

Abstract
Light‐activated ruthenium polypyridyl anticancer prodrugs often suffer from poor water solubility, poor selectivity, and/or ill‐defined intracellular targets. Coordination of the d‐ or l‐glucose thioether ligand 3 (2‐(2‐(2‐(methylthio)ethoxy)ethoxy)ethyl‐β‐glucopyranoside) to the highly lipophilic ruthenium complex [Ru(tpy)(dppn)(H2O)]2+ ([1]2+; dppn=benzo[i]dipyrido‐[3,2‐a:2′,3′‐c]phenazine, tpy=2,2′:6′,2′′‐terpyridine) solved all these problems at once. The two enantiomers of [Ru(tpy)(dppn)(3)][PF6]2, [d‐2][PF6]2 and [l‐2][PF6]2, were soluble in water, which allowed the influence of the chirality of the glucose moiety on uptake, toxicity, and intracellular localization of the prodrug to be probed without changing any other physicochemical properties. Both compounds showed mild, but different, cytotoxicity in A549 (human lung carcinoma) and MCF‐7 (human breast adenocarcinoma) cancer cells in the dark, whereas following low doses of visible light irradiation (3.1 J cm−2 at λ = 454 nm), a similar, but high cytotoxicity (EC50 < 1 μm), was observed. Irrespective of the chirality, both slightly emissive Ru complexes were found in the mitochondria, and two modes of action may contribute to light‐induced cell death: 1) the glucose thioether ligand is photosubstituted by water, thus [1]2+, which interacts with DNA at an exceptionally high 400:1 base pair/Ru ratio, is released; 2) both [1]2+ and [2]2+ produce massive amounts of singlet oxygen, which leads to very efficient photodynamic DNA cleavage.
Keywords: glucose transport, glycoconjugates, mitochondria, photoactivated chemotherapy, ruthenium
Introduction
One of the major challenges in the development of new anticancer drugs is to improve their selectivity. A common strategy to better differentiate normal proliferating cells from malignant cells is to develop drugs that target specific hallmarks of cancer cells, such as aerobic glycolysis. First described by Warburg,1 cancer cells use glycolysis for their energy production, and therefore have a higher demand for simple sugars such as d‐glucose. The cell membrane is impermeable to polar molecules so specific membrane transporters control glucose uptake (GLUT and SGLT). These transporters are overexpressed in many types of cancer cell, which aids d‐glucose penetration into the cell, and also provides a method to target imaging or therapeutic compounds to cancer cells.2 For example, 2‐deoxy‐2‐(18F)fluoro‐d‐glucose (18‐FDG) is a common radiotracer used in clinics to image tumors in vivo.3
Conjugating d‐glucose or other GLUT substrates to organic3, 4 or inorganic5 anticancer compounds is a rapidly expanding cancer‐targeting strategy. Several methods have been proposed to assess the benefits of d‐glucose functionalization of an anticancer drug. Enhanced uptake is usually demonstrated indirectly, for example by comparing glucose‐functionalized drugs to their aglycon counterparts,6 or by competitive inhibition experiments with d‐glucose.7 However, these methods usually ignore the difference in water solubility between the glycoconjugates and their aglycon analogues, which can have a major influence on the drug uptake, localization, and/or mode‐of‐action for many compounds. Comparing the biological effects of a glycoconjugate on different cell lines with different expressions of glucose transporters is another alternative.8 However, depending on the cell line different glucose transporters may be overexpressed,9 which complicates the interpretation of such experiments. Finally, adding glucose‐transporter inhibitors to switch off the uptake of glucose‐functionalized compounds is also possible.10 However, synergies between biologically active compounds have been demonstrated on multiple occasions,11 and it may be difficult to distinguish impaired drug uptake due to the inhibitors from the cytotoxicity of the inhibitor itself.12
To solve these biases, we propose a new approach that directly compares the activity of the d‐ and l‐glucose conjugates of achiral, highly lipophilic ruthenium compound [Ru(tpy)(dppn)(H2O)]2+ ([1]2+; dppn=benzo[i]dipyrido‐[3,2‐a:2′,3′‐c]phenazine, tpy=2,2′:6′,2′′‐terpyridine). Unlike d‐glucose, l‐glucose is not a substrate for the glucose‐transport system.13 Our basic assumption was that an l‐glucose‐modified drug would have the same structural properties and water solubility as its d‐glucose analogue, but would not be recognized by cellular enzymes, therefore it may be possible to probe the biological effects of the d‐glucose functional group without the experimental biases mentioned above. Compound [1]2+ has another interesting property: it belongs to a family of metallodrugs that can be activated by visible‐light irradiation.14 Light‐activatable anticancer compounds may improve the selectivity of anticancer treatments by an external trigger that can limit the toxicity of the treatment to the time and place of light irradiation.15
We designed two light‐activatable prodrugs [d‐2]2+ and [l‐2]2+ ([Ru(tpy)(dppn)(d‐3)]2+ and [Ru(tpy)(dppn)(l‐3)]2+, respectively; 3 is a thioether ligand covalently linked to d‐ or l‐glucose that binds to ruthenium via a thermally stable Ru⋅⋅⋅S coordination bond, see Figure 1). The synthesis, photochemistry, and biological evaluation of these enantiomeric ruthenium compounds is reported, and their cytotoxicity, cellular distribution, and mode‐of‐action are discussed.
Figure 1.
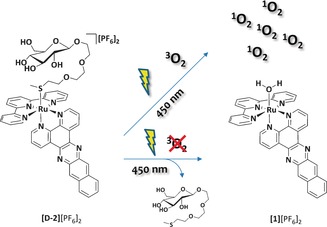
Schematic representation of the light‐induced dual mode‐of‐action for glycoconjugated [d‐2]2+, in which d‐3 is a thioether‐glucose conjugate. For clarity the l enantiomers are omitted.
Results and Discussion
Synthesis
The thioether glucose conjugates d‐3 and l‐3 (Scheme 1) were synthesized from d‐ and l‐glucose, respectively, according to Schmidt's methodology (Scheme S1 in the Supporting Information).16 As expected both ligands had the same physical and spectroscopic properties, except for their opposite sign of optical rotation (d‐3: [α] =−10.0 (c=1.00 in MeOH); l‐3: [α] =+11.8 (c=1.00 in MeOH)) and their different retention times for chiral HPLC (Figures S17 and S18 in the Supporting Information). Next, the ligands d‐3 or l‐3 were coordinated to ruthenium (Scheme 1). Isolation of the aqua complex [1][PF6]2 was necessary, and further reaction with a three‐fold excess of d‐3 or l‐3 under mild conditions (acetone, 50 °C) afforded the d‐ or l‐glucose conjugates, [d‐2][PF6]2 and [l‐2][PF6]2, respectively, in moderate yields. Unlike [1][PF6]2 and [Ru(tpy)(dppn)Cl]Cl ([4][Cl]), which are virtually insoluble in water, [d‐2][PF6]2 and [l‐2][PF6]2 can be dissolved in water, even in absence of DMSO.
Scheme 1.
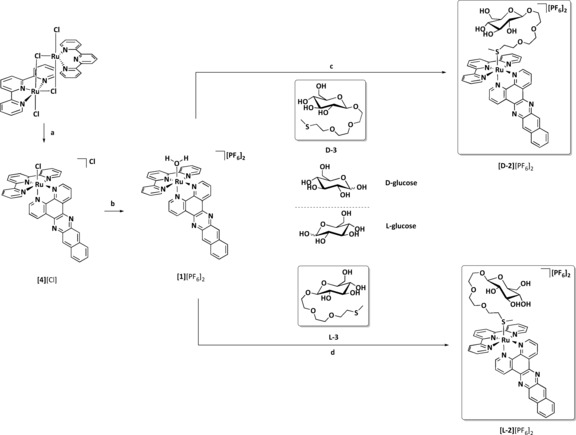
Synthesis of [d‐2][PF6]2 and [l‐2][PF6]2. a) Dppn (0.5 equiv), ethylene glycol, 5 h, 100 °C, 75 %; b) i) AgNO3 (1.0 equiv), acetone/water (3:1), 50 °C, ii) NH4PF6, 84 %; c) d‐3 (2.66 equiv), acetone, 50 °C, 24 h, 35 %; d) l‐3 (2.66 equiv), acetone, 50 °C, 24 h, 32 %.
Photochemistry
The photoreactivity of the water‐soluble thioether complex [d‐2][PF6]2 was tested under different conditions. Figure 2 shows the evolution of the UV/Vis spectrum of [d‐2][PF6]2 upon blue‐light irradiation (λ exc=450 nm) in demineralized water under an argon atmosphere. The initial absorption maximum at λ=460 nm decreased, and a metal‐to‐ligand charge transfer (MLCT) band at λ=474 nm increased over time; a clear isosbestic point was also observed, which showed that a single photoproduct was obtained under these conditions. According to mass spectrometry analysis, the photoproduct was [1]2+ (m/z calcd: 342.5; m/z found: 342.4). Photosubstitution of the thioether glucose conjugate by water proceeds with a quantum yield (Φ 450) of 0.00095 (±0.00002) in deoxygenated water (Table 1). Usually, photosubstitution processes significantly quench the emission of ruthenium polypyridyl complexes. Indeed, the phosphorescence of [d‐2][PF6]2 in phosphate buffered saline (PBS) (pH 7.4) at λ exc=450 nm in air was very weak; the wavelength of the emission maximum was λ=648 nm, and a phosphorescence quantum yield (Φ P) of 3.7×10−5 was measured (Table 1). Under prolonged blue‐light irradiation in air, the wavelength of the emission band shifted from λ=648 to 690 nm, with consecutive formation of the weakly emissive photoproduct [1]2+ (Φ P=3.2×10−5; Table 1). NIR emission spectroscopy was also performed at λ exc=450 nm to check whether irradiation of [d‐2]2+ in air would produce singlet oxygen (1O2). In water, PBS, or D2O, no emission at λ=1270 nm was detected because of the very short lifetime of 1O2 in aqueous solution. However, in CD3OD an intense emission peak at λ=1270 nm, characteristic of 1O2, was observed upon blue‐light irradiation of [d‐2][PF6]2 (Figure S4 in the Supporting Information). The quantum yield of 1O2 production (Φ Δ) for [d‐2][PF6]2 in CD3OD was 0.71 (Table 1), in other words, [d‐2][PF6]2 generates 1O2 very efficiently in air (Figure S4 in the Supporting Information). The photoproduct [Ru(tpy)(dppn)(CD3OD)]2+, which was obtained after extensive blue‐light irradiation of [d‐2]2+ in CD3OD, also generated 1O2 with a high Φ Δ value (0.43; Table 1).
Figure 2.
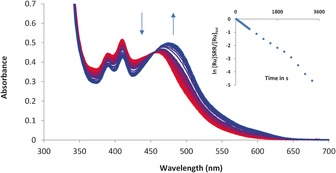
Electronic absorption spectra of [d‐2][PF6]2 in deoxygenated H2O irradiated at for 60 min. Spectra were taken every 30 s for the first 10 min, and then every 5 min until 60 min. T=298 K, [Ru]tot=4.23×10−5, λ exc=450 nm, photon flux=1.77×10−7 Einstein s−1. Insert depicts the evolution of ln[RuSRR]/[Ruu]tot vs. irradiation time (s), where [RuSRR] is the concentration of [2]2+ at irradiation time t and [Ru]tot the total ruthenium concentration.
Table 1.
Lowest‐energy absorption maxima and quantum yields for photosubstitution (Φ 450), 1O2 production (Φ Δ), and phosphorescence (Φ p) at 298 K.
| Complex | λ abs [nm][a] (ϵ [m −1 cm−1]) | Φ 450 [a] | Φ Δ [b] | Φ P [c] |
|---|---|---|---|---|
| [d‐2] 2+ | 458 (11 619) | 0.00095±0.00002 | 0.71 | 0.000037 |
| [1] 2+ | 475 (12 643) | – | 0.43 | 0.000032 |
[a] In water under argon. [b] In CD3OD in air. [c] In PBS in air.
The photosubstitution of the thioether ligand d‐3 in [d‐2]2+ contrasts with recent work from the Turro group, who demonstrated that the analogous complex [Ru(tpy)(dppn)(pyridine)]2+ did not undergo photosubstitution of the pyridine ligand in organic solvents. Instead, the complex was found to efficiently produce 1O2 (Φ Δ=0.98) due to the presence of low‐lying π–π* excited states centered on the dppn ligand.17 Therefore, the nature of the monodentate ligand plays an important role in the photoreactivity of this family of dppn‐based ruthenium complexes. Although photosubstitution, phosphorescence, and 1O2 generation often represent competing pathways in ruthenium photochemistry, when [d‐2]2+ is irradiated with blue light all these processes may occur depending on the O2 concentration. Under deoxygenated conditions photosubstitution of the thioether ligand to form the aqua complex is preferred, whereas in air, efficient generation of 1O2 becomes a competing pathway and is observed both before and after photosubstitution.
Cytotoxicity assay
The cytotoxic properties of [d‐2][PF6]2 and its enantiomer [l‐2][PF6]2 were first tested in the dark on two human cancer cell lines, A549 (human lung carcinoma) and MCF‐7 (human breast adenocarcinoma).9 In parallel, considering the dual photoreactivity of [d‐2][PF6]2, the phototoxicity of [d‐2][PF6]2 and [l‐2][PF6]2 was also tested under a low dose of blue light (5 min at λ=454±11 nm, 10.5±0.7 mW cm−2, 3.2±0.2 J cm−2; see Experimental for full protocol). Briefly, cells were seeded at t=0 (5×103 and 8×103 cells/well for A549 and MCF‐7, respectively), treated with a concentration series of either [d‐2][PF6]2 or [l‐2][PF6]2 24 h after seeding, and then irradiated or maintained in the dark after media refreshment 48 h after seeding. Cell viability was assayed by using sulforhodamine B (SRB) 96 h after seeding. The dose–response curves and effective concentrations (EC50; the concentration of drug that gives a half‐maximum effect) are shown in Figure 3 and Table 2, respectively. Images of the dark and irradiated samples of cells treated with A549 and MCF‐7 (20 μm, [d‐2][PF6]2) 96 h after seeding are shown in Figures S15 and S2 (see the Supporting Information).
Figure 3.
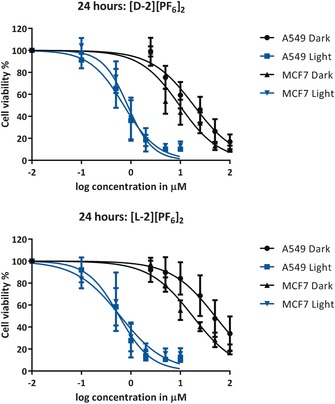
Cell viability of A549 and MCF‐7 cells versus the log(concentration) of [d‐2][PF6]2 and [l‐2][PF6]2 under dark and irradiated conditions. Data points are the mean of three independent experiments (±SD error bars).
Table 2.
(Photo)cytotoxicity of [d‐2][PF6]2 and [l‐2][PF6]2 expressed as effective concentrations (EC50) in the dark and after irradiation with blue light, and photocytotoxicity index (PI) values versus A549 and MCF‐7 cells. +95% and −95 % confidence intervals are also indicated (in μm).
| A549 | MCF‐7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | EC50 dark[a] | EC50 454 nm[b] | PI[c] | EC50 dark[a] | EC50 454 nm[b] | PI[c] | ||||
| [d‐2](PF6)2 | 19 | +4.0 | 0.72 | +0.16 | 26 | 9.6 | +2.9 | 0.86 | +0.21 | 11 |
| −3.3 | −0.13 | −2.3 | −0.17 | |||||||
| [l‐2](PF6)2 | 50 | +17 | 0.58 | +0.13 | 86 | 18 | +3.8 | 0.61 | +0.28 | 30 |
| −13 | −0.11 | −3.1 | −0.19 | |||||||
[a] Cells were incubated with the Ru complex for 24 h. [b] Cells were incubated with the Ru complex for 24 h, and the media was refreshed before irradiation with blue light (5 min at λ exc=450 nm with (3.2±0.2) J cm−2). [c] PI=EC50(dark)/EC50(450nm).
In the dark the cytotoxicity of [d‐2][PF6]2 and [l‐2][PF6]2 was significantly different for the A549 and MCF‐7 cells. For [d‐2][PF6]2 the effective concentration was 2.6 and 1.9 times lower for the A549 and MCF‐7 cells, respectively, than for [l‐2][PF6]2 (Table 2). Both compounds were activated upon irradiation and showed similarly high cytotoxicity, which was characterized by submicromolar EC50 values. According to these results, the chiral nature of the glucose functional group seems to have an effect on the cytotoxicity of the non‐irradiated prodrugs. In addition, upon light irradiation either the release of the photoproduct [1]2+ or efficient 1O2 generation by both enantiomers of the prodrug [2]2+ and the activated drug [1]2+, or both, results in a highly cytotoxic combination.18
Cellular localization and in vitro imaging
Contrary to the many ruthenium complexes capable of ligand photosubstitution, [d‐2][PF6]2 and [l‐2][PF6]2 were slightly emissive within the cells, which allowed uptake and localization studies to be performed (Figure 4; and Figures S9–S14 in the Supporting Information). Microscopy imaging was performed for A549 cells treated in the dark with [d‐2][PF6]2 or [l‐2][PF6]2 for 4, 6, and 24 h (λ exc=488 nm; Figure S9 in the Supporting Information). These images revealed that, independent of the incubation time, [d‐2][PF6]2 and [l‐2][PF6]2 displayed no significant difference in localization or emission intensity. In addition, both complexes were clearly localized outside the nucleus (Figures S9 and S10 in the Supporting Information). Co‐localization experiments performed with MitoTracker Deep Red (MTDR, λ exc=639 nm) were attempted after 6 h incubation (Figure S13 in the Supporting Information). Due to the weaker emission of [1]2+ relative to MTDR, and the absorption of MTDR at λ=488 nm, it was impossible to quantitatively co‐localize the ruthenium compound and the dye unequivocally. However, when added separately the ruthenium compound and MTDR gave qualitatively very similar images at λ=488 or 639 nm, respectively (Figure 4). This suggested that the ruthenium compound might localize in the mitochondria. To confirm localization in the mitochondria, an experiment was designed in which the cells were treated with [d‐2][PF6]2 (25 μm) in the presence of MTDR. MTDR images taken at λ exc=639 nm showed normal mitochondrial morphology (Figures S13B and S13E in the Supporting Information). More images were taken at λ=488 nm (Figures S13C and S13F in the Supporting Information), followed by a second set of MTDR images (at λ=639 nm, see Figures S13D and S13G in the Supporting Information). The mitochondria of the cells irradiated at λ exc=488 nm were altered compared to the untreated cells and showed bubble‐like structures. Thus, treating the cells with a combination of ruthenium complex and λ=488 nm light modified the mitochondria structure. Mitochondria are known to have a highly negative inner‐membrane potential, which can be targeted by cationic, lipophilic compounds.18 Considering the positive charge and lipophilic nature of [d‐2][PF6]2 and [l‐2][PF6]2 and the experimental facts highlighted above, we propose that both complexes target the mitochondria upon crossing the plasma membrane.
Figure 4.

Microscope images of an A549 cell treated with: a) [d‐2][PF6]2 (λ exc=488 nm, c final=25 μm), b) MitoTracker deep red (λ exc=639 nm, c final=1.1 μm), and c) [l‐2][PF6]2 (λ exc=488 nm, c final=25 μm).
Due to the lack of selective uptake of [d‐2][PF6]2 versus [l‐2][PF6]2 and the proposed localization in the mitochondria, a final imaging experiment with sodium azide (NaN3) was performed. Sodium azide is known to inhibit all energy‐dependent uptake mechanisms. Cells treated with NaN3 and [d‐2][PF6]2 or [l‐2][PF6]2 did not show significant differences in uptake or localization (Figure S12 in the Supporting Information) relative to cells that were only treated with ruthenium complex. Altogether, although the cytotoxicity of both enantiomers depends on the chirality of the glucose moiety, these results support a glucose‐transporter‐ and energy‐independent19 uptake mechanism in vitro.
Photochemistry with DNA
The mitochondria house double‐stranded circular DNA, which was very interesting given the mitochondrial localization and the DNA light‐switch capabilities of dppz (dipyrido‐[3,2‐a:2′,3′‐c]phenazine) analogues of [1]2+.20 Therefore, the photochemistry of [d‐2][PF6]2 with calf thymus DNA (CT‐DNA) and pUC19 plasmid DNA was studied in more detail. As explained above, in PBS under blue‐light excitation (λ exc=450 nm) in air the emission maximum of [d‐2][PF6]2 shifted from λ=648 to 690 nm, which was attributed to the formation of [1]2+. Under these conditions there was no significant change in the emission intensity over the irradiation time (Figure S6 in the Supporting Information). However, under the same irradiation conditions, but in the presence of CT‐DNA, the emission shifted from λ=648 to 700 nm and the intensity increased 10‐fold over 25 min of irradiation (Figure S5B in the Supporting Information). Under similar conditions, the presence of micelles (Pluronic F‐127), 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) or 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine (DMPC) liposomes, l‐cysteine, l‐histidine, l‐glutathione, l‐lysine, l‐tryptophan, and 5′‐guanosine monophosphate (5′‐GMP) (5 mm in PBS) did not enhance the emission after irradiation. These data confirm a specific DNA light‐switch interaction of [1]2+ with CT‐DNA and further support that the emission observed in the cells is the result of interaction with DNA.
The emission study suggested different interactions of [d‐2]2+ and [1]2+ with DNA. To investigate this further, the interaction between [d‐2][PF6]2 and pUC19 plasmid DNA was analyzed by agarose gel electrophoresis. pUC19 was supplied as a 2686 base pairs (bp) plasmid, of which ≈95 % was in the supercoiled (SC) form. A single nick in one of the SC strands, caused by 1O2 for example, results in the open circular (OC) form, which migrates more slowly through the gel than the SC form. Staining and visualization of the DNA with ethidium bromide (EtBr), a known DNA intercalator, was sensitive to the ratio of DNA base pairs relative to the metal complex (bp/MC; see Figure S7 in the Supporting Information). Thus, [d‐2]2+ and EtBr compete for the same DNA binding sites. To determine the photobinding and photocleaving ability of [d‐2]2+, a 400:1 bp/MC ratio was used. At this low metal‐complex concentration (≈5 μm), [d‐2]2+ displayed minimal binding in the dark and allowed DNA visualization by treatment with EtBr (Figure S7 in the Supporting Information). However, with increasing light doses (λ exc=450 nm, 1 min, 0.6 J cm−2 to 15 min, 9.5 J cm−2) two phenomena were observed. First, an increased volume of the OC form was observed at higher light doses, which is a sign of 1O2 generation (Figure S8 in the Supporting Information). Second, migration retardation of the SC form was clearly observed. In our case, a limitation of DNA gel electrophoresis was that the specific mode of interaction (covalent modification versus intercalation) could not be specified. However, it did show that photoinduced association of [1]2+ or [d‐2]2+ with the SC form occurred. Taking into account both sets of DNA experiments, we propose that the photoproduct [1]2+ can interact with mitochondrial DNA either by intercalation or coordination, which results in increased emission of the metal complex and DNA photocleavage by formation of 1O2. Both DNA binding and cleavage occur at very high bp/MC ratios relative to previously reported DNA light switches,20, 21 which highlights the exceptional photodynamic properties of [1]2+.
Conclusions
Glycoconjugation dramatically improves the hydrophilicity of a compound, which also improves the drug properties of lipophilic compounds such as [1]2+. Additionally, combining glycoconjugation with the light‐induced uncaging properties of ruthenium‐based polypyridyl compounds, the hydrophobic active molecule [1]2+ can be recovered inside the cell if the glycoconjugated compound can still cross the cell membrane. For [d‐2][PF6]2 and [l‐2][PF6]2 this approach was very successful: the dppn ligand is lipophilic enough to counterbalance the hydrophilicity of the glucose moiety, which allows passive uptake to take place. Upon irradiation with a low dose of visible light (λ exc=454 nm, 3.2 J cm−2) a very high cytotoxic activity, characterized by submicromolar EC50 values, was obtained. The significant phototoxic indices of these compounds may be a consequence of at least two photochemical reactions occurring in the mitochondria. First, photosubstitution of the thioether glucose ligand in [d‐2]2+ by water occurs, which allows better interaction of the photoproduct [1]2+ with biomolecules. In particular, mitochondrial DNA seems a likely target of the achiral photoproduct [1]2+ because it interacts with plasmid DNA at a particularly high base pair/Ru ratio. Second, both [2]2+ and [1]2+ efficiently generate 1O2, which leads to extensive DNA photocleavage for adducts of mitochondrial DNA and [1]2+. To the best of our knowledge, these results represent the first practical demonstration that photosubstitution and 1O2 generation can combine in vitro to create a dual mode of action, resulting in highly efficient light‐induced cancer cell death.
The second function of glycoconjugation is to introduce specific interactions between the (pro)drug and glucose‐sensitive enzymes. When increased cytotoxicities are measured for d‐glucose‐appended drugs, for example versus their aglycon analogues, they are often interpreted as a sign of glucose‐transporter‐mediated uptake. A similar interpretation would have led us to conclude that the higher dark cytotoxicity of [d‐2][PF6]2 versus [l‐2][PF6]2 was the result of the d‐glucose functional group in [d‐2][PF6]2 targeting the glucose transporters. However, in vitro imaging showed no difference in uptake or cellular localization between the two enantiomers, and addition of sodium azide indicated energy‐independent drug uptake. These results demonstrate that GLUT or SGLT are not involved in the uptake of these compounds, and thus other enzymes (for example, efflux pumps and/or glucosidases)22 must be responsible for the two‐fold greater cytotoxicity of [d‐2][PF6]2 relative to [l‐2][PF6]2 in the dark. This work also had unexpected consequences: although [d‐2][PF6]2—the complex conjugated to the natural d‐glucose moiety—would be expected to be the most interesting “targeted” enantiomer, [l‐2][PF6]2—the complex conjugated to the non‐natural l‐glucose moiety—actually shows a higher phototoxic index because of its lower cytotoxicity in the dark. Ultimately, l‐glucose derivatization showed a better pharmacological outcome than functionalization with d‐glucose.
Experimental Section
Synthesis
General: Reagents were purchased from Sigma–Aldrich and used without further purification. Benzo[i]dipyrido‐[3,2‐a:2′,3′‐c]phenazine (dppn) was synthesized according to a literature procedure.23 2,2′:6′,2′′‐Terpyridine (tpy) was purchased from ABCR GmbH & Co. Dry solvents were collected from a Pure Solve MD5 dry solvent dispenser from Demaco. For all inorganic reactions solvents were deoxygenated by bubbling argon gas through the solution for 30 min. Flash chromatography was performed on silica gel (Screening devices B. V.) with a particle size of 40–64 μm and a pore size of 60 Å. TLC analysis was performed on TLC aluminum foils with a silica gel matrix (Supelco silica gel 60, 56524); plates were visualized under a UV lamp (λ=254 nm) after spraying with 10 % H2SO4 in ethanol or with solution of NH4Mo7O24 ⋅4 H2O (25 g L−1) and NH4CeSO44 ⋅H2O (10 g L−1) in 10 % H2SO4, followed by charring at ≈250 °C on a heating plate. Optical rotation measurements were performed with a Propol automated polarimeter (sodium d‐line, λ=589 nm) with a sample concentration of 10 mg mL−1 (c=1), unless stated otherwise. IR spectra were recorded with a PerkinElmer universal attenuated total reflectance (UATR; Single Reflection Diamond) Spectrum Two instrument ( =4000–700 cm−1; resolution=4 cm−1). 1H and 13C NMR were recorded in [D6]acetone, [D6]DMSO, MeOD, or CDCl3 with chemical shifts (δ) reported relative to the solvent peak. HRMS were recorded by direct injection (2 μL of a 2 μm solution in 50:50 water/acetonitrile and 0.1 % formic acid) into a mass spectrometer (Thermo Finnigan LTQ Orbitrap) equipped with an electrospray ion source in positive mode (source voltage 3.5 kV, sheath gas flow 10, capillary temperature 250 °C) with resolution (R)=60 000 at m/z 400 (mass range m/z 150–2000) and dioctyl phthalate (m/z 391.28428) as a lock mass. The high‐resolution mass spectrometer was calibrated prior to measurements with a calibration mixture (Thermo Finnigan). Combustion analysis was performed at Kolbe Mikrolab (Germany).
Ligand synthesis: A full description of the ligand syntheses and the characterization data for the ligands is provided in the Supporting Information.
Complex [4][Cl]: Ruthenium dimer [{RuCl2(tpy)}2]⋅H2O24 (300 mg, 0.347 mmol) and dppn23 (231 mg, 0.695 mmol) were dissolved in a deoxygenated solution of ethylene glycol (17 mL), and the mixture was heated at 100 °C under an argon atmosphere for 5 h. The resulting purple solution was filtered over Celite. Et2O (50 mL) was added to the filtrate, and the resulting precipitate was collected on a glass frit, thoroughly washed with water and Et2O, and then dried under high vacuum to give [4][Cl] (383 mg, 0.473 mmol, 75 %) as a purple powder. R f=0.31 (10 % MeOH/CH2Cl2); 1H NMR (400 MHz, [D6]DMSO): δ=10.42 (d, J=4.7 Hz, 1 H; 1), 9.75 (d, J=8.0 Hz, 1 H; 3), 9.21–9.06 (m, 3 H; 8, 22, 15), 8.90 (d, J=8.1 Hz, 2 H; T3’, T5’), 8.75 (d, J=8.1 Hz, 2 H; T6, T6’’), 8.56 (dd, J=8.1, 5.4 Hz, 1 H; 2), 8.38 (d, J=9.5 Hz, 1 H; T3, T3’’), 8.30 (t, J=8.1 Hz, 1 H; T4’’), 7.99 (t, J=8.1 Hz, 2 H; T5, T5’’), 7.83 (d, J=5.4 Hz, 1 H; 20), 7.78–7.65 (m, 4 H; 10, 13, T4, T4’’), 7.53 (dd, J=8.1, 5.5 Hz, 1 H; 21), 7.32 ppm (t, J=6.7 Hz, 2 H; 11, 12); 13C NMR (100 MHz, [D6]DMSO): δ=158.4 (Cq), 157.6 (Cq), 154.0 (CH 1), 153.80 (CH 20), 152.6 (Cq), 152.5 (CH 10, CH 13), 150.5 (Cq), 141.5 (Cq), 141.0 (Cq), 137.9 (Cq), 137.8 (Cq), 137.2 (CH T5, CH T5’’), 134.5 (Cq), 134.4 (Cq), 134.2 (CH T4’), 131.9 (CH 3), 130.8 (CH 8), 130.1 (Cq), 129.6 (Cq), 128.5 (CH T3), 128.5, 128.5 (CH T3’’), 127.9 (CH 22), 127.8 (CH 15), 127.3 (CH T4, T4’’), 127.2 (CH 11, CH 12), 126.5 (CH 21), 123.7 (CH 21), 122.80 ppm (CH T3’, CH T5’); HRMS: m/z calcd (%) for [C37H23N7ClRu−Cl−]: 702.07415; found: 702.07439; elemental analysis calcd (%) for C37H23N7Ru⋅4 H2O: C 54.89, H 3.86, N 12.11; found: C 53.63, H 3.83, N 11.61.
Complex [1][PF6]2: AgNO3 (39.0 mg, 0.230 mmol) was added to a solution of [4][Cl] (73.0 mg, 0.099 mmol) in 3:1 acetone/water (10 mL). This mixture was stirred under an argon atmosphere at 50 °C for 16 h, and then filtered over Celite. A saturated aqueous solution of NH4PF6 (2 mL) was added to the filtrate, and the resulting brown precipitate was collected on a glass frit then washed with H2O (×3) and Et2O (×3) to afford [1][PF6]2 (60 mg, 0.083 mmol, 84 %) as a brown precipitate, which was used without further purification. R f=0.5 (100:80:20 acetone/water/saturated aqueous KPF6).
Complex [d‐2][PF6]2: [1][PF6]2 (60.0 mg, 0.0668 mmol) and ligand d‐3 (61.0 mg, 0.178 mmol) were dissolved in deoxygenated acetone, and the mixture was stirred at 50 °C for 24 h under an argon atmosphere in the dark. The resulting brown/orange solution was concentrated under vacuum at 30 °C in the dark. The crude material was purified by column chromatography on silica gel (acetone/water/saturated aqueous KPF6 100:0:0 to 80:20:0 to 100:80:20), followed by further purification by column chromatography on Sephadex LH‐20 (acetone). The orange fraction was collected and the volume was reduced to about 10 % then Et2O was added. The resulting precipitate was collected by filtration on a Whatman RC60 membrane filter then washed with EtOAc (×3), Et2O (×3), and n‐hexane (×3) to afford [d‐2][PF6]2 (30 mg, 23 μmol, 35 %) as an orange powder. R f=0.36 (16:4:1 acetone/water/saturated aqueous KPF6); 1H NMR (400 MHz, [D6]acetone): δ=10.39 (d, J=5.4 Hz, 1 H; 1), 10.09 (d, J=8.2 Hz, 1 H; 3), 9.60 (d, J=8.3 Hz, 1 H; 22), 9.27 (s, 1 H; 8), 9.15 (s, 1 H; 15), 9.03 (d, J=8.2 Hz, 2 H; T4’, T2’), 8.84 (d, J=8.0 Hz, 2 H; T5, T1’’), 8.73 (dd, J=8.3, 5.3 Hz, 1 H; 2), 8.61 (t, J=8.2 Hz, 1 H; T3’), 8.44 (dd, J=18.7, 9.0 Hz, 2 H; 11, 12), 8.19 (t, J=7.9 Hz, 2 H; T4, T2’’), 8.13 (t, J=3.7 Hz, 2 H; T2, T4’’), 8.06 (d, J=5.4 Hz, 1 H; 20), 7.84–7.76 (m, 3 H; 21, 10, 13), 7.47 (t, J=6.6 Hz, 2 H; T3, T2’’), 4.31 (d, J=7.7 Hz, 1 H; H‐1), 4.23 (dd, J=12.9, 3.5 Hz, 1 H; CHH H‐6), 3.99–3.92 (m, 1 H; OCHH), 3.86–3.76 (m, 1 H; OCHH), 3.71–3.51 (m, 9 H; CHH H‐6, 2×OCHH, 3×OCH 2), 3.41–3.23 (m, 3 H; H‐3, H‐4, H‐5), 3.11 (td, J=8.2, 3.5 Hz, 1 H; H‐2), 2.26–2.18 (m, 2 H; OCH 2SMe), 1.66 ppm (s, 3 H); 13C NMR (100 MHz, [D6]acetone): δ=159.0 (Cq), 158.5 (Cq), 155.0 (CH 1), 153.2 (CH 1, CH T2, CH T4’’), 139.4 (CH T4, CH T2’’), 138.3 (CH T3’), 136.0 (Cq), 135.0 (CH 3) 134.7 (CH 22), 132.5 (Cq), 131.4 (Cq), 129.5 (CH T3, CH T2’’), 129.5, 129.5 (CH 11, CH 12) 129.1 (CH 8, CH 15), 128.9 (CH 11, CH 5, CH 8), 128.0 (Harom), 126.1 (CH T5, CH T1’’), 125.4 (CH T4’, CH T2’), 104.2 (C‐1), 78.0 (C‐3), 77.5 (C‐4), 74.8 (C‐2), 71.7 (C‐5), 71.0 (2×OCH2), 70.9 (OCH2), 69.3 (OCH2), 68.2 (OCH2), 62.9 (C‐6), 35.6 (OCH2SMe), 15.6 ppm (OCH2SMe); HRMS: m/z calcd for [C50H49O8N7SRu−2 PF6]: 504.61979; found: 504.61993; elemental analysis calcd (%): C 46.23, H 3.80, N 7.55; found: C 46.26, H 3.81, N 7.53.
Complex [l‐2][PF6]2: [l‐2][PF6]2 was synthesized from ligand l‐3 (94.0 mg, 0.0964 mmol) and purified according to the procedure described for above for [d‐2][PF6]2. This procedure afforded [l‐2][PF6]2 (40 mg, 0.031 mmol, 32 %) as an orange powder. 1H NMR and HRMS data matched that reported for [d‐2][PF6]2. Elemental analysis calcd (%) for [l‐2][PF6]2 ⋅H2O: C 45.60, H 3.90, N 7.44; found: C 45.70, H 4.06, N 7.32.
Photochemistry
A stock solution of [d‐2][PF6]2 in demineralized water (c=4.23×10−5 m, 3.00 mL) was transferred to a quartz fluorescence cuvette (1 cm width) that contained a stirrer bar. This solution was deoxygenated for 15 min with argon then kept at a constant temperature (25 °C) and irradiated with light from a blue LED (λ exc=450 nm, full‐width at half‐maximum (FWHM)=19 nm) with photon flux=1.77×10−7 Einstein s−1. During this period UV/Vis spectra were recorded at intervals of 30 s for the first 10 min then every 5 min until 1 h. ESI‐MS spectra were recorded after the irradiation experiment to confirm the formation of the aqua species [Ru(tpy)(dppn)(H2O)]2+. The quantum yield of photosubstitution (Φ i) was calculated according to Equation (1) by using λ=490 and 410 nm as reference wavelengths, with ϵ 490=7220 and 12 000 m −1 cm−1 and ϵ 410=12 660 and 10 720 m −1 cm−1 for [d‐2][PF6]2 and [1][PF6]2, respectively Eqn. (1).
| (1) |
in which k is the first‐order photosubstitution rate constant, n is the number of moles of ruthenium complex in the cuvette, Φ is the photon flux determined by standard ferrioxalate actinometry, and A e is the absorbance of the solution at the irradiation wavelength. The quantum yield Φ i was determined as an average over two experiments (± standard deviation (SD)).
The 1O2 measurements and emission spectroscopy are described in full detail in the Supporting Information.
Cell culturing and cytotoxicity assay
General: Human cancer cell lines A549 (human lung carcinoma) and MCF‐7 (human breast adenocarcinoma) were distributed by the European Collection of Cell Cultures (ECACC), and purchased from Sigma Aldrich. Dulbecco's Modified Eagle Medium (DMEM, with and without phenol red, without glutamine), Glutamine‐S (GM; 200 mm), trichloroacetic acid (TCA), glacial acetic acid, sulforhodamine B (SRB), and tris(hydroxylmethyl)aminomethane (Tris base) were purchased from Sigma Aldrich. Fetal calf serum (FCS) was purchased from Hyclone. Penicillin and streptomycin were purchased from Duchefa and were diluted to give a penicillin/streptomycin solution (P/S; 100 mg mL−1). Trypsin and Opti‐MEM (without phenol red) were purchased from Gibco Life Technologies. Trypan blue (0.4 % in 0.81 % sodium chloride and 0.06 % potassium phosphate dibasic solution) was purchased from BioRad. Plastic disposable flasks and 96‐well plates were purchased from Sarstedt. Cells were counted by using a BioRad TC10 automated cell counter with Biorad cell‐counting slides. UV/Vis measurements for analysis of 96‐well plates were performed with a M1000 Tecan Reader. Cells were inspected with an Olympus IX81 microscope.
Cell culturing: Cells were cultured in DMEM complete (DMEM with phenol red, supplemented with FCS (8.0 % v/v), P/S solution (0.2 % v/v), GM (0.9 % v/v)). Cells were cultured under humidified conditions (37 °C atmosphere containing 7.0 % CO2) in 75 cm2 flasks and subcultured (1:3–1:6) upon reaching 70–80 % confluency (approximately once per week). Media was refreshed every 2 d; cells were passaged for 4–8 weeks.
Cell‐irradiation setup: The cell‐irradiation system consisted of a Ditabis thermostat (980923001) fitted with two flat‐bottomed microplate thermoblocks (800010600) and a 96‐LED array fitted to a standard 96‐well plate. The λ=454 nm LED (OVL‐3324), fans (40 mm, 24 V DC, 9714839), and power supply (EA‐PS 2042‐06B) were obtained from Farnell. See Hopkins et al.25 for a full description of the cell‐irradiation setup.
Cytotoxicity assay: Cells were seeded at t=0 in 96‐well plates at a density of 5×103 (A549) and 8×103 cells/well (MCF‐7) in Opti‐MEM complete without phenol red (100 μL) and incubated for 24 h at 37 °C/7 % CO2. After this period, aliquots (100 μL) of six different concentrations (1–100 μm for dark plates, 0.1–10 μm for irradiated plates) of freshly prepared stock solutions of [d‐2][PF6]2 and [l‐2][PF6]2 in Opti‐MEM were added to the wells in triplicate. Plates were incubated in the dark for an additional 24 h. After this period the media in each well was refreshed, and half of the plates were irradiated for 5 min with blue light (λ=454±11 nm, power density = 10.5±0.7 mW cm−2, light dose = 3.2±0.2 J cm−2) and the other half were kept in the dark under otherwise identical conditions. After irradiation all the plates were placed incubated for an additional 48 h. The cells were fixed by adding cold TCA (10 % w/v; 100 μL) in each well. Next, TCA was removed from the wells, and the plates were gently washed with water (×5), air dried, stained with SRB (0.6 % w/v in acetic acid (1 % v/v; 100 μL) for 30–45 min, washed with acetic acid (1 % v/v; ≈300 μL), and air dried. The SRB dye was solubilized with Tris base (10 mm; 200 μL), and the absorbance in each well was read at λ=510 nm by using a M1000 Tecan Reader.
The SRB absorbance data was used to calculate the fraction of viable cells in each well (Excel and GraphPad Prism software). The absorbance data were averaged from triplicate wells per concentration. Relative cell viabilities were calculated by dividing the average absorbance of the treated wells by the average absorbance of the untreated wells. Three independent biological replicates were completed for each cell line (three different passage numbers per cell line). The average cell viability of the three biological replicates was plotted versus log(concentration) [μm], with the SD error of each point. By using the dose–response curve for each cell line under dark‐ and irradiated conditions, the effective concentration (EC50) was calculated by fitting the curves to a non‐linear regression function with fixed y maximum (100 %) and minimum (0 %) (relative cell viability) and a variable Hill slope, which resulted in the simplified two‐parameter Hill‐slope equation [Eq. (2)]:
| (2) |
The DNA photointeraction studies and (emission) microscopy are described in full detail in the Supporting Information.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Dutch Organization for Scientific Research (NWO‐CW) via a VIDI grant to S.B. The European Research Council is kindly acknowledged for a Starting Grant to S.B. Prof. E. Bouwman is gratefully acknowledged for her support and input. The COST action CM1105 “Functional metal complexes that bind to biomolecules” is gratefully acknowledged for stimulating scientific discussion. Gerwin Spijksma is gratefully acknowledged for performing the HRMS measurements. W. Pomp and Prof. T. Schmidt are gratefully acknowledged for access to their microscopy setup.
L. N. Lameijer, S. L. Hopkins, T. G. Brevé, S. H. C. Askes, S. Bonnet, Chem. Eur. J. 2016, 22, 18484.
References
- 1.
- 1a. Warburg O., Science 1956, 123, 309; [DOI] [PubMed] [Google Scholar]
- 1b. Deng D., Xu C., Sun P., Wu J., Yan C., Hu M., Yan N., Nature 2014, 510, 121–125; [DOI] [PubMed] [Google Scholar]
- 1c. Alfarouk K. O., Verduzco D., Rauch C., Muddathir A. K., Adil H. H. B., Elhassan G. O., Ibrahim M. E., David Polo Orozco J., Cardone R. A., Reshkin S. J., Harguindey S., Oncoscience 2014, 1, 777–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Welling M. M., Alberto R., Nucl. Med. Commun. 2010, 31, 239–248; [DOI] [PubMed] [Google Scholar]
- 2b. Ferreira C. L., Bayly S. R., Green D. E., Storr T., Barta C. A., Steele J., Adam M. J., Orvig C., Bioconjugate Chem. 2006, 17, 1321–1329. [DOI] [PubMed] [Google Scholar]
- 3. Calvaresi E. C., Hergenrother P. J., Chem. Sci. 2013, 4, 2319–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Calvaresi E. C., Granchi C., Tuccinardi T., Di Bussolo V., Huigens 3rd R. W., Lee H. Y., Palchaudhuri R., Macchia M., Martinelli A., Minutolo F., Hergenrother P. J., ChemBioChem 2013, 14, 2263–2267; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Pelageev D. N., Dyshlovoy S. A., Pokhilo N. D., Denisenko V. A., Borisova K. L., Keller-von Amsberg G., Bokemeyer C., Fedorov S. N., Honecker F., Anufriev V. P., Eur. J. Med. Chem. 2014, 77, 139–144. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Basu U., Khan I., Hussain A., Gole B., Kondaiah P., Chakravarty A. R., Inorg. Chem. 2014, 53, 2152–2162; [DOI] [PubMed] [Google Scholar]
- 5b. Law W. H., Lee L. C., Louie M. W., Liu H. W., Ang T. W., Lo K. K., Inorg. Chem. 2013, 52, 13029–13041; [DOI] [PubMed] [Google Scholar]
- 5c. Liu P., Lu Y., Gao X., Liu R., Zhang-Negrerie D., Shi Y., Wang Y., Wang S., Gao Q., Chem. Commun. 2013, 49, 2421–2423; [DOI] [PubMed] [Google Scholar]
- 5d. Schibli R., Dumas C., Petrig J., Spadola L., Scapozza L., Garcia-Garayoa E., Schubiger P. A., Bioconjugate Chem. 2005, 16, 105–112; [DOI] [PubMed] [Google Scholar]
- 5e. Bowen M. L., Orvig C., Chem. Commun. 2008, 5077–5091. [DOI] [PubMed] [Google Scholar]
- 6. Banik B., Somyajit K., Hussain A., Nagaraju G., Chakravarty A. R., Dalton Trans. 2014, 43, 1321–1331. [DOI] [PubMed] [Google Scholar]
- 7. Park J., Um J. I., Jo A., Lee J., Jung D. W., Williams D. R., Park S. B., Chem. Commun. 2014, 50, 9251–9254. [DOI] [PubMed] [Google Scholar]
- 8. Patra M., Johnstone T. C., Suntharalingam K., Lippard S. J., Angew. Chem. Int. Ed. 2016, 55, 2550–2554; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2596–2600. [Google Scholar]
- 9. Barron C., Tsiani E., Tsakiridis T., BMC Proceedings 2012, 6, P4. [Google Scholar]
- 10. Pohl J., Bertram B., Hilgard P., Nowrousian M. R., Stuben J., Wiessler M., Cancer Chemother. Pharmacol. 1995, 35, 364–370. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Liu Y., Cao Y., Zhang W., Bergmeier S., Qian Y., Akbar H., Colvin R., Ding J., Tong L., Wu S., Hines J., Chen X., Mol. Cancer Ther. 2012, 11, 1672–1682; [DOI] [PubMed] [Google Scholar]
- 11b. Miskimins W. K., Ahn H. J., Kim J. Y., Ryu S., Jung Y. S., Choi J. Y., PLoS One 2014, 9, e85576; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Dilip A., Cheng G., Joseph J., Kunnimalaiyaan S., Kalyanaraman B., Kunnimalaiyaan M., Gamblin T. C., Anticancer Drugs 2013, 24, 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Venturelli L., Nappini S., Bulfoni M., Gianfranceschi G., Dal Zilio S., Coceano G., Del Ben F., Turetta M., Scoles G., Vaccari L., Cesselli D., Cojoc D., Sci. Rep. 2016, 6, 21629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng D., Sun P., Yan C., Ke M., Jiang X., Xiong L., Ren W., Hirata K., Yamamoto M., Fan S., Yan N., Nature 2015, 526, 391–396. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Goldbach R. E., Rodriguez-Garcia I., van Lenthe J. H., Siegler M. A., Bonnet S., Chem. Eur. J. 2011, 17, 9924–9929; [DOI] [PubMed] [Google Scholar]
- 14b. Howerton B. S., Heidary D. K., Glazer E. C., J. Am. Chem. Soc. 2012, 134, 8324–8327; [DOI] [PubMed] [Google Scholar]
- 14c. Respondek T., Garner R. N., Herroon M. K., Podgorski I., Turro C., Kodanko J. J., J. Am. Chem. Soc. 2011, 133, 17164–17167; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14d. van Rixel V. H. S., Siewert B., Hopkins S. L., Askes S. H. C., Busemann A., Siegler M. A., Bonnet S., Chem. Sci. 2016, 22, 10960–10968; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14e. Sgambellone M. A., David A., Garner R. N., Dunbar K. R., Turro C., J. Am. Chem. Soc. 2013, 135, 11274–11282; [DOI] [PubMed] [Google Scholar]
- 14f. Zayat L., Calero C., Albores P., Baraldo L., Etchenique R., J. Am. Chem. Soc. 2003, 125, 882–883. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Crespy D., Landfester K., Schubert U. S., Schiller A., Chem. Commun. 2010, 46, 6651–6662; [DOI] [PubMed] [Google Scholar]
- 15b. Schatzschneider U., Eur. J. Inorg. Chem. 2010, 1451–1467; [Google Scholar]
- 15c. Mari C., Pierroz V., Ferrari S., Gasser G., Chem. Sci. 2015, 6, 2660–2686; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Farrer N. J., Salassa L., Sadler P. J., Dalton Trans. 2009, 10690–10701; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15e. Presa A., Brissos R. F., Caballero A. B., Borilovic I., Korrodi-Gregório L., Pérez-Tomás R., Roubeau O., Gamez P., Angew. Chem. Int. Ed. 2015, 54, 4561–4565; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4644–4648. [Google Scholar]
- 16. Zhu X., Schmidt R. R., Angew. Chem. Int. Ed. 2009, 48, 1900–1934; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1932–1967. [Google Scholar]
- 17. Knoll J. D., Albani B. A., Turro C., Chem. Commun. 2015, 51, 8777–8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang H., Yu B., Zhang P., Huang J., Chen Y., Gasser G., Ji L., Chao H., Angew. Chem. Int. Ed. 2015, 54, 14049–14052; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14255–14258. [Google Scholar]
- 19. Komor A. C., Barton J. K., Chem. Commun. 2013, 49, 3617–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Friedman A. E., Chambron J.-C., Sauvage J.-P., Turro N. J., Barton J. K., J. Am. Chem. Soc. 1990, 112, 4960–4962; [Google Scholar]
- 20b. Niyazi H., Hall J. P., O'Sullivan K., Winter G., Sorensen T., Kelly J. M., Cardin C. J., Nat. Chem, 2012, 4, 621–628. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Song H., Kaiser J. T., Barton J. K., Nat. Chem. 2012, 4, 615–620; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Sun Y., Joyce L. E., Dickson N. M., Turro C., Chem. Commun. 2010, 46, 2426–2428. [DOI] [PubMed] [Google Scholar]
- 22. Seker H., Bertram B., Burkle A., Kaina B., Pohl J., Koepsell H., Wiesser M., Br. J. Cancer 2000, 82, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McConnell A. J., Lim M. H., Olmon E. D., Song H., Dervan E. E., Barton J. K., Inorg. Chem. 2012, 51, 12511–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marelius D. C., Bhagan S., Charboneau D. J., Schroeder K. M., Kamdar J. M., McGettigan A. R., Freeman B. J., Moore C. E., Rheingold A. L., Cooksy A. L., Smith D. K., Paul J. J., Papish E. T., Grotjahn D. B., Eur. J. Inorg. Chem. 2014, 676–689. [Google Scholar]
- 25. Hopkins S. L., Siewert B., Askes S. H., Veldhuizen P., Zwier R., Heger M., Bonnet S., Photochem. Photobiol. Sci. 2016, 15, 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary