

ABSTRACT
Autosomal Dominant Leukodystrophy (ADLD), a fatal adult onset demyelinating disorder, is the only human disease that has been linked to mutations of the nuclear lamina protein, lamin B1, and is primarily caused by duplications of the LMNB1 gene. Why CNS myelin is specifically targeted and the mechanisms underlying ADLD are unclear. Recent work from our group has demonstrated that over expression of lamin B1 in oligodendrocytes, the myelin producing cells in the CNS, resulted in age dependent epigenetic modifications, transcriptional down-regulation of lipogenic gene expression and significant reductions of myelin-enriched lipids. Given the high lipid content of meylin, we hypothesize that lipid loss is one of the primary drivers of the demyelination phenotype. These results can, at least partially, explain the age dependence and cell type specificity in ADLD and are discussed in the context of the existing literature, in an attempt to delineate potential pathways underlying the disease phenotype.
KEYWORDS: ADLD demyelination, epigenetic modifications, lipid synthesis, leukodystrophy, Lamin B1, nuclear lamina, nuclear structure, transcription
Introduction
The nuclear lamina is a critical structural component of the nuclear envelope found adjacent to the inner nuclear membrane in all multicellular organisms.1 Nuclear lamins are a class of intermediate filament proteins and in vertebrates they are composed of 2 main types: the A and B type lamins. The A type lamins, lamins A & C are alternatively spliced products of the same gene LMNA. Two separate genes, LMNB1 and LMNB2 code for the major B type lamins.1 The A and B type lamins closely interact in the nuclear lamina meshwork although they have been shown to form independent microdomains.2
While originally identified as having a structural role in maintaining nuclear architecture, the nuclear lamina is now thought to play a critical role in multiple cellular processes including transcription, DNA replication, DNA repair and epigenetic regulation.1 Genes associated with the nuclear lamina are thought to be transcriptionally silenced and enriched in specific repressive histone marks.3,4 In addition, the nuclear lamina has been shown to play an important role in both normal and pathological aging.5,6
Autosomal Dominant Leukodystrophy is caused by mutations involving the Lamin B1 gene
While at least 12 distinct disorders have been associated with mutations in the LMNA gene, only one disease, Autosomal Dominant Leukodystrophy (ADLD), has so far been linked to mutations involving lamin B1.7,8 The majority of ADLD cases are caused by duplications involving the lamin B1 gene locus that results in increased lamin B1 protein expression.8,9 A recent report described a deletion upstream of the lamin B1 gene that resulted in a variant ADLD phenotype.10 Lamin B1 expression was also found to be increased in these patient samples suggesting that increased lamin B1 expression is the common pathway that leads to the disease phenotype.
ADLD is a slowly progressive and fatal demyelinating disorder presenting in the 4th or 5th decade of life.11,12 It is characterized clinically by early autonomic abnormalities, pyramidal and cerebellar dysfunction, muscle wasting and symmetrical demyelination of the CNS with the brain stem and spinal cord showing early involvement.11,12 ADLD is also unique as it is one of the few leukodystrophies that has a purely adult onset.12
ADLD Pathology-location and timing: Clues from mouse models
That lamin b1 is critical for CNS development has been well documented using both germ line and conditional knockouts.13 Mice lacking a functional lamin B1 protein survived embryonic development but died immediately after birth with lung, bone and neurodevelopmental defects.14 Conditional knockouts with a deletion of lamin b1 in forebrain specific neurons exhibited normal longevity but had significantly small cerebral cortices, disorganized layering of cortical neurons with a smaller number of neurons and neuronal migration defects.15 Lamin B1 has also been shown to be required for normal dendrite development in primary mouse cortical neurons.16 While the lamin b1 appears to be essential for the proper development of neuronal lineages, its role is glial cells such as astrocytes and oligodendrocyte is still unknown.
Given the widespread nature of lamin B1 expression, the specific involvement of myelin and the late age onset of the disease process have been puzzling features. However, recently developed mouse models for ADLD are providing insights into the disease process that may help answer both these questions. Previous work by Heng et al., (2013) suggested that oligodendrocyte and not neuron or astrocyte specific overexpression of lamin B1 was sufficient to produce a late age onset motor dysfunction reminiscent of ADLD.17 In this report, myelin defects were identified in the brain stem only demonstrable by electron microscopy while biochemical analysis revealed a reduction of the myelin protein, PLP1.17 However, as PLP1 knock out mice do not exhibit demyelination18 this suggested that other mechanisms were responsible for the pathology observed in the lamin b1 over expressing mice.
To further understand these mechanisms, our group, in a report by Rolyan et al. (2015) characterized independently derived mouse lines that expressed a FLAG tagged lamin B1 in oligodendrocytes.19 These FLAG-PLP-LMNB1 transgenic (TG) mice exhibited a robust overexpression of lamin B1 that was highest in the spinal cord and an age dependent muscle wasting, kyphosis and motor dysfunction that culminated in premature mortality by 13–15 months of age (Fig. 1a). Interestingly, the spinal cord exhibited the most severe pathological alterations with the cervical region showing the most extensive demyelination (Fig. 1b). Cervical involvement was also consistent with forelimb paralysis as the being earliest presenting phenotype.
Figure 1.

Spinal cord degeneration in a mouse model of ADLD. (A) PLP-FLAG-LMNB1 transgenic mice (TG), with oligodendrocyte specific overexpression of lamin B1, show age dependent degenerative phenotypes including kyphosis (black arrow), forelimb paralysis (white arrowhead) and muscle wasting at 13 months while wild type (WT) littermates show no obvious phenotypes. (B) Cervical spinal cords section of TG mice show significant vacuolar degeneration involving the white matter (arrows). No such alterations are observed in spinal cord sections from WT littermates. Top panel – H&E staining, Bottom panel – Fluormyelin staining (Fluromyelin is a fluorescent dye that specifically stains white matter). Reproduced with permission from Rolyan et al., (2015).19
The pathology in the spinal cord of the TG mice shared a number of similarities with the histo-pathological alterations in ADLD patient CNS tissue. Both exhibited a vacuolar degenerative phenotype primarily involving the white matter tracts with a preservation of oligodendrocytes.20,21 This suggested that the over expression of lamin B1 is not lethal to oligodendrocytes and that the demyelination is not the result of oligodendrocyte loss. Recent reports have linked lamin b1 to pathways of cellular proliferation and senescence.22,23 However, results from both human patients and our mouse model suggest that oligodendrocyte proliferation and survival are unaffected, suggesting that these pathways might not play a role in the ADLD phenotype. Another similarity was the finding that in ADLD patents axonal loss was minimal while in the mice axonal loss was present only in the terminal stages, secondary to the demyelination.19,20
Some differences were also present between the mouse model and human disease. The mouse model showed significant astrocytosis and microglial infiltration, with some degree of microglial infiltration present even before the onset of demyelination in the mice. While patient brain sections exhibited a modest degree of reactive gliosis, no astrocytic proliferation was observed, although the astrocytes in affected regions were found to have abnormal and shortened processes.20,21 It is unclear whether these differences represent species-specific responses to lamin B1 overexpression. However, it is important to remember that ADLD has a relatively slow disease progression with death occurring at least a 10-20 y after the onset of symptoms. As the pathological studies in ADLD have been carried out on post mortem brain samples it is possible that the demyelination lesions are no longer active sites of inflammation and thus do not show a cellular infiltrate.
Another notable difference between mouse and human diseases was the location of the pathology. The PLP-FLAG-LMNB1 mouse model displayed the most severe pathology in the spinal cord while other regions such as the brain stem showed minimal involvement.19 In ADLD patients, in addition to the spinal cord, the brain stem, cerebellum and cerebral lobes all show white matter loss.20,21 We attributed this difference to the expression pattern of the transgenic FLAG tagged LMNB1 protein which was maximal in the spinal cord. Support for this assertion comes from a recently published report that describes a variant form of ADLD where an enhancer adoption mechanism resulted in increased expression of lamin B1 in the frontal lobe, the region that exhibited the most severe pathology by MRI.10 In the patient sample brain sample studied, the cerebellum was unaffected and this coincided with lamin b1 levels that were comparable with controls. The precise location of lamin b1 over expression thus may play an important role in determining the specific areas of the CNS that are impacted. This is especially relevant as ADLD patients from independent families have unique duplications and the differing regulatory sequences upstream of the lamin B1 gene in these families may contribute to altered spatial patterns of lamin B1 overexpression and a varied clinical presentation.10 The specific locations of the pathology, both in mice and humans, may also raise the possibility of differential susceptibility of oligodendrocytes to lamin b1 over expression. A recent report has suggested that the developmental origin of oligodendrocytes determines the response to demyelination and susceptibility to age-associated functional decline and it would be tempting to speculate whether a similar mechanism also holds true for oligodendrocyte function due to lamin B1 overexpression.24
Epigenetic and transcriptional pathways link lipid synthesis and demyelination in ADLD
Given the importance of lamin B1 in regulating chromatin, Rolyan et al., (2015) interrogated specific histone modifications in oligodendrocytes from the FLAG-PLP-LMNB1 spinal cord sections and demonstrated that there were age dependent increases in repressive histone marks (H3K9me3 and H3K27me3) and decreases in activating histone marks (AcH3 and AcH4). This suggested that lamin B1 overexpression promotes transcriptional repression in older animals that might be a driving force in oligodendrocyte dysfunction (Fig. 2). The nuclear lamina is known to undergo age dependent alterations6 and whether the increased epigenetic impact of Lamin B1 overexpression reflects a greater sensitivity to perturbations in nuclear structure at a later age is yet to be determined. Age is a particularly important factor in the case of oligodendrocyte function as remyelination efficiency has been shown to decline with age through mechanisms also mediated by chromatin modification.25
Figure 2.
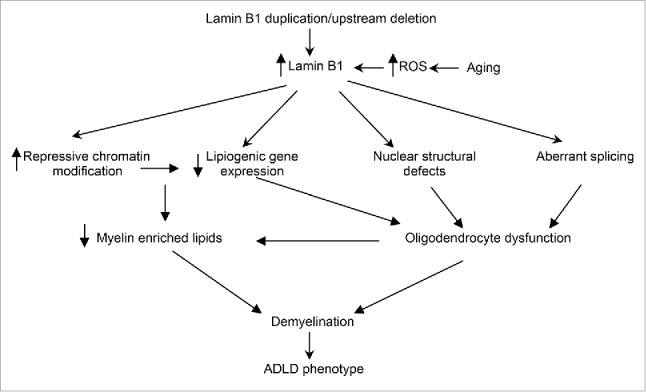
ADLD disease mechanism. Potential disease mechanisms underlying ADLD. Results from our group suggest that lipid dysregulation mediated by age dependent epigenetic alterations in oligodendrocytes are a major driver for the demyelination observed in an ADLD mouse model. However, other pathways such nuclear structural defects or aberrant splicing may also contribute to oligodendrocyte dysfunction. Findings that elevated reactive oxygen species (ROS) promote an accumulation of lamin B1 may provide an alternative hypothesis to explain the late age onset of the disease phenotype.
To identify genes that might be impacted by chromatin alterations in the ADLD mouse model we carried out a genome wide trancriptomics analysis of spinal cord tissue isolated from TG and WT mice at 2 different time points, one before the onset of the disease (3 months) and one after (13 months). Interestingly, genes encoding myelin proteins were not found to be significantly reduced either at the RNA or protein level in the TG mice. As predicted from the histone alterations, there was a dramatic increase in the percentage of genes that were down regulated in the older TG mice from 8.4% (3 months) to 48.4% (13 months) suggesting that the chromatin alterations do indeed lead to a global transcriptional repression in an age dependent manner.19
Up regulated genes primarily belonged to inflammatory pathways and were thought to indicate a secondary response to the demyelination phenotype derived from astrocytes or microglia. Only 12 genes that were common to the 2 time points were downregulated. Intriguingly, a closer analysis of these genes revealed that 9 of them belonged to lipid synthesis pathways (Table 1). Furthermore, 8 of these 9 genes were those known to be enriched in myelinating oligodendrocytes. Consistent with these results a lipidomics analysis revealed significant reductions in myelin enriched lipids in TG mice spinal cord extracts when compared to WT mice, even at time points prior to the onset of pathological alterations. Lipid dysregulation is a particularly attractive hypothesis in ADLD as myelin is composed of ∼70% lipids and mutations in lipid synthesis genes have resulted in myelin pathology.26 Further evidence supporting the lipid hypothesis is the similarity in histo-pathological features we have observed and the mouse models where specific myelin enriched lipids such as galactolipids have been disrupted.27 The lipid dysregulation we have identified in the ADLD mouse model also provides a rationale to explain the involvement of oligodendrocytes in the disease pathology. As they require an extremely robust lipid biosynthetic process, oligodendrocytes might be acutely sensitive to the reductions in lipogenic gene expression that are caused by lamin B1 over expression to the extent that other cell types such as astrocytes or neurons in the CNS are not.
Table 1.
Lipid synthesis genes downregulated in 3 and 13 month transgenic PLP-FLAG-LMNB1 mice.
| Fold change relative to wild types |
||||
|---|---|---|---|---|
| Gene Symbol | Full name | 3 month | 13 month | Enriched in myelinating oligodendrocyte |
| Ldlr | Low-Density Lipoprotein (LDL) Receptor | 0.71 | 0.31 | Yes |
| Cyp51 | Lanosterol 14-demethylase (CYP51) | 0.75 | 0.37 | Yes |
| Hmgcs1 | 3-Hydroxy-3-Methylglutaryl-CoA Synthase 1 | 0.65 | 0.38 | Yes |
| Sqle | Squalene Epoxidase | 0.75 | 0.41 | No |
| Sc4mol | Methylsterol Monooxygenase | 0.71 | 0.41 | Yes |
| Hmgcr | 3-Hydroxy-3-Methylglutaryl-CoA Reductase | 0.74 | 0.55 | Yes |
| Scd1 | Stearoyl-CoA desaturase-1 | 0.62 | 0.55 | Yes |
| Dhcr7 | DHCR7 7-dehydrocholesterol reductase | 0.73 | 0.58 | Yes |
| Elovl7 | ELOVL Fatty Acid Elongase | 0.74 | 0.69 | Yes |
Our results linking lamin B1 and lipid biosynthesis confirms a distinct yet poorly understood role for the nuclear lamina in lipid regulation. Mutations in lamin A cause lipodystrophies, a class of disorders characterized by a selective loss of adipose tissue.5 While the mechanisms in Lamin A associated lipodystrophies are unclear, previous studies have suggested that mutant lamin A sequesters the critical lipogenic transcription factor Sterol regulatory Binding Protein 1 (SREBP 1) at the nuclear periphery thereby reducing availability of the protein for transcriptional activation.28 Rolyan et al, (2015) also demonstrated a reduced expression of the SREBP1 and 2 genes in tissues from the Lamin B1 overexpressing mice. It remains to be determined whether Lamin B1 regulates the activity of these genes through a chromatin mediated transcriptional mechanism or whether post-translational mechanisms such as nuclear sequestration also play a role.
While our results indicate that age dependent epigenetic alterations and transcriptional pathways are a critical component of the pathology in ADLD, other potential mechanisms can also contribute to the disease (Fig. 2). Recent reports have linked lamin b1 to oxidative stress pathways and suggested that elevated reactive oxygen species (ROS) lead to an accumulation of lamin B1.23 As elevated ROS levels are associated with the aging brain, it is also possible that lamin b1 accumulation in ADLD patients is accelerated by age dependent ROS to critical levels that disrupt oligodendrocyte function, thus providing an alternate mechanism to explain the age dependence of the disease. Fibroblasts from ADLD patients exhibit increased nuclear rigidity and nuclear abnormalities and this has been proposed to alter nuclear signaling, although the exact pathways are unclear.29 Altered mRNA processing has been demonstrated in ADLD fibroblasts and it is thought that this may contribute to an aberrant regulation of myelin specific genes.30
In conclusion, the recent report by Rolyan et al., (2015) provides evidence that lamin B1 over expression can down regulate the expression of lipid synthesis genes and myelin enriched lipids though age dependent epigenetic pathways. This model provides a mechanistic framework that can, at least partially, explain some aspects of the age dependence and cell type specificity in ADLD. Other mechanisms such as oxidative stress response, nuclear shape alterations and altered splicing may also contribute, either in concert or through independent pathways to the disease phenotype (Fig. 2).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The author would like to thank members of the Padiath laboratory for helpful discussions.
Funding
This work was supported by National Institutes of Health Grants (R01NS095884, R21AG046897) and National Multiple Sclerosis Society Grant (RG5045A1) to QSP.
References
- [1].Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem 2015; 84:131-64; PMID:25747401; http://dx.doi.org/ 10.1146/annurev-biochem-060614-034115 [DOI] [PubMed] [Google Scholar]
- [2].Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, et al.. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev 2008; 22:3409-21; PMID:19141474; http://dx.doi.org/ 10.1101/gad.1735208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stancheva I, Schirmer EC. Nuclear envelope: connecting structural genome organization to regulation of gene expression. Adv Exp Med Biol 2014; 773:209-44; http://dx.doi.org/ 10.1007/978-1-4899-8032-8_10 [DOI] [PubMed] [Google Scholar]
- [4].Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al.. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008; 453:948-51; PMID:18463634; http://dx.doi.org/ 10.1038/nature06947 [DOI] [PubMed] [Google Scholar]
- [5].Ghosh S, Zhou Z. Genetics of aging, progeria and lamin disorders. Curr Opin Genetics Dev 2014; 26C:41-6; PMID:25005744; http://dx.doi.org/ 10.1016/j.gde.2014.05.003 [DOI] [PubMed] [Google Scholar]
- [6].Haithcock E, Dayani Y, Neufeld E, Zahand AJ, Feinstein N, Mattout A, Gruenbaum Y, Liu J. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc Natl Acad Sci U S A 2005; 102:16690-5; http://dx.doi.org/ 10.1073/pnas.0506955102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chojnowski A, Ong PF, Dreesen O. Nuclear lamina remodelling and its implications for human disease. Cell Tissue Res 2015; 360:621-31; PMID:25532872; http://dx.doi.org/ 10.1007/s00441-014-2069-4 [DOI] [PubMed] [Google Scholar]
- [8].Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 2006; 38:1114-23; PMID:16951681; http://dx.doi.org/ 10.1038/ng1872 [DOI] [PubMed] [Google Scholar]
- [9].Giorgio E, Rolyan H, Kropp L, Chakka AB, Yatsenko S, Di Gregorio E, Lacerenza D, Vaula G, Talarico F, Mandich P, et al.. Analysis of LMNB1 duplications in autosomal dominant leukodystrophy provides insights into duplication mechanisms and allele-specific expression. Hum Mutat 2013; 34:1160-71; PMID:23649844; http://dx.doi.org/ 10.1002/humu.22348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Giorgio E, Robyr D, Spielmann M, Ferrero E, Di Gregorio E, Imperiale D, Vaula G, Stamoulis G, Santoni F, Atzori C, et al.. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD). Hum Mol Genetics 2015; 24:3143-54; PMID:25701871; http://dx.doi.org/ 10.1093/hmg/ddv065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nahhas N, Sabet Rasekh P, Vanderver A, Padiath Q. Autosomal Dominant Leukodystrophy with Autonomic Disease In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, et al., eds. GeneReviews(R). Seattle (WA), 1993 [Google Scholar]
- [12].Padiath QS, Fu YH. Autosomal dominant leukodystrophy caused by lamin B1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol 2010; 98:337-57; http://dx.doi.org/ 10.1016/S0091-679X(10)98014-X [DOI] [PubMed] [Google Scholar]
- [13].Young SG, Jung HJ, Lee JM, Fong LG. Nuclear lamins and neurobiology. Mol Cell Biol 2014; 34:2776-85; PMID:24842906; http://dx.doi.org/ 10.1128/MCB.00486-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A 2004; 101:10428-33; PMID:15232008; http://dx.doi.org/ 10.1073/pnas.0401424101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Coffinier C, Jung HJ, Nobumori C, Chang S, Tu Y, Barnes RH 2nd, Yoshinaga Y, de Jong PJ, Vergnes L, Reue K, et al.. Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol Biol Cell 2011; 22:4683-93; http://dx.doi.org/ 10.1091/mbc.E11-06-0504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Giacomini C, Mahajani S, Ruffilli R, Marotta R, Gasparini L. Lamin B1 protein is required for dendrite development in primary mouse cortical neurons. Mol Biol Cell 2016; 27:35-47; PMID:26510501; http://dx.doi.org/ 10.1091/mbc.E15-05-0307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Heng MY, Lin ST, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptacek LJ, et al.. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J Clin Invest 2013; 123:2719-29; PMID:23676464; http://dx.doi.org/ 10.1172/JCI66737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Klugmann M, Schwab MH, Puhlhofer A, Schneider A, Zimmermann F, Griffiths IR, Nave KA. Assembly of CNS myelin in the absence of proteolipid protein. Neuron 1997; 18:59-70; PMID:9010205; http://dx.doi.org/ 10.1016/S0896-6273(01)80046-5 [DOI] [PubMed] [Google Scholar]
- [19].Rolyan H, Tyurina YY, Hernandez M, Amoscato AA, Sparvero LJ, Nmezi BC, Lu Y, Estecio MR, Lin K, Chen J, et al.. Defects of lipid synthesis are linked to the age-dependent demyelination caused by Lamin B1 overexpression. J Neurosci 2015; 35:12002-17; PMID:26311780; http://dx.doi.org/ 10.1523/JNEUROSCI.1668-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Melberg A, Hallberg L, Kalimo H, Raininko R. MR characteristics and neuropathology in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. AJNR Am J Neuroradiol 2006; 27:904-11 [PMC free article] [PubMed] [Google Scholar]
- [21].Sundblom J, Melberg A, Kalimo H, Smits A, Raininko R. MR imaging characteristics and neuropathology of the spinal cord in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. AJNR Am J Neuroradiol 2009; 30:328-35; PMID:18945794; http://dx.doi.org/ 10.3174/ajnr.A1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, Goldman RD. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 2011; 25:2579-93; PMID:22155925; http://dx.doi.org/ 10.1101/gad.179515.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Barascu A, Le Chalony C, Pennarun G, Genet D, Imam N, Lopez B, Bertrand P. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J 2012; 31:1080-94; http://dx.doi.org/ 10.1038/emboj.2011.492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Crawford AH, Tripathi RB, Richardson WD, Franklin RJ. Developmental origin of oligodendrocyte lineage cells determines response to demyelination and susceptibility to age-associated functional decline. Cell Rep 2016; pii: S2211-1247(16)30358-8; PMID:27149850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, Casaccia-Bonnefil P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci 2008; 11:1024-34; PMID:19160500; http://dx.doi.org/ 10.1038/nn.2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chrast R, Saher G, Nave KA, Verheijen MH. Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res 2011; 52:419-34; http://dx.doi.org/ 10.1194/jlr.R009761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dupree JL, Suzuki K, Popko B. Galactolipids in the formation and function of the myelin sheath. Microscopy Res Technique 1998; 41:431-40; PMID:9672425; http://dx.doi.org/ 10.1002/(SICI)1097-0029(19980601)41:5%3c431::AID-JEMT9%3e3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- [28].Duband-Goulet I, Woerner S, Gasparini S, Attanda W, Konde E, Tellier-Lebegue C, Craescu CT, Gombault A, Roussel P, Vadrot N, et al.. Subcellular localization of SREBP1 depends on its interaction with the C-terminal region of wild-type and disease related A-type lamins. Exp Cell Res 2011; 317:2800-13; PMID:21993218; http://dx.doi.org/ 10.1016/j.yexcr.2011.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ferrera D, Canale C, Marotta R, Mazzaro N, Gritti M, Mazzanti M, Capellari S, Cortelli P, Gasparini L. Lamin B1 overexpression increases nuclear rigidity in autosomal dominant leukodystrophy fibroblasts. FASEB J 2014; 28(9):3906-18; PMID:24858279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bartoletti-Stella A, Gasparini L, Giacomini C, Corrado P, Terlizzi R, Giorgio E, Magini P, Seri M, Baruzzi A, Parchi P, et al.. Messenger RNA processing is altered in autosomal dominant leukodystrophydagger. Hum Mol Genetics 2015; 24:2746-56; http://dx.doi.org/ 10.1093/hmg/ddv034 [DOI] [PMC free article] [PubMed] [Google Scholar]