increased glutamic acid level in NOD mice, genotyping was performed in the NOD‐like and B6‐like (B6×NOD)F2 mice. Ninety‐two markers distributed throughout the genome, including the previously defined Idd regions, were used. As shown in Table 1, six chromosomes showed significant association (P < 0·05), of which three regions were positioned within Idd regions, i.e. Idd22, Idd2 and Idd4. The other six regions that showed significant association or close to significant association were positioned on chromosomes 1, 15 and 19. In all cases the NOD‐derived alleles did contribute to the NOD‐like phenotype.
Table 1.
Genetic association of increased glutamic acid levels
| Marker | Region | B6‐like | NOD‐like | P‐value | ||||
|---|---|---|---|---|---|---|---|---|
| B | F | N | B | F | N | |||
| D1Mit117 | 10 | 7 | 3 | 0 | 17 | 3 | 0·001 | |
| D8Mit80 | Idd22 | 7 | 12 | 1 | 2 | 12 | 6 | 0·042 |
| D9Mit105 | 7 | 12 | 1 | 3 | 8 | 9 | 0·012 | |
| D9Mit182 | Idd2 | 6 | 13 | 1 | 2 | 9 | 9 | 0·010 |
| D11Mit38 | Idd4 | 8 | 11 | 1 | 2 | 12 | 6 | 0·027 |
| D15Mit175 | 9 | 10 | 1 | 4 | 9 | 7 | 0·039 | |
| D15Mit206 | 6 | 14 | 0 | 1 | 12 | 7 | 0·005 | |
| D19Mit117 | 11 | 9 | 0 | 6 | 10 | 4 | 0·063 | |
| D19Mit100 | 3 | 17 | 0 | 3 | 9 | 8 | 0·005 | |
In total, 92 microsatellite markers were tested for association to NOD‐like or B6‐like glutamic acid levels. Markers with P < 0·05 were considered significant. Genotypic distribution within the chosen cohort is displayed where B represents the B6 parent, N represents the NOD parent and F represents the (NOD×C57BL/6)F1. Genotype–phenotype association was analysed by χ 2 analysis.
Candidate gene analysis
To further understand the factors controlling the glutamic acid levels, we performed a detailed in silico analysis of the glutamic acid metabolism. Enzymes, receptors and transporters harboured in and around the mapped regions were retrieved from the KEGG and MGI databases. Using this criterion, nine enzymes, two transport proteins and two receptors were found to lie in and around the significantly mapped regions (Table 2). The corresponding genes for these enzymes were analysed for potential genetic differences between NOD and B6 mice, i.e. SNPs, using the dbSNP database.31 The search from the dbSNP database yielded 145 SNPs among the significantly mapped genes, of which four SNPs were in the coding regions, 15 in the 5′, 3′ untranslated regions and the remaining 126 SNPs were present in the intron regions (Table 2).
Table 2.
Candidate gene analysis
| Protein | Gene/Function | Full name/SNPs observed (non‐synonymous, synonymous, intron, upstream, splice site) | P‐value | Idd | Marker |
|---|---|---|---|---|---|
| (Transport) | |||||
| GLAST | Slc1a3 | Glial high‐affinity glutamate transporter, member 3 (0,0,2,0,0) | 0·005 | D15Mit175 | |
| EAAC1 | Slc1a1 | Neuronal/epithelial high‐affinity glutamate transporter, member 1 (0,0,0,0,0) |
0·063 0·005 |
D19Mit117 D19Mit100 | |
| (Receptors) | |||||
| Grik4 | Grik4 | Ionotropic glutamate receptor, kainate 4 (0,0,59,0,0) | 0·012 | D9Mit105 | |
| Grm2 | Grm2 | Metabotropic glutamate receptor 2 (0,1,12,12,0) | 0·010 | Idd2 | D9Mit182 |
| (Enzymes) | |||||
| Eprs | Eprs | Glutamyl‐prolyl‐tRNA synthetase (0,1,2,0,0) | 0·001 | D1Mit117 | |
| Aadat | Aadat | Aminoadipate aminotransferase (0,1,21,1,0) | 0·042 | Idd22 | D8Mit80 |
| Gpt2 | Gpt2 | Glutamic pyruvate transaminase (alanine aminotransferase) 2 (0,0,0,0,0) | 0·042 | Idd22 | D8Mit80 |
| Got2 | Got2 | Glutamatic‐oxaloacetic transaminase 2, mitochondrial (0,0,0,1,0) | 0·042 | Idd22 | D8Mit80 |
| Gclc | Gclc | Glutamate‐cysteine ligase, catalytic subunit (0,0,16,0,0) | 0·012 | D9Mit105 | |
| 0·010 | Idd2 | D9Mit182 | |||
| Pfas | Pfas | Phosphoribosylformylglycinamidine synthase (0,0,0,0,0) | 0·027 | Idd4 | D11Mit38 |
| Ggt6 | Ggt6 | γ‐Glutamyltransferase 6 (0,0,0,0,0) | 0·027 | Idd4 | D11Mit38 |
| Aldh18a1 | Aldh18a1 | Aldehyde dehydrogenase 18 family, member A1 (0,0,0,0,0) | 0·005 | D19Mit100 | |
| Got1 | Got1 | Glutamic‐oxaloacetic transaminase 1, soluble (0,1,14,1,0) | 0·005 | D19Mit100 |
Genes corresponding to glutamic acid metabolism that are present in the significantly associated loci were analysed.
To analyse the possible effect of genetic variation affecting gene regulation for the candidate genes, we used the Ensemble Regulatory Build. As displayed in Table 3, multiple regulatory regions were identified for eight of the candidate genes.
Table 3.
Number of regulatory region in candidate genes
| Promoter | Open chromatin sites | Promoter flanking region | CTCF/TF binding site | Enhancer | |
|---|---|---|---|---|---|
| Slc1a3 | 1 | 6 | 5 | 6 | 1 |
| Grm2 | 0 | 1 | 0 | 6 | 0 |
| Grik4 | 1 | 14 | 5 | 23 | 4 |
| Eprs | 1 | 0 | 4 | 6 | 0 |
| Aadat | 1 | 0 | 4 | 0 | 0 |
| Gclc | 1 | 0 | 1 | 6 | 3 |
| Aldh18a1 | 1 | 0 | 1 | 4 | 1 |
| Got1 | 2 | 1 | 2 | 5 | 0 |
Next, the presence of SNPs within or in the vicinity of the regulatory regions presented in Table 3 was analysed. As shown in Table 4, multiple SNPs were present in or around the regulatory regions.
Table 4.
Number of single nucleotide polymorphisms identified in the regulatory regions of the candidate genes
| Promoter | Open chromatin sites | Promoter flanking region | CTCF/TF binding site | Enhancer | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Within | Vicinity | Within | Vicinity | Within | Vicinity | Within | Vicinity | Within | Vicinity | |
| Slc1a3 | 0 | 2 | 0 | 1 | 0 | 2 | 1 | 4 | 0 | 2 |
| Grm2 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 5 | 0 | 0 |
| Grik4 | 0 | 0 | 0 | 1 | 3 | 0 | 1 | 5 | 0 | 1 |
| Eprs | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 0 | 0 |
| Aadat | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| Gclc | 0 | 0 | 0 | 0 | 1 | 2 | 1 | 1 | 1 | 2 |
| Aldh18a1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Got1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
Finally, we analysed what specific transcription factors and DNA modification enzymes were associated with the above‐mentioned SNP‐altered regions. As shown in Table 5, a diverse set of factors were associated with these different candidate regions.
Table 5.
Enzymes and proteins associated with gene specific regulatory regions containing single nucleotide polymorphisms
| Enzymes/Proteins | Promoter | Open chromatin sites | Promoter flanking region | CTCF/TF binding site | Enhancer |
|---|---|---|---|---|---|
| H3K36me3 | Aadat | Slc1a3, Got1, Grik4 | Gclc, Got1 | Slc1a3, Eprs, Gclc | Gclc |
| JUN | Slc1a3 | ||||
| DNase1 | Slc1a3, Aadat | Slc1a3, Got1, Grik4 | Gclc, Got1, Grik4 | Slc1a3, Grm2, Eprs, Aadat, Gclc, Grik4 | Gclc |
| H3K4me3 | Slc1a3, Aadat | ||||
| H3K27ac | Slc1a3, Aadat | Got1, Grik4 | Gclc, Got1, Grik4 | Slc1a3, Aadat, Gclc | Gclc |
| SIN3A | Slc1a3 | Slc1a3 | |||
| MXI1 | Slc1a3 | Slc1a3 | |||
| BHLHE40 | Slc1a3 | Slc1a3, Grm2 | |||
| MYC | Slc1a3 | Slc1a3 | |||
| NELFe | Slc1a3 | Slc1a3 | |||
| Max | Slc1a3 | Slc1a3 | |||
| ZMIZ1 | Slc1a3 | Slc1a3 | |||
| H3K9ac | Slc1a3, Aadat | Got1, Grik4 | Gclc, Got1 | Slc1a3, Aadat, Gclc | |
| PolII | Slc1a3, Aadat | Slc1a3, Aadat | |||
| ZNF384 | Slc1a3 | ||||
| TCF12 | Grm2 | ||||
| SMC3 | Aadat | Gclc | Grm2, Aadat, Gclc, Grik4 | ||
| Rad21 | Aadat | Gclc | Grm2, Aadat, Gclc, Grik4 | ||
| H3K4me1 | Grik4 | Grik4 | |||
| MAZ | Grik4 | ||||
| H3K27me3 | Grik4 | Grm2, Grik4 |
Increased expression of EAAC1 in NOD liver
The in silico candidate gene analysis revealed important enzymes and high‐affinity transporters including alanine aminotransferase (ALAT), glutamic‐oxaloacetic transaminase (ASAT), excitatory amino acid transporters 1 and 3 [EAAT1 (GLAST) and EAAT3 (EAAC1)]. Prompted by this, we analysed the expression of the GLAST and EAAC1 by Western blot using whole organ lysates from liver and kidneys, important sites of amino acid transport. We found increased expression of EAAC1 in liver of NOD mice compared with the control B6 mice (Fig. 3). However, no difference in expression of EAAC1 was observed in the kidney from NOD and B6 mice (data not shown). Moreover, no detectable expression of GLAST (data not shown) was observed in these two organs, confirming previous studies.32, 33, 34, 35
Figure 3.
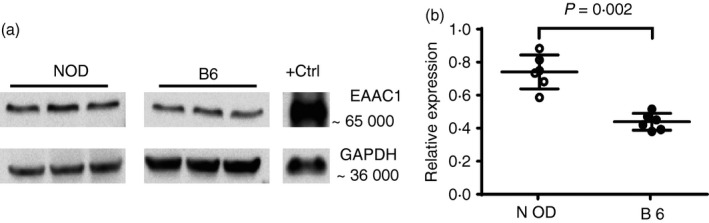
Increased expression of EAAC1 in non‐obese diabetic (NOD) mouse liver. (a) Whole liver lysates were resolved on SDS–PAGE and probed with EAAC1‐specific antibodies. Bands corresponding to EAAC1 and GAPDH were observed at the expected positions. Gel represents one of four experiments with similar results. (b) Relative expression of EAAC1 in livers of NOD and C57BL/6 (B6) mice (n = 6 per group). Band intensity was quantified and normalized to GAPDH expression.
Increased levels of glutamic acid induce apoptosis in healthy islets
Glutamic acid is the most common neurotransmitter in the mammalian brain. Glutamic acid has previously been shown to exert neurotoxic affects and is associated with neurodegenerative diseases.25 Glutamic acid has also been shown to be toxic to human beta cells at physiological concentrations.21 To test the effect of glutamic acid on primary beta cells of mouse origin, we isolated Langerhans’ islets from NOD.Rag2 −/− mice. Islets from these mice are free from autoimmune cellular infiltrates and therefore allow for scoring of islet apoptosis per se. Forty to fifty islets per well were cultured with increasing amounts of glutamic acid and stained for apoptosis. Induction of apoptosis was observed already at 1 mm glutamic acid (Fig. 4a). Beta cell specificity of the toxic effects of glutamic acid was analysed using flow cytometry. Cultured islets that had been analysed in microscope were dispersed and stained with insulin antibodies. As shown in Figure 4(b), increased numbers of apoptotic cells were observed among the insulin‐positive cells compared with the insulin‐negative cells. In addition, insulin‐positive cells appeared to be more sensitive to glutamic acid, as an increase in TUNEL‐positive cells was only observed in the insulin‐negative cells, whereas the insulin‐positive cells reached maximum apoptosis already at 1 mm of glutamic acid.
Discussion
Glutamic acid is one of the most distinguishing metabolites in children progressing to T1D, as well as in pre‐diabetic NOD mice.7, 14 In this study we focused on glutamic acid as a potential pathogenic metabolite and analysed both genetic and phenotypic factors controlling its elevated levels in NOD. We found that the elevated glutamic acid level in NOD was under multigenic control, with a significant genetic association to nine chromosomal regions including Idd2, Idd4 and Idd22. Other regions that were significantly associated were present on chromosomes 1, 9, 15 and 19. These significantly associated regions not only harbour important enzymes involved in glutamic acid metabolism, they also contain proteins and receptors associated with glutamic acid. Among the important glutamic acid transporters that showed significant association were GLAST and EAAC1. Both are important glutamic acid transporters expressed in the nervous system as well as in the epithelium. EAAC1 has been shown to be the only glutamate transporter present in intestinal epithelium,36 whereas GLAST is responsible for glutamate transport in other organs including brain and retina.37, 38 GLT1, expressed in the pancreas, was not significantly associated, suggesting the increased glutamic acid levels to be a result of impaired transporter function in other organs. Indeed, our Western blot results indicated an increased expression of EAAC1 in livers of NOD mice. An enhanced expression of EAAC1 could lead to an imbalance of glutamate in the system. It is also plausible that EAAC1 is up‐regulated in response to the increased glutamic acid level.
In addition to the glutamic acid transporters, important enzymes involved in glutamic acid metabolism were identified, including ALAT and ASAT. Both of these enzymes were considered to be important because they serve as a critical link between glutamic acid and the tricarboxylic acid cycle via the formation of α‐ketoglutarate.39 Soluble forms of these enzymes are present in blood and have been used to alter glutamate levels in blood.40 Our in silico analysis through various phenome databases showed significantly lower expression of ALAT in NOD mice.41, 42 Although below average levels of ALAT and ASAT are not considered pathogenic in clinical practice, the effect of chronic low levels of these enzymes in the body, and in particular in T1D is still to be revealed. We hypothesize that low levels of ALAT and ASAT sustain the enhanced glutamic levels observed in NOD and could also explain the lower levels of α‐ketoglutarate in NOD mice that we have reported previously.14 Moreover, bioinformatic analysis using the JAX Phenome database, as well as the Ensembl Regulatory Build, revealed the presence of multiple SNPs between the two strains. As no non‐synonymous SNP was identified, we focused on SNPs present in regulatory regions. Indeed, SNPs were present within or in the vicinity of regulatory regions for the selected genes. Based on this we hypothesize that this could affect transcription efficiency as well as splicing of the nascent RNA.43 However, additional analysis is required to truly pinpoint the possible mechanism.
Initial triggers in T1D have not been fully understood, although some studies have suggested excessive beta cell apoptosis as the possible trigger.17 Apoptosis, widely seen as a natural mechanism to maintain cellular homeostasis, can lead to a break in tolerance and can activate the immune system. Indeed, this has been seen in NOD mice, where anti‐DNA antibodies have been observed in the pancreatic infiltrate.19 Apoptotic cells can not only break tolerance but have the ability to induce an interferon response from the plasmacytoid dendritic cells.44 Interferon signature has in fact been observed to precede islet autoimmunity in children and NOD mice.45, 46 In this study, we report increased apoptosis among NOD beta cells after culture with glutamic acid. It is plausible that the increased glutamic acid levels leading to enhanced apoptosis acts as one of the triggers in T1D. We acknowledge that increased glutamic acid levels alone are not sufficient to trigger the disease. However, it is tempting to speculate that in conjunction with other deviations seen in NOD mice, such as defective clearance of apoptotic cells by macrophages, this phenotype could play a significant role in the initial events leading to the disease development.47 Interestingly, a single dose of Z‐VAD, a pan‐caspase inhibitor, protected NOD from T1D development.19
In conclusion, we propose that increased glutamic acid contributes to the initiation of T1D in the NOD mouse. It is possible that a similar mechanism contributes to human T1D, as it has been shown that this particular metabolite is increased in genetically susceptible individuals progressing to T1D.
Disclosures
The authors report no conflict of interest.
Acknowledgements
VB and KL designed and performed the experiments, analysed the data and wrote the paper. This study was supported by Barndiabetesfonden, Diabetesfonden, Insamlingsstiftelsen at Umeå University and Stiftelsen JC Kempe.
References
- 1. Vehik K, Dabelea D. The changing epidemiology of type 1 diabetes: why is it going through the roof? Diabetes Metab Res Rev 2011; 27:3–13. [DOI] [PubMed] [Google Scholar]
- 2. Todd JA. Etiology of type 1 diabetes. Immunity 2010; 32:457–67. [DOI] [PubMed] [Google Scholar]
- 3. Coppieters KT, Wiberg A, von Herrath MG. Viral infections and molecular mimicry in type 1 diabetes. APMIS 2012; 120:941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Holick MF. Vitamin D: importance in the prevention of cancers, type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr 2004; 79:362–71. [DOI] [PubMed] [Google Scholar]
- 5. Lefebvre DE, Powell KL, Strom A, Scott FW. Dietary proteins as environmental modifiers of type 1 diabetes mellitus. Annu Rev Nutr 2006; 26:175–202. [DOI] [PubMed] [Google Scholar]
- 6. Taplin CE, Barker JM. Autoantibodies in type 1 diabetes. Autoimmunity 2008; 41:11–8. [DOI] [PubMed] [Google Scholar]
- 7. Oresic M, Simell S, Sysi‐Aho M, Nanto‐Salonen K, Seppanen‐Laakso T, Parikka V, et al Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J Exp Med 2008; 205:2975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pflueger M, Seppanen‐Laakso T, Suortti T, Hyotylainen T, Achenbach P, Bonifacio E, et al Age‐ and islet autoimmunity‐associated differences in amino acid and lipid metabolites in children at risk for type 1 diabetes. Diabetes 2011; 60:2740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non‐obese, diabetic strain of mice. Jikken Dobutsu 1980; 29:1–13. [DOI] [PubMed] [Google Scholar]
- 10. Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol 2011; 33:67–87. [DOI] [PubMed] [Google Scholar]
- 11. Lamhamedi‐Cherradi SE, Luan JJ, Eloy L, Fluteau G, Bach JF, Garchon HJ. Resistance of T‐cells to apoptosis in autoimmune diabetic (NOD) mice is increased early in life and is associated with dysregulated expression of Bcl‐x. Diabetologia 1998; 41:178–84. [DOI] [PubMed] [Google Scholar]
- 12. D'Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, et al The defect in T‐cell regulation in NOD mice is an effect on the T‐cell effectors. Proc Natl Acad Sci USA 2008; 105:19857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leijon K, Hammarstrom B, Holmberg D. Non‐obese diabetic (NOD) mice display enhanced immune responses and prolonged survival of lymphoid cells. Int Immunol 1994; 6:339–45. [DOI] [PubMed] [Google Scholar]
- 14. Madsen R, Banday VS, Moritz T, Trygg J, Lejon K. Altered metabolic signature in pre‐diabetic NOD mice. PLoS ONE 2012; 7:e35445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sysi‐Aho M, Ermolov A, Gopalacharyulu PV, Tripathi A, Seppanen‐Laakso T, Maukonen J, et al Metabolic regulation in progression to autoimmune diabetes. PLoS Comput Biol 2011; 7:e1002257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol 2010; 6:280–9. [DOI] [PubMed] [Google Scholar]
- 17. Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Perspectives in diabetes neonatal beta‐cell apoptosis a trigger for autoimmune diabetes? Diabetes 2000; 49:7. [DOI] [PubMed] [Google Scholar]
- 18. O'Brien BA, Harmon BV, Cameron DP, Allan DJ. Apoptosis is the mode of beta‐cell death responsible for the development of IDDM in the Nonobese Diabetic (NOD) mouse. Diabetes 1997; 46:7. [DOI] [PubMed] [Google Scholar]
- 19. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al Crosstalk between neutrophils, B‐1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med 2013; 19:65–73. [DOI] [PubMed] [Google Scholar]
- 20. Scaglia L, Cahill CJ, Finegood DT, Bonner‐Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997; 138:1736–41. [DOI] [PubMed] [Google Scholar]
- 21. Di Cairano ES, Davalli AM, Perego L, Sala S, Sacchi VF, La Rosa S, et al The glial glutamate transporter 1 (GLT1) is expressed by pancreatic beta‐cells and prevents glutamate‐induced beta‐cell death. J Biol Chem 2011; 286:14007–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 2000; 130:1007s–15s. [DOI] [PubMed] [Google Scholar]
- 23. Seal RP, Amara SG. Excitatory amino acid transporters: a family in flux. Annu Rev Pharmacol Toxicol 1999; 39:431–56. [DOI] [PubMed] [Google Scholar]
- 24. Mitani A, Watanabe M, Kataoka K. Functional change of NMDA receptors related to enhancement of susceptibility to neurotoxicity in the developing pontine nucleus. J Neurosci 1998; 18:7941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meldrum B, Garthwaite J. Excitatory amino‐acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci 1990; 11:379–87. [DOI] [PubMed] [Google Scholar]
- 26. Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al Beta‐lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005; 433:73–7. [DOI] [PubMed] [Google Scholar]
- 27. Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. The ensembl regulatory build. Genome Biol 2015; 16:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Darvasi A, Soller M. Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor Appl Genet 1992; 85:353–9. [DOI] [PubMed] [Google Scholar]
- 29. Lander ES, Botstein D. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 1989; 121:185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang BE, Lin DY. Efficient association mapping of quantitative trait loci with selective genotyping. Am J Hum Genet 2007; 80:567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berger UV, Hediger MA. Distribution of the glutamate transporters GLT‐1 (SLC1A2) and GLAST (SLC1A3) in peripheral organs. Anat Embryol (Berl) 2006; 211:595–606. [DOI] [PubMed] [Google Scholar]
- 33. Gegelashvili G, Schousboe A. Cellular distribution and kinetic properties of high‐affinity glutamate transporters. Brain Res Bull 1998; 45:233–8. [DOI] [PubMed] [Google Scholar]
- 34. Broer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev 2008; 88:249–86. [DOI] [PubMed] [Google Scholar]
- 35. Brosnan JT. Interorgan amino acid transport and its regulation. J Nutr 2003; 133:2068S–72S. [DOI] [PubMed] [Google Scholar]
- 36. Iwanaga T, Goto M, Watanabe M. Cellular distribution of glutamate transporters in the gastrointestinal tract of mice: an immunohistochemical and in situ hybridization approach. Biomed Res 2005; 26:271–8. [DOI] [PubMed] [Google Scholar]
- 37. Rauen T, Taylor WR, Kuhlbrodt K, Wiessner M. High‐affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST‐1 in transmitter clearance. Cell Tissue Res 1998; 291:19–31. [DOI] [PubMed] [Google Scholar]
- 38. Ullensvang K, Lehre KP, Storm‐Mathisen J, Danbolt NC. Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur J Neurosci 1997; 9:1646–55. [DOI] [PubMed] [Google Scholar]
- 39. Karmen A, Wroblewski F, Ladue JS. Transaminase activity in human blood. J Clin Invest 1955; 34:126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boyko M, Gruenbaum SE, Gruenbaum BF, Shapira Y, Zlotnik A. Brain to blood glutamate scavenging as a novel therapeutic modality: a review. J Neural Transm 2014; 121:971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harrill A. Drug study: Clinico‐pathological survey of the liver and adverse reactions in response to isoniazid treatment in females of 34 inbred strains of mice. Mouse phenome database web site, 2011.
- 42. The Jackson Laboratory . Blood chemistry survey of 11 strains of mice. MPD:22815. Mouse Phenome Database web site, The Jackson Laboratory, Bar Harbor, Maine, USA.
- 43. Duan J, Sanders AR, Molen JE, Martinolich L, Mowry BJ, Levinson DF, et al Polymorphisms in the 5′‐untranslated region of the human serotonin receptor 1B (HTR1B) gene affect gene expression. Mol Psychiatry 2003; 8:901–10. [DOI] [PubMed] [Google Scholar]
- 44. Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L. Induction of interferon‐α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum 2004; 50:1861–72. [DOI] [PubMed] [Google Scholar]
- 45. Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, et al A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 2014; 63:2538–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Q, McDevitt HO. The role of interferon α in initiation of type I diabetes in the NOD mouse. Clin Immunol 2011; 140:3–7. [DOI] [PubMed] [Google Scholar]
- 47. O'Brien BA, Huang Y, Geng X, Dutz JP, Finegood DT. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes 2002; 51:2481–8. [DOI] [PubMed] [Google Scholar]