

Abstract
Purpose
To review advances made in the treatment of age-related macular degeneration (AMD) and share perspectives on the future of AMD treatment.
Methods
Review of published clinical and experimental studies.
Results
Inhibitors of vascular endothelial growth factor (VEGF) truly revolutionized the treatment of AMD. However, available results from longer-term studies suggest that a degenerative process is unveiled, and continues to occur, even when neovascularization is controlled. Furthermore, anti-VEGF therapy may play a role in the development of atrophic changes. We have proposed using neuroprotection to prevent atrophy, and multiple models of retinal degeneration have shown that it is necessary to block both apoptotic and necrotic cell death pathways. Despite the success of anti-VEGF therapy and the promise of neuroprotection, neither addresses the underlying cause of AMD. It has been postulated that in early AMD, the retention and abnormal accumulation of lipids in Bruch's membrane and below the retinal pigmented epithelium (RPE) lead to drusen. Thus, it is conceivable to target the retained lipoproteins and seek to remove them. In a case study and pilot multicenter clinical trial, we observed significant regression of drusen and an improvement in visual acuity in patients taking high-dose statin therapy. These results, though preliminary, warrant further investigation.
Conclusion
Future treatment of AMD should be based on biology, which will require continued elucidation of the pathogenic mechanisms of AMD development. Neuroprotection represents a potential therapeutic approach, and other promising targets include immune pathways (e.g., inflammation, complement, and inflammasomes) and lipid/lipoprotein accumulation. Finally, due to the heterogeneity of AMD, future progress in therapy will benefit from improved phenotyping and classification.
It is a real honor to deliver the Weisenfeld Lecture—especially to be the first woman to do so. Mildred Weisenfeld was diagnosed with retinitis pigmentosa at age 15, and lost all of her vision by age 23. She decided that patients with blinding diseases needed more than vision aids—a dog, a cane, and Braille texts—and she thought that we should provide hope through eye research. In 1946 she founded the nonprofit that became Fight for Sight, and she campaigned for the founding of the National Eye Institute.
Advances in Age-Related Macular Degeneration Therapy
In this lecture, I will review some of the advances we have made in the treatment of age-related macular degeneration (AMD), and share some of my perspectives on where I think we should be headed next. Age-related macular degeneration remains an important cause of blindness throughout the world. According to the World Health Organization, it is the third leading cause of blindness worldwide (after cataract and glaucoma) and the leading cause of blindness in industrialized countries.1 As clinicians, we recognize AMD by looking into the eye, and seeing deposits (drusen) in the macula, pigmentary changes, or, in the more advanced forms, geographic atrophy or neovascular AMD (Fig. 1). We have made some advances in the treatment of AMD—a little progress in the early and intermediate stages, with vitamin and mineral supplementation based on studies such as the Age-Related Eye Disease Study (AREDS)—but we placed more focus on late neovascular AMD. This began with laser photocoagulation, followed by photodynamic therapy, with a brief foray into surgical treatments, such as removal and translocation of choroidal neovascularization (CNV), and also intravitreal steroids.
Figure 1.

Top left: normal macula. Top center: macula with intermediate AMD showing intermediate (>63 μm, black arrow), large (>125 μm, green arrow), and very large (>250 μm, white arrow) drusen. Top right: hyperpigmentation (black arrows) and focal atrophy in an eye with intermediate AMD. Bottom left: geographic atrophy; bottom middle: neovascular AMD; and bottom right: disciform scar. Reprinted with permission from Miller JW. Age-related macular degeneration revisited--piecing the puzzle: the LXIX Edward Jackson memorial lecture. Am J Ophthalmol. 2013;155:1–35.e13. Copyright 2013 Elsevier, Inc.
Anti-VEGF Therapy
However, AMD treatment was truly revolutionized with the development of inhibitors of vascular endothelial growth factor (VEGF). We are familiar with the Phase III findings of the anti-VEGF therapy ranibizumab (Lucentis) for AMD—indeed, many retina surgeons have the vision data imprinted in their brains—but it was truly remarkable to achieve sustained improvement of vision in patients with neovascular AMD compared to treatments available at the time.2,3
With anti-VEGF therapy, more than 90% of patients avoid moderate vision loss, and approximately one-third achieve vision of 20/40 or better.2 With the CATT,4,5 IVAN,6 and other trials,7–10 we have demonstrated that we can achieve good results, and very similar vision outcomes, with a variety of anti-VEGF agents. Today, multiple anti-VEGF therapies are available to the clinician—pegaptanib (Macugen), ranibizumab (Lucentis), bevacizumab (Avastin), and aflibercept (Eylea)—and we can offer different treatments as needed.
Longer-Term Results of Anti-VEGF Therapy
But what about the longer-term results of anti-VEGF therapy? Prospective randomized controlled clinical trials (RCTs) have largely analyzed treatment outcomes up to 24 months at most.2,5,11 Although there have been some extension studies, such as SEVEN-UP,12 and retrospective studies from retina practice groups like Peden and colleagues13 and work at Massachusetts Eye and Ear (Roh M, et al. IOVS 2002;43:ARVO E-Abstract 1415), these have been limited by patient numbers and variation in treatment protocols and drugs utilized. Still, it is worth reviewing the information that we have available. In the current economic and health care environment, it seems unlikely that we will have large, prospective trials that will give us definitive answers on the longer-term results of anti-VEGF therapy. On the other hand, we may obtain important information from registries that are in development, which may provide actual long-term outcomes of treatment in large populations.
Available results from longer-term studies reveal vision outcomes at 4 to 7 years. These range from 37% to 66% achieving 20/70 or better, 23% to 47% achieving 20/40 or better, and 22% to 37% achieving 20/200 or worse.12–14 Anatomically, fluorescein angiography suggests active disease in 48% to 97%.12 Optical coherence tomography (OCT) indicates fluid or at least degenerative cysts in 72%, and, perhaps most importantly, fundus autofluorescence demonstrates macular atrophy in almost all patients (up to 98.2%).14
Unveiling of the Degenerative Process
So what happens when the neovascular process is controlled? I would postulate that the major event is an unveiling of the degenerative process that we know is occurring in AMD—and hence the development of geographic atrophy. There may also be progression of atrophic changes secondary to poor perfusion, and anti-VEGF therapy may actually play a role.
Regarding the degenerative process, loss of cones, rods, and retinal pigment epithelium (RPE) occurs in the atrophic (dry) form of AMD in early and late stages (Fig. 2). Presumably, this occurs in the neovascular form of AMD as well—particularly when neovascularization is controlled.
Figure 2.

Upper right: loss of cones over drusen. Lower right: The retinal pigment epithelium (RPE) and rods are lost in geographic atrophy. Reprinted with permission from Milam AH. The Human Retina in Health and Disease [CD-ROM]. Philadelphia: Scheie Eye Institute, University of Pennsylvania. Copyright by Scheie Eye Institute, University of Pennsylvania.
Indeed, in earlier studies, Green15 demonstrated photoreceptor and RPE atrophy in CNV lesions and disciform scars (Fig. 3). More recently, results from the CATT showed that geographic atrophy growth rates in treated neovascular AMD were similar to the rates in nonneovascular AMD.16 Clearly, this degeneration is a pathologic process that needs to be targeted.
Figure 3.
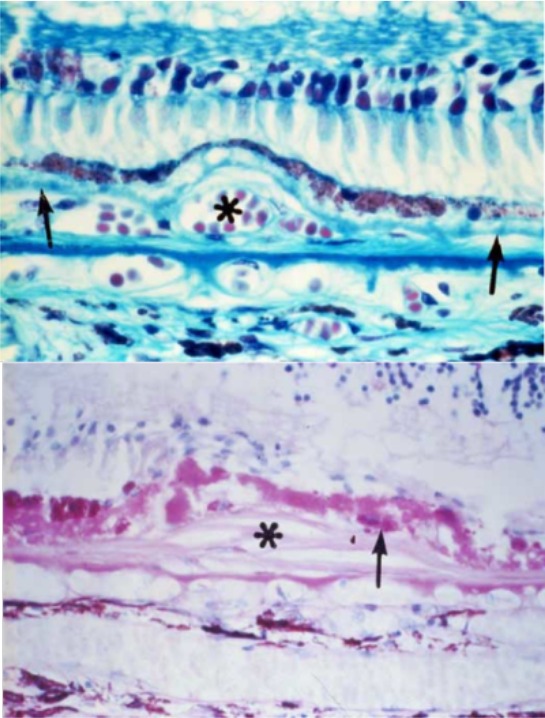
Upper: The retinal pigment epithelium (RPE) is intact over the choroidal neovascularization (CNV), indicated by the asterisk, but photoreceptor degeneration is occurring. Lower: Photoreceptor degeneration is more pronounced and accompanied by loss of the RPE. Reprinted with permission from Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27; originally published in Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100(10):1519–1535. Copyright 1993 American Academy of Ophthalmology, Inc.
It is also possible that the progression of atrophy is due to poor perfusion. As seen in Figure 4, as CNV grows, there is loss of normal choriocapillaris.17 As clinicians, we treat the CNV and initiate its regression—but in doing so, we actually may be eliminating the only remaining blood supply for the outer retina. Therefore, it would not be surprising that anti-VEGF therapy would have secondary atrophic effects. Finally, VEGF has known neurotrophic effects, and blocking it may accelerate atrophy.18 While this is scientifically plausible, the clinical evidence is currently lacking.
Figure 4.

Top: loss of choriocapillaris with growth of choroidal neovascularization (CNV). Bottom: (A) Loss of choriocapillaris adjacent to a region of CNV (right). Reprinted with permission from McLeod DS, Taomoto M, Otsuji T, Green WR, Sunness JS, Lutty GA. Quantifying changes in RPE and choroidal vasculature in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2002;43:1986–1993. Copyright 2002 the Association for Research in Vision and Ophthalmology.
Neuroprotection
We have proposed that we might intervene in the degenerative process using neuroprotection, and thus prevent these atrophic changes. We have suggested neuroprotective adjuvant therapy along with anti-VEGF therapy to prevent photoreceptor cell death. Through this approach, we believe that we can improve vision outcomes, both short- and long-term. We and others have studied neuroprotection using a model of retinal detachment,19,20 and while this may seem distantly related to AMD, separation of photoreceptors from RPE, or a retinal detachment, occurs in various retinal disorders—including neovascular AMD, diabetic retinopathy, and rhegmatogenous retinal detachment.21 Moreover, retinal detachment can be readily modeled in small animals.19
Using this retinal detachment model, we and others found evidence that apoptosis is involved, with activation of caspases and known upstream ligands, including TNF-alpha and Fas ligand (FasL).19,22–24 However, inhibition of caspase activation with a pan-caspase inhibitor was insufficient in preventing photoreceptor cell death.19 Therefore, we investigated whether photoreceptor cell death might involve other cell death pathways.
In published literature from the 1970s, three cell death modes were described based on morphology.25 Type I cell death shows cellular condensation and fragmentation. Type II is associated with the formation of numerous autophagic vacuoles, and type III exhibits a cellular and organelle swelling and plasma membrane rupture. These cell death types are now referred as apoptosis, autophagy, and necrosis, respectively. Apoptosis is the best characterized programmed cell death, and caspases have been established as a central regulator of apoptosis. Recent studies also have identified that autophagy-related proteins (ATG) play a key role in the induction of autophagy. Necrosis was believed to be an unregulated form of cell death. However, recent studies indicate that necrosis can be regulated, induced by regulated signal transduction pathways such as receptor-interacting protein (RIP) kinases.26,27
We looked for evidence of programmed necrosis, or necroptosis, in photoreceptor cell death. In the retinal detachment model, in situ hybridization showed that Rip3 expression increased after retinal detachment, especially in the outer nuclear layer.28 We tried to inhibit photoreceptor cell death using either a necrosis inhibitor (Nec-1) or an apoptosis inhibitor (ZVAD). Treatment with Nec-1 or ZVAD alone showed no effect on photoreceptor loss after experimental retinal detachment. In contrast, combined treatment with Nec-1 and ZVAD significantly reduced the photoreceptor death. Using electron microscopy to examine modes of cell death in the retinal detachment model, we observed that caspase inhibition alone led to increased necrotic cell death. Thus, cells have alternative death pathways through RIP kinase activation; in order to prevent photoreceptor cell death after retinal detachment, it is necessary to block both apoptotic and necrotic pathways.
We wondered whether similar mechanisms might occur in other models of AMD, such as the dsRNA model of retinal degeneration; double-stranded RNA (dsRNA) is a component of drusen and a ligand for Toll-like receptor 3 (TLR3), which mediates innate immune response and cell death.29 Subretinal injection of polyinosinic-polycytidylic acid [poly(I:C)], a synthetic analogue of dsRNA, induced TLR3-dependent retinal degeneration, resulting in areas of subretinal atrophy, loss of RPE cells, and TUNEL-positive cells in the outer nuclear layer and inner nuclear layer.29 Thus, we used this model to examine the mechanism of cell death in the RPE and photoreceptors, as well as to investigate the role of inflammation in AMD. We found that photoreceptor cell death occurred predominantly by apoptosis, whereas RPE death occurred mainly by necrosis, showing almost exclusively necrotic features.30 Thus, apoptosis and necroptosis are indeed active in a dsRNA model of AMD, and a combination of apoptosis and necrosis inhibition is effective in preventing photoreceptor and RPE cell degeneration.
More recently, we found further evidence linking immune responses, necroptosis, and photoreceptor cell death. In patients with photoreceptor injury associated with retinal detachment, we found increased levels of cleaved interleukin 1 beta (IL1β), a downstream product of inflammasome activation.31 In rodents with experimental retinal detachment, infiltrating macrophages were the primary source of IL1β, and photoreceptor cell death led to inflammasome activation in a macrophage- and RIP3-dependent manner.31 Additionally, we found that resident microglia and infiltrating macrophages express FasL, which triggered photoreceptor death in the membrane-bound form (mFasL) yet had neuroprotective properties in the soluble form (sFasL).32 We thus believe that neuroprotection may provide a broad-based treatment approach for a wide variety of retinal disorders, including AMD. It could be conceived as an adjuvant therapy with anti-VEGF for neovascular AMD. It could also be initiated sooner, to treat early and intermediate AMD; this will require long-term delivery, with gene therapy as a potential delivery platform.
Treatment of AMD: Biology Based
Despite its promise, neuroprotection still does not address the underlying cause of AMD—and if the goal is to intervene early in the disease, we will need to attack a key pathway. It is worth emphasizing that the success in treating neovascular AMD was based on such a strategy: targeting the VEGF pathway as a key mediator of angiogenesis and permeability. Targeted therapies for early AMD will require better understanding of AMD pathogenesis. Historically, insights into AMD pathogenesis have been derived from clinical observation and imaging, epidemiology, and histopathology—and more recently from genetics and molecular biology. Considering the available evidence, the pathogenesis of AMD may be narrowed down to six major pathways (Table).33
Table.
Pathogenesis of AMD

Angiogenesis has been treated effectively with anti-VEGF agents, and neuroprotection would potentially address the last pathway: cellular stress and toxicity, which leads to cell death. However, even if introduced early, neuroprotection does not address the underlying cause of AMD. Therefore, early biologically based treatment would ideally interfere with an upstream pathway in AMD pathogenesis to limit disease development and progression.
Lipid and Lipoprotein Metabolism and Transport
Regarding lipid transport and metabolism, similarities have been observed between AMD and atherosclerosis, with Bruch's membrane acting like vascular endothelium. Curcio and colleagues34 have postulated that lipoproteins, such as apolipoprotein B, deliver cholesterol to tissues and become “retained” in Bruch's membrane and sub-RPE space. These retained lipids lead to a lipid wall, basal linear deposits (BlinD), and drusen (Fig. 5A).
Figure 5.

Early AMD pathogenesis. In the outer retina, Bruch's membrane (BrM) consists of a layer of elastin (EL) sandwiched between two layers of collagen and basal lamina (BL), and underlies the retinal pigment epithelium (RPE). The neural retina (not shown) is above the RPE. (A) Development of the lipid wall, leading to basal linear deposits (BlinD), basal laminar deposits (BlamD), and drusen. (B) Complement activation. See text for details. Adapted from Miller JW. Age-related macular degeneration revisited--piecing the puzzle: the LXIX Edward Jackson memorial lecture. Am J Ophthalmol. 2013;155:1–35.e13. Copyright 2013 Elsevier, Inc.
The RPE plays a key role in metabolizing lipoproteins that originate from the photoreceptor outer segments and the systemic circulation. The RPE itself synthesizes lipoproteins as well. With age, the RPE starts to accumulate lipofuscin, and a lipid wall develops along Bruch's membrane in the sub-RPE space, resulting in formation of BlinD, basal laminar deposits (BlamD), and ultimately drusen, which are visible upon clinical observation. Considering attacking this abnormal lipid accumulation, obvious targets would be the processes of lipid transport or metabolism, as well as associated genes, many of which have been identified. However, it is also conceivable to target the retained lipoproteins and seek to remove them.
Inflammation and Immunity
Inflammation and immunity are attractive targets because they appear to be central to all stages of AMD—not only in its development, but also in progression to the intermediate and advanced stages. It appears that early in the disease process, lipoprotein accumulation results in a smoldering and chronic inflammatory response that is directed to the RPE, choriocapillaris, and Bruch's membrane. This includes deposition of complement components (Fig. 5B), as well as recruitment and activation of inflammatory cells (including circulating leukocytes, resident microglia, and infiltrating macrophages). Additionally, inflammasome activation is a growing area of research and therapeutic development, although groups differ in their approach to this target. Important gene associations have been identified for complement and inflammatory pathways.33
Progression to Advanced AMD
As AMD progresses, lipoprotein retention and inflammation can lead to angiogenesis, which is associated with dissolution of Bruch's membrane and disturbances in the extracellular matrix. This, in turn, leads to the advanced neovascular form of AMD (Fig. 6A). Alternatively, RPE injury and subsequent RPE and photoreceptor cell death result in atrophic changes, which underlie the pathogenesis of advanced atrophic AMD or geographic atrophy (Fig. 6B).
Figure 6.

Progression to advanced age-related macular degeneration (AMD). (A) Dissolution of Bruch's membrane (BrM), disruption of the extracellular matrix, and angiogenesis (advanced neovascular AMD). (B) Injury of the retinal pigment epithelium (RPE) and subsequent death of the RPE and photoreceptors in geographic atrophy (advanced atrophic AMD). Adapted from Miller JW. Age-related macular degeneration revisited--piecing the puzzle: the LXIX Edward Jackson memorial lecture. Am J Ophthalmol. 2013;155:1–35.e13. Copyright 2013 Elsevier, Inc.
Statin Therapy for AMD
Numerous investigations over the past few decades have explored the therapeutic potential of statins for AMD, not only for their well-known lipid-lowering effects but also for their potential anti-inflammatory effects. Previous investigations on whether statins could affect AMD status or alter progression showed mixed results, and a 2015 Cochrane systematic review and meta-analysis concluded that the available evidence is insufficient to support a role for statins in preventing or delaying onset of AMD, or in progression of AMD.35 Indeed, some studies show effectiveness for statins in AMD therapy, while others do not. Recently, Guymer and colleagues36 conducted a prospective, randomized, placebo-controlled study with 114 subjects, and found that treatment with oral simvastatin (Zocor) 40 mg daily may slow progression of nonadvanced AMD, especially in those with the complement factor H (CFH) risk allele. Regarding the association between serum lipids and AMD risk, VanderBeek and colleagues37 showed that increased serum low-density lipoprotein (LDL), increased serum triglycerides, and more than 1 year of statin use led to increased risk of neovascular AMD; while one might conclude that statins might promote AMD, the authors postulated that study patients had lipid profiles that were resistant to statin treatment and thus were at increased risk of AMD.37 Cougnard-Gregoire et al.38 demonstrated that increased serum high-density lipoprotein (HDL) increases AMD risk in the ALIENOR study. Conversely, a meta-analysis of three cohorts by Klein and colleagues39 showed no association of AMD incidence or progression with serum lipids, statin use, or lipid pathway genes.
The variability in these studies may be explained in part by the intrinsic heterogeneity of AMD. Even the term “intermediate AMD” covers a wide spectrum from large drusen to confluent soft drusen and a variety of atrophic changes. Moreover, studies conducted to date have involved not only variable statin dosing, but also variable activity among different statins; for example, 40-mg simvastatin (Zocor) is approximately equivalent to 20-mg atorvastatin (Lipitor),40 and researchers should be aware of this variability when surveying the literature. Finally, the wide use of statins in the general population complicates research in the AMD patient population.
Nonetheless, the cardiovascular disease literature may inform the potential use of statins for AMD. Pitt et al.41 demonstrated that in patients with coronary artery disease, high-dose (80 mg daily) atorvastatin (Lipitor) prevented restenosis, and additional studies by Nissen et al.,42 Zhao et al.,43 and others using high statin doses have confirmed protective effects and even demonstrated resorption of atherosclerotic plaque. These studies suggest that intensive statin therapy may reverse the “retained” lipid in early AMD. In a single patient treated by Demetrios Vavvas, we observed complete disappearance of large, soft, confluent macular drusen (without accompanying atrophy of the RPE) and gains in visual acuity in a patient with AMD after 6 months of 80 mg atorvastatin daily (Fig. 7).44
Figure 7.

Regression of drusen in a patient receiving high-dose oral atorvastatin. (A) An otherwise healthy patient with age-related macular degeneration (AMD) with bilateral, large, soft, confluent macular drusenoid pigment epithelial detachments and pigmentary alterations on color fundus photography (upper figure parts), and decreasing visual acuity with significant distortion. (B) Spectral-domain optical coherence tomography (SD-OCT) showing the significant extent of these deposits and the overlying retinal pigment epithelium (RPE) and photoreceptor architectural distortion (upper figure parts). The patient was started on atorvastatin 10 mg and escalated to 80 mg over 9 months. Six months after 80 mg atorvastatin, visual acuity improved by 12 letters to 20/20. Fundus photographs and SD-OCT revealed complete disappearance of the drusen without accompanying atrophy of the retinal pigment epithelium (lower figure parts). Reprinted with permission from Vavvas DG, Daniels AB, Kapsala ZG, et al. Regression of some high-risk features of age-related macular degeneration (AMD) in patients receiving intensive statin treatment. EBioMedicine. 2016;5:198–203. Copyright 2016 by Vavvas DG, Daniels AB, Kapsala ZG, et al.
Based on the promising results of this case study, we initiated a pilot prospective interventional study in two centers of high-dose atorvastatin therapy in AMD patients with bilateral large soft drusen/drusenoid pigment epithelial detachments (PEDs) in both eyes, without significant geographic atrophy or CNV in either eye.44 Of 23 patients who completed follow-up of at least 12 months, 10 showed drusen regression without atrophy; of these patients, visual acuity improved by an average of three letters, and regression was nearly complete in eight patients. No patients progressed to neovascular AMD.
When we have seen drusen regression previously, it is accompanied by atrophy and vision loss. However, the evidence in our study suggests that drusen regression with high-dose atorvastatin occurs without any development of atrophy or neovascularization. Possible mechanisms include altering RPE metabolism or creating a gradient to allow efflux of lipids from the outer retina and/or the infiltrating macrophages. In addition, statins have anti-inflammatory and antiangiogenic effects. While this is a pilot study, high-dose atorvastatin is a tantalizing prospect for treating AMD, and warrants further investigation.
Future Treatment of AMD
Future treatment of AMD should be based on biology, and this will require continuing to elucidate the interconnections between the pathogenic mechanisms in AMD development (Table). Therapeutic targets include inflammation, the complement pathway, and inflammasomes; accordingly, there are many clinical trials under way in this space, so we will be learning if these are effective therapeutic strategies. Neuroprotection represents another promising area of research and therapeutic development.
Because the heterogeneity of AMD creates challenges to developing effective treatments for early and intermediate disease, future progress in therapy will benefit from improvements in phenotyping and classification. We need to use our findings from imaging and dark adaptation and perhaps combine that with metabolomics and genotyping in order to tease out the subtypes within this heterogeneous patient population.
Acknowledgments
I thank the following individuals for their contributions to the work presented here. Long term anti-VEGF therapy study: Ivana Kim, Demetrios Vavvas, Marina Braschler, Thomas Braschler, Miin Roh, and John Lowenstein (Massachusetts Eye and Ear, Harvard Medical School). Neuroprotection studies: Demetrios Vavvas, Yusuke Murakami, George Trichonas, Hidetaka Matsumoto, Maki Kayama, and Keiko Kataoka (Massachusetts Eye and Ear, Harvard Medical School). Pilot multicenter clinical study of high-dose atorvastatin: Demetrios Vavvas and Anthony Daniels (Massachusetts Eye and Ear, Harvard Medical School); Miltiadis Tsilimbaris and Zoi Kapsala (University of Crete). Administrative and editorial support: Wendy Chao (Massachusetts Eye and Ear, Harvard Medical School). Illustrations: Alexander Coster Scott (Boston, Massachusetts).
Presented at the annual meeting of the Association for Research in Vision and Ophthalmology, Denver, Colorado, United States, May 4, 2015.
Supported by National Eye Institute (NEI)/National Institutes of Health (NIH) P30 EY014104 to Massachusetts Eye and Ear, Yeatts Retina Research Fund, Research to Prevent Blindness, Massachusetts Lions Research Fund, and Neovascular Research Funds.
Disclosure: J.W. Miller, Alcon (C), Amgen, Inc. (C), Biogen Idec, Inc. (C), KalVista Pharmaceuticals Ltd. (C), Maculogix, Inc. (C), P; ONL Therapeutics, LLC, P; Valeant Pharmaceuticals, P
References
- 1. Priority Eye Diseases. World Health Organization website. Available at: http://www.who.int/blindness/causes/priority. Accessed July 1, 2015.
- 2. Rosenfeld PJ,, Brown DM,, Heier JS,, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006; 355: 1419–1431. [DOI] [PubMed] [Google Scholar]
- 3. Brown DM, Kaiser PK, Michels M,et al.; ANCHOR Study Group. . Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006; 355: 1432–1444. [DOI] [PubMed] [Google Scholar]
- 4. Martin DF, Maguire MG, Ying GS,et al.; CATT Research Group. . Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011; 364: 1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martin DF, Maguire MG, Fine SL,et al.; CATT Research Group. . Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012; 119: 1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chakravarthy U, Harding SP, Rogers CA,et al.; IVAN Study Investigators. . Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology. 2012; 119: 1399–1411. [DOI] [PubMed] [Google Scholar]
- 7. Kodjikian L,, Souied EH,, Mimoun G,, et al. Ranibizumab versus bevacizumab for neovascular age-related macular degeneration: results from the GEFAL Noninferiority Randomized Trial. Ophthalmology. 2013; 120: 2300–2309. [DOI] [PubMed] [Google Scholar]
- 8. Krebs I,, Schmetterer L,, Boltz A,, et al. A randomised double-masked trial comparing the visual outcome after treatment with ranibizumab or bevacizumab in patients with neovascular age-related macular degeneration. Br J Ophthalmol. 2013; 97: 266–271. [DOI] [PubMed] [Google Scholar]
- 9. Schauwvlieghe AM,, Dijkman G,, Hooymans JM,, et al. Ranibizumab versus bevacizumab in the Netherlands: comparing the efficacy of bevacizumab to ranibizumab in patients with exudative age-related macular degeneration – the BRAMD Study. Ophthalmologica. 2013; 230 (suppl 1): 2–3. [Google Scholar]
- 10. Berg K,, Pedersen TR,, Sandvik L,, Bragadottir R. Comparison of ranibizumab and bevacizumab for neovascular age-related macular degeneration according to LUCAS treat-and-extend protocol. Ophthalmology. 2015; 122: 146–152. [DOI] [PubMed] [Google Scholar]
- 11. Brown DM,, Michels M,, Kaiser PK,, et al. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study. Ophthalmology. 2009; 116: 57–65.e55. [DOI] [PubMed] [Google Scholar]
- 12. Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K; SEVEN-UP Study Group. . Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology. 2013; 120: 2292–2299. [DOI] [PubMed] [Google Scholar]
- 13. Peden MC,, Suner IJ,, Hammer ME,, Grizzard WS. Long-term outcomes in eyes receiving fixed-interval dosing of anti-vascular endothelial growth factor agents for wet age-related macular degeneration. Ophthalmology. 2015; 122: 803–808. [DOI] [PubMed] [Google Scholar]
- 14. Bhisitkul RB,, Mendes TS,, Rofagha S,, et al. Macular atrophy progression and 7-year vision outcomes in subjects from the ANCHOR, MARINA, and HORIZON studies: the SEVEN-UP study. Am J Ophthalmol. 2015; 159: 915–924. [DOI] [PubMed] [Google Scholar]
- 15. Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999; 5: 27. [PubMed] [Google Scholar]
- 16. Grunwald JE,, Pistilli M,, Ying GS,, et al. Growth of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2015; 122: 809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McLeod DS,, Taomoto M,, Otsuji T,, Green WR,, Sunness JS,, Lutty GA. Quantifying changes in RPE and choroidal vasculature in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2002; 43: 1986–1993. [PubMed] [Google Scholar]
- 18. Saint-Geniez M,, Maharaj AS,, Walshe TE,, et al. Endogenous VEGF is required for visual function: evidence for a survival role on Müller cells and photoreceptors. PLoS One. 2008; 3: e3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hisatomi T,, Sakamoto T,, Murata T,, et al. Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. Am J Pathol. 2001; 158: 1271–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsumoto H,, Miller JW,, Vavvas DG. Retinal detachment model in rodents by subretinal injection of sodium hyaluronate. J Vis Exp. 2013; 79: 50660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murakami Y,, Notomi S,, Hisatomi T,, et al. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog Retin Eye Res. 2013; 37: 114–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zacks DN,, Hanninen V,, Pantcheva M,, Ezra E,, Grosskreutz C,, Miller JW. Caspase activation in an experimental model of retinal detachment. Invest Ophthalmol Vis Sci. 2003; 44: 1262–1267. [DOI] [PubMed] [Google Scholar]
- 23. Zacks DN,, Zheng QD,, Han Y,, Bakhru R,, Miller JW. FAS-mediated apoptosis and its relation to intrinsic pathway activation in an experimental model of retinal detachment. Invest Ophthalmol Vis Sci. 2004; 45: 4563–4569. [DOI] [PubMed] [Google Scholar]
- 24. Nakazawa T,, Kayama M,, Ryu M,, et al. Tumor necrosis factor-alpha mediates photoreceptor death in a rodent model of retinal detachment. Invest Ophthalmol Vis Sci. 2011; 52: 1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schweichel JU,, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973; 7: 253–266. [DOI] [PubMed] [Google Scholar]
- 26. Chan FK,, Shisler J,, Bixby JG,, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003; 278: 51613–51621. [DOI] [PubMed] [Google Scholar]
- 27. Degterev A,, Huang Z,, Boyce M,, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005; 1: 112–119. [DOI] [PubMed] [Google Scholar]
- 28. Trichonas G,, Murakami Y,, Thanos A,, et al. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci U S A. 2010; 107: 21695–21700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang Z,, Stratton C,, Francis PJ,, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008; 359: 1456–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murakami Y,, Matsumoto H,, Roh M,, et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 2014; 21: 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kataoka K,, Matsumoto H,, Kaneko H,, et al. Macrophage- and RIP3-dependent inflammasome activation exacerbates retinal detachment-induced photoreceptor cell death. Cell Death Dis. 2015; 6: e1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsumoto H,, Murakami Y,, Kataoka K,, et al. Membrane-bound and soluble Fas ligands have opposite functions in photoreceptor cell death following separation from the retinal pigment epithelium. Cell Death Dis. 2015; 6: e1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller JW. Age-related macular degeneration revisited--piecing the puzzle: the LXIX Edward Jackson memorial lecture. Am J Ophthalmol. 2013; 155: 1–35.e13. [DOI] [PubMed] [Google Scholar]
- 34. Curcio CA,, Johnson M,, Rudolf M,, Huang JD. The oil spill in ageing Bruch membrane. Br J Ophthalmol. 2011; 95: 1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gehlbach P,, Li T,, Hatef E. Statins for age-related macular degeneration. Cochrane Database Syst Rev. 2015; 2: CD006927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guymer RH,, Baird PN,, Varsamidis M,, et al. Proof of concept, randomized, placebo-controlled study of the effect of simvastatin on the course of age-related macular degeneration. PLoS One. 2013; 8: e83759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. VanderBeek BL,, Zacks DN,, Talwar N,, Nan B,, Stein JD. Role of statins in the development and progression of age-related macular degeneration. Retina. 2013; 33: 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cougnard-Gregoire A,, Delyfer MN,, Korobelnik JF,, et al. Elevated high-density lipoprotein cholesterol and age-related macular degeneration: the Alienor study. PLoS One. 2014; 9: e90973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klein R,, Myers CE,, Buitendijk GH,, et al. Lipids, lipid genes, and incident age-related macular degeneration: the three continent age-related macular degeneration consortium. Am J Ophthalmol. 2014; 158: 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rogers SL,, Magliano DJ,, Levison DB,, et al. A dose-specific meta-analysis of lipid changes in randomized controlled trials of atorvastatin and simvastatin. Clin Ther. 2007; 29: 242–252. [DOI] [PubMed] [Google Scholar]
- 41. Pitt B,, Waters D,, Brown WV,, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med. 1999; 341: 70–76. [DOI] [PubMed] [Google Scholar]
- 42. Nissen SE,, Tuzcu EM,, Schoenhagen P,, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med. 2005; 352: 29–38. [DOI] [PubMed] [Google Scholar]
- 43. Zhao XQ,, Dong L,, Hatsukami T,, et al. MR imaging of carotid plaque composition during lipid-lowering therapy a prospective assessment of effect and time course. JACC Cardiovasc Imaging. 2011; 4: 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vavvas DG,, Daniels AB,, Kapsala ZG,, et al. Regression of some high-risk features of age-related macular degeneration (AMD) in patients receiving intensive statin treatment. EBioMedicine. 2016; 5: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]