

ABSTRACT
We herein report retargeting of T-helper (Th) cells against the universal cancer antigen telomerase for use in adoptive cell therapy. The redirected Th cells may counter tumor tolerance, transform the inflammatory milieu, and induce epitope spreading and cancer senescence. We have previously conducted a series of trials evaluating vaccination with telomerase peptides. From long-term survivors, we isolated >100 CD4+ Th-cell clones recognizing telomerase epitopes. The clones were characterized with regard to HLA restriction, functional avidity, fine specificity, proliferative capacity, cytokine profile, and recognition of naturally processed epitopes. DP4 is the most prevalent HLA molecule worldwide. Two DP4-restricted T-cell clones with different functional avidity, C13 and D71, were selected for molecular T-cell receptor (TCR) cloning. Both clones showed a high proliferative capacity, recognition of naturally processed telomerase epitopes, and a polyfunctional and Th1-weighted cytokine profile. TCR C13 and D71 were cloned into the retroviral vector MP71 together with the compact and GMP-applicable marker/suicide gene RQR8. Both TCRs were expressed well in recipient T cells after PBMC transduction. The transduced T cells co-expressed RQR8 and acquired the desired telomerase specificity, with a polyfunctional response including production of TNFa, IFNγ, and CD107a. Interestingly, the DP4-restricted TCRs were expressed and functional both in CD4+ and CD8+ T cells. The findings demonstrate that the cloned TCRs confer recipient T cells with the desired hTERT-specificity and functionality. We hypothesize that adoptive therapy with Th cells may offer a powerful novel approach for overcoming tumor tolerance and synergize with other forms of immunotherapy.
KEYWORDS: Adoptive cell therapy, cancer, DP4, HLA II, immunotherapy, long-term survivor, retargeted T cell, T-cell receptor, telomerase, T-helper cell
Introduction
Most cancer vaccines have focused on recruiting CD8+ cytotoxic T cells (CTLs). We have investigated the potential of recruiting CD4+ T-helper (Th) cells, which are known to be key modulators of the immune response. To this aim, we have conducted a series of clinical trials evaluating vaccination with long peptides designed to recruit Th cells. This includes studies in melanoma, non-small cell lung cancer (NSCLC), and pancreatic cancer patients evaluating vaccination with the telomerase peptide GV1001.1-4
Human telomerase reverse transcriptase (hTERT) is overexpressed in most human cancers and thus a widely applicable target for cancer therapy.5,6 Because hTERT expression is important for tumor growth, the risk of tumor escape is presumably limited. In our GV1001 vaccine trials, we have observed immune response rates ranging from 50% to 80%, combined with low toxicity.1-4 None of the patients experienced serious adverse events. Interestingly, the GV1001 immune response was associated with improved survival in all trials. This effect was particularly evident at the tail of the survival curve, similar to what has been observed for checkpoint inhibitors. In the advanced NSCLC patients, the mean survival for immune responders was 54 mo, compared to 13 mo for those not developing an immune response.7 Six NSCLC patients were still alive at the last clinical update, all belonging to the immune responders. Telomerase-based vaccines carry a putative risk of bone marrow toxicity. We therefore closely monitored hematologic counts and also conducted hematopoietic progenitor cell assays on bone marrow samples,8 without detecting any hematologic toxicity.
Adoptive cell therapy with retargeted T cells has mainly been tested with chimeric antigen receptors (CARs) or HLA class I restricted TCRs, both designed to directly bind tumor cells. CAR T-cell therapy is showing remarkable clinical efficacy against hematological malignancies.9,10 In solid tumors, however, there are concerns that retargeted T cells may be unable to overcome established tumor tolerance.11,12 In this study, we investigate adoptive cell therapy using hTERT-specific TCRs from CD4+ Th cells. These TCRs recognize antigen in the context of HLA class II. We hypothesize that the engraftment of tumor and lymph nodes with hTERT-specific Th cells may transform the micro-environment, target key cell types, and mobilize the patient's immune system to mount a broad response. The Th cells may directly engage dendritic cells and tumor-infiltrating macrophages, class II positive endothelial cells in the tumor vasculature, and other class II positive stromal cells. Most malignant cells are HLA class II negative, but some upregulate HLA class II under the influence of IFNγ. Further, Th cells can mediate bystander killing of class II negative cancer cells.
The clinical efficacy from checkpoint inhibitor therapy has highlighted the potential of modulating the inflammatory tumor milieu.13-15 It is well documented that Th cells are key orchestrators of immunity and that their activity is necessary for effective antitumor responses. Th cells promote survival of CD8+ T cells and development of memory CD8+ T cells.16,17 Th cells are also able to counter the suppressive activity of T regulatory cells (Tregs) and myeloid-derived suppressor cells (MDSCs)18 and to produce Th1 cytokines that drive cancer into senescence.19 Further, Th cells can induce epitope spreading, i.e., responses against a broad spectrum of antigens. Even with vaccines targeting defined CTL antigens, there is evidence that clinical efficacy may depend on epitope spreading rather than a response against the vaccine antigen.20,21 In our hTERT vaccine studies, we (Inderberg et al.) have observed that clinical response is associated with intramolecular epitope spreading.22,23 A Th response may recruit host T cells recognizing the patient's individual spectrum of neoantigens. A number of recent reports have suggested that the most effective anticancer responses are directed against these unknown targets,24-27 whereas only a low proportion of tumor-infiltrating lymphocytes recognize shared antigens.28
In this study, we characterize Th-cell clones to identify candidates for molecular TCR cloning and use in adoptive cell therapy. We focus on T-cell clones from patients with a durable immune response, apparent clinical benefit, and long-term survival. In addition to the possible clinical advantage from their response, these patients have been monitored for years without development of side effects, amid an ongoing hTERT-specific immune response. As reported below, we have identified TCRs with favorable characteristics, cloned the TCRs into retroviral constructs, and tested their ability to confer the desired specificity and functionality to recipient T cells.
Results
T-cell clones from two long-term survivors showed diverse avidity
We have generated >100 CD4+ Th-cell clones from patients vaccinated with the hTERT peptide GV1001.1-4 This includes a series of T-cell clones from two long-term survivors, patients C and D, in the NSCLC trial CTN-2000.2,29 Both patients developed a de novo GV1001-specific response after vaccination, associated with tumor regression and extended survival. Patient C had inoperable stage III NSCLC and developed a complete tumor response after vaccination. A durable GV1001-specific T cell response was demonstrated, detectable at all 31 time points tested from week 37 onwards (last immune assay after 8 y).2 The patient is alive to date (14 y) and had no evidence of relapse at the last clinical follow-up (12 y). He received a total of 44 vaccine injections over a period of 11 y without any side effects. Patient D survived for 46 mo. She received a total of 16 vaccine injections and developed a durable T-cell memory response, without notable adverse effects.2,7
We investigated the functional avidity of the T-cell clones by use of peptide titration in 3H-thymidine proliferation assays. A striking diversity was observed. Among GV1001-specific clones generated from different patients, the minimum peptide concentration required varied 100,000-fold, with a range from 10−5 M to 10−10 M (Fig. 1,4,22,29 and unpublished). For the clones from patients C and D, the peptide concentration producing a half-maximal response (EC50) ranged from 10−7 to 10−9 M (Fig. 1A and B and data not shown). Most T-cell clones had a threshold peptide concentration where a plateau for maximum proliferation response was reached. Further, the high-avidity clones mounted somewhat lower responses at the highest peptide concentrations (Fig. 1). The latter finding is consistent with the notion that high-avidity T cells may be susceptible to exhaustion if exposed to high antigen loads, as discussed below.
Figure 1.
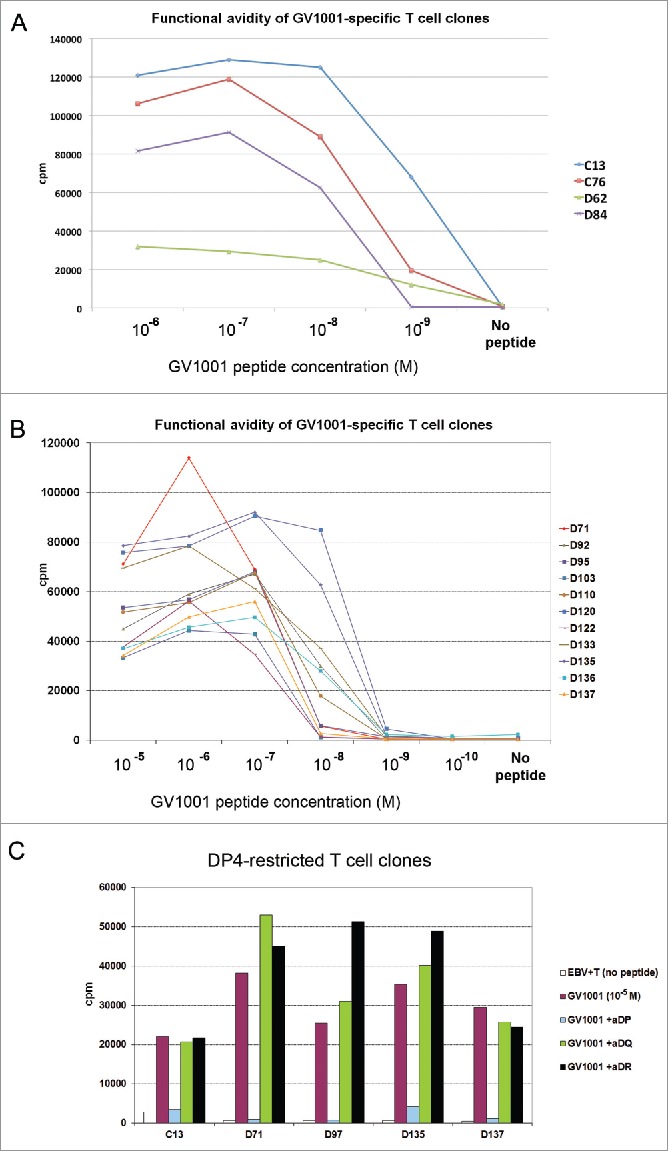
Functional avidity and HLA restriction of GV1001-specific T-cell clones. T-cell clones from NSCLC patients C and D, both long-term survivors after GV1001 vaccination, were tested for proliferation after stimulation with irradiated EBV-transformed cells +/− GV1001 peptide. Panels (A)–(C) show mean cpm of triplicate wells. (A, B) Functional avidity of T-cell clones determined by titration of GV1001. (C) HLA restriction determined by blockage with mAbs against DP, DQ, or DP-molecules.
HLA restriction of GV1001-specific T-cell clones
The HLA restriction of a TCR is important for its applicability in the patient population. To determine the HLA restriction, we genotyped the patients and analyzed the clones in proliferation assays with mAbs blocking either DP, DQ, or DR molecules. Finally, the clones were tested against panels of APCs (EBV cells) covering the relevant HLA-molecules. The most common HLA class II allele is DP4, which is prevalent in many races and ethnic groups and estimated to be present in 76% of Caucasians.30 We therefore focused on DP4 restricted TCRs as lead candidates for molecular cloning. Patients C and D were genotyped as DP04*01/DP04*02 and DP04*01/DP05*01, respectively. Fig. 1c shows HLA-restriction assays for five DP-restricted clones from patients C and D. These clones were shown to be DP4-restricted in subsequent tests with EBV cells covering alternative HLA molecules (data not shown). In studies of GV1001-specific clones from other patients, we observed that the clones covered a wide range of HLA-DP, DQ, and DR alleles (Refs. [1,4,22,29] and unpublished). This observation suggests that it may be possible to develop a panel of GV1001-specific TCRs covering most patients.
Fine-specificity and recognition of naturally processed epitopes
We investigated the fine-specificity of the T-cell clones by stimulation with a series of truncated peptides that cover the GV1001 sequence. The results revealed that the T-cell clones recognize different core epitopes (Fig. 2A). Taken together with the above-mentioned HLA-restriction promiscuity, this observation points to a diverse T-cell response that may be less vulnerable to tumor escape. On the other hand, the different fine-specificity among the T-cell clones suggests that each clone should be evaluated for its ability to recognize naturally processed epitopes.
Figure 2.
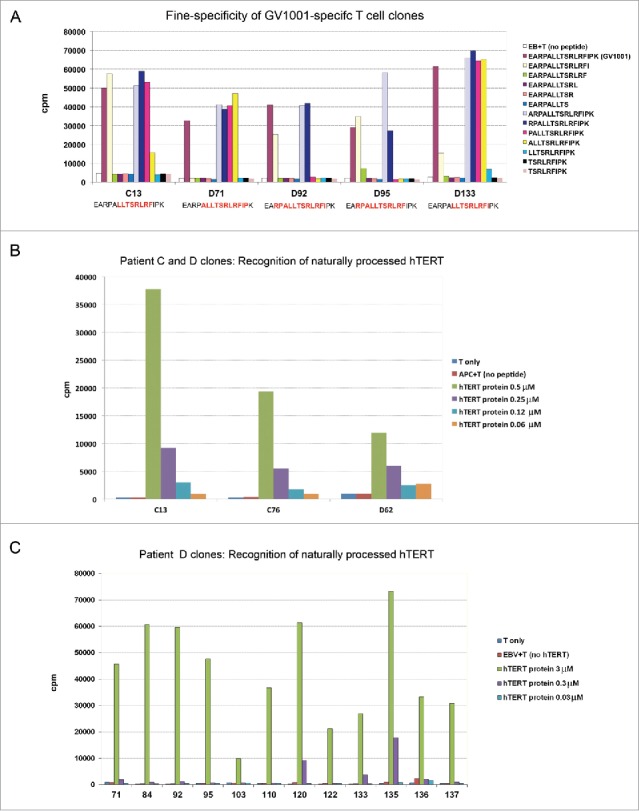
Fine specificity of T-cell clones and recognition of naturally processed hTERT epitopes. T-cell clones from patients C and D were tested for proliferation after stimulation with irradiated EBV-transformed cells +/− antigen. Panels (A)–(C) show mean cpm of triplicate wells. (A) Fine specificity analysis by stimulation with truncated peptides covering the GV1001 sequence (aa sequences given in right text box). The core sequence recognized by each clone is different and highlighted (red) in text boxes bellow chart. (B, C) Recognition of naturally processed antigens. T-cell clones were stimulated with EBV-transformed cells incubated with a 173 aa recombinant hTERT protein fragment. The concentration of the protein fragment was titrated as indicated.
A peptide-specific response is of no clinical relevance if the peptide is not naturally processed. We screened the GV1001-specific clones for their ability to recognize naturally processed epitopes by loading a 173 aa hTERT protein fragment, which comprises the GV1001 sequence, on autologous APCs (PBMCs or EBV cells). The results demonstrated that about 90% of our GV1001-specific T-cell clones responded to stimulation with this hTERT protein fragment. In general, the high avidity clones responded best. Fig. 2 shows the response for a panel of T-cell clones from patients C and D.
Cytokine profile
The cytokine profile is likely to be of importance for Th-cell therapy. Analyses of supernatants may be vulnerable to variations over time, due to dynamics in cytokine production and consumption. To address this issue, we analyzed supernatants collected from four GV1001-specific T-cell clones every day for one week (Fig. 3). The clones were obtained from patient C and a subject from a GV1001 trial in pancreatic cancer (patient E29). The results revealed that some cytokines, in particular IL-2, peaked at day 1 and disappeared from day 2 or 3 onwards. The TNFα and IL-4 levels were highest at days 1 and 2, and dropped gradually thereafter. By contrast, other cytokines gradually increased over the first days and remained at high levels thereafter. The GV1001 cultures maintained their high levels of INFγ, IL-13, and MIP-1β throughout, suggesting that these cytokines could be used for reliable classification of the clones at any time point. We concluded that day 2 supernatants may be suitable for assessing the cytokine profile. In day 2 supernatants, most GV1001-specific T-cell clones showed a polyfunctional cytokine profile, which is considered to be important for effective immunity, and secreted high levels of INFγ, TNFa, IL-13, MIP-1α, MIP-1β, and eotaxin (Fig. 3 and Ref. [29]). We detected highly variable levels of IL-6, IL-17, IL-8, IP-10, MCP-1, and GM-CSF (Fig. 3 and Ref. [29]).
Figure 3.
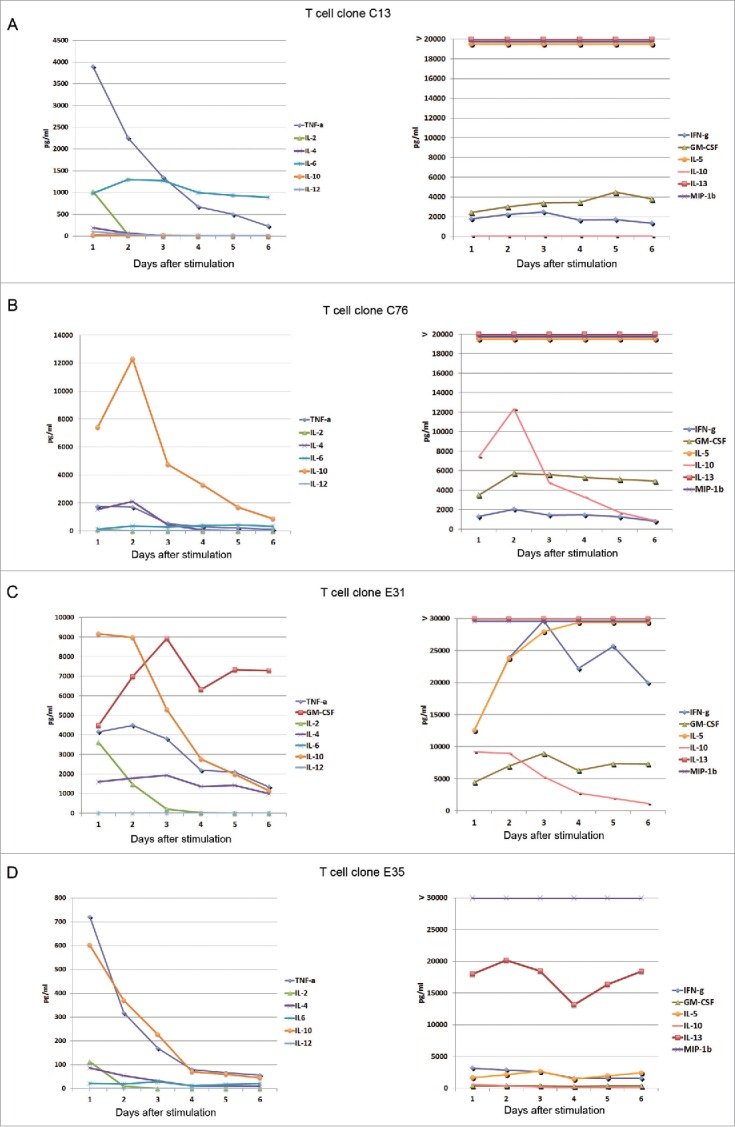
Cytokine secretion by GV1001-specific T-cell clones monitored over 6 d. T-cell clones from immune responders in GV1001 vaccine trials were stimulated with irradiated EBV-transformed cells +/− peptide GV1001. Supernatants from duplicate cultures were collected each day for 6 d and analyzed by Bioplex cytokine assays. Panels (A) and (B) show data from T-cell clones from patient C. Panels (C) and (D) show T-cell clones from patient E. Mean cytokine concentrations (pg/mL) from cultures with peptide are displayed. The controls without peptide had only negligible cytokine levels for all T-cell clones at all time points, below 1% of the levels measured in cultures with peptide. IL-13 was measured at >90,000 pg/mL at all time points for T-cell clones C13, C76, and E 31 (A–C), and at 13,000–20,000 pg/mL for E35 (D). MIP-1b was measured at >30,000 pg/mL at all time points for all four T-cell clones.
Selection of two DP-4 restricted T-cell clones for molecular TCR cloning
We selected T-cell clones for molecular TCR cloning, based on the following criteria: (i) evidence of clinical response in the host patient; (ii) durable immune responses in the host, without evidence of bone marrow toxicity or other adverse effects; (iii) high specificity of T-cell clones in the in vitro assays and no signs of cross reactivity; (iv) high or moderate avidity in functional assays; (v) recognition of the most common HLA-molecule (DP4); (vi) recognition of naturally processed epitopes from hTERT; and (vii) favorable cytokine profile. We identified seven DP4-restricted T-cell clones from patients C and D (long-term survivors), of which three clones also fulfilled the other criteria. Two of these T-cell clones, C13 and D71, were confirmed to be monoclonal by TCR clonotype mapping29 and selected as lead candidates for molecular cloning.
T-cell clones C13 and D71 were among the clones with the highest proliferation counts in thymidine assays, performed as simultaneous screening of large numbers of clones (100 nm to 10 μM GV1001; Fig. 1 and data not shown). They recognized different core motives (Fig. 2A) and did not show any cross-reactivity against allogeneic cells (data not shown). Clone C13 recorded the highest functional avidity among the clones from patients C and D, with a peak proliferation response at about 10−7 M and EC50 at about 10−9 M (Fig. 1A). Clone D71 displayed moderate functional avidity with peak response at 10−6 M and EC50 at 10−7 M (Fig. 1B). Both T-cell clones responded at the maximum peptide concentrations tested, but showed slightly lower proliferation counts at the highest peptide concentrations (Fig. 1A and B).
In experiments with naturally processed hTERT protein (173 aa fragment), Th clone C13 again showed potent proliferative capacity and high avidity, with peak proliferation response at 3 μM and responses at protein concentrations as low as 0.3 μM (Fig. 2B). T-cell clone D71 responded at hTERT concentrations ≥0.3 μM. By comparison, most other T-cell clones tested in our assays required a minimum hTERT protein concentration of 1 to 3 μM (Fig. 2 and data not shown).
The Bioplex multiplex assays demonstrated that clones C13 and D71 secreted a wide range of chemokines and other cytokines upon stimulation with peptide GV1001, consistent with a polyfunctional cytokine profile (Ref. [29] and data not shown). Both C13 and D71 had a high IFNγ/IL-10 ratio, suggesting a favorable balance between immunity and tolerance, and a high INFγ/IL-4 ratio consistent with a Th1-directed profile.29 The IFNγhigh, TNFαhigh, IL-4low, IL-10low profile remained intact throughout daily assessments of clone C13 for one week after antigen stimulation (Fig. 3).
Contrary to supernatant-based assays, flow cytometry provide information at the single cell level. We stimulated T-cell clones C13 and D71 with DP4-positive EBV cells with/without peptide GV1001 or a 173 aa hTERT protein fragment. The results demonstrated that a high fraction of both C13 and D71 T cells co-produced TNFα, CD107a, and Granzyme B (Fig. 4 and data not shown). No production of the Th2-cytokine IL-4 was detected. Some Th cells are reported to harbor cytotoxic capacity, whereas other Th cells do not. Here, we noted that both clones C13 and D71 produced CD107a upon stimulation with GV1001, whereas only C13 produced CD107a after stimulation with hTERT protein (Fig. 4B). This may be related to the higher functional avidity of clone C13, with an EC50 of 10−9 M compared to about 10−7 M for clone D71 (Fig. 1).
Figure 4.

T-helper clones C13 and D71 respond to naturally processed hTERT epitopes with secretion of INFγ, TNFα, and CD107a. T-cell clones C13 and B71 were stimulated with irradiated EBV-transformed cells +/− peptide GV1001 or a 173 aa hTERT protein fragment. The cultures were incubated overnight and analyzed by flow cytometry. Top panels in (A) show INFγ/TNFα staining for clone C13. Bottom panels in (A) show INFγ/TNFα staining for clone D71. Panel (B) shows CD107 a secretion for clones C13 and D71. The bar chart (right) shows the percentage of CD107a+ T cells, as determined from the CD107a+ cell region indicated in the histograms.
Molecular cloning of TCR C13 and D71 together with suicide/sorter gene RQR8
The TCR sequences of T-cell clones C13 and D71 were identified as described previously.31 The TCRs were codon optimized and modified to express mouse TCR constant regions, in order to improve surface expression and facilitate easy detection of the expressed TCR on human T cells. We molecularly cloned the TCRs into the retroviral vector MP71, together with the suicide/marker/sorter gene RQR8.32 The RQR8 construct has been developed by our lab (M Pule) and is a compact gene combining minimal target epitopes from CD34 and CD20. The RQR8 protein allows for large-scale purification of transduced T cells with the clinically approved CliniMACS CD34 system. We have previously reported that administration of the anti-CD20 antibody rituximab mediates effective deletion of RQR8-exopressing T cells in vitro and in vivo.32
TCR/RQR8 expression in transduced SupT1 cells and binding to HLA class II tetramers
The T-cell line SupT1 was transduced with the TCR-C13_RQR8 and TCR-D71_RQR8 plasmids. Both TCR C13 and TCR D71 were expressed in >90% of transduced SupT1 cells (Fig. 5A and data not shown). The TCR constructs were considerably larger than the CAR constructs that we had originally co-expressed with RQR8. We still observed that both TCRs C13 and D71 were efficiently co-expressed with protein RQR8.
Figure 5.
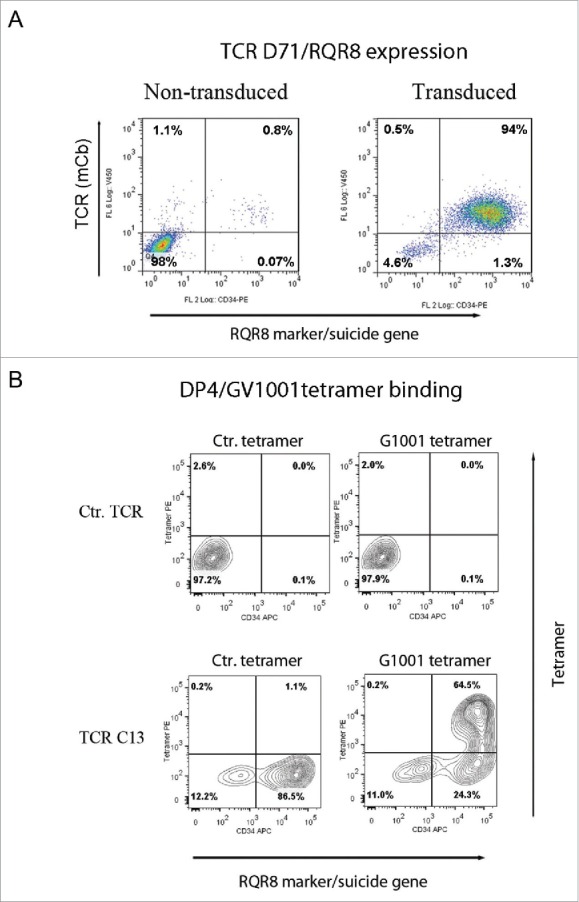
TCR expression in SupT1 cells and binding to DP4/GV1001 tetramer. SupT1 T cells were transduced with TCR-C13_RQR8 or TCR-D71_RQR8 and analyzed by flow cytometry. The expression of TCR C13/D71 was measured with a mAb recognizing the murine constant beta region (mCb) incorporated in the TCRs. The expression of marker/suicide gene RQR8 was measured with the mAb QBen10. (A) Expression of D71_RQR8 in T cell line SupT1. Left and right plots show non-transduced and transduced cells, respectively. Percentage of cells in each quartile is given. A similar TCR and RQR8 expression was observed for TCR-C13_RQR8 (not shown). (B) SupT1 T cells transduced with TCR-C13_RQR8 or a control TCR (ctr TCR) were stained with an HLA-DP4 tetramer loaded with peptide GV1001. A HLA-DP4 tetramer with irrelevant peptide was included as negative control. Top part shows non-transduced cells. Bottom part shows transduced cells. The percentage of cells in each quartile is given.
Tetramers represent useful tools for confirming binding of HLA class I restricted TCRs, but HLA class II tetramers are generally less stable than their class I counterparts. We obtained GV1001/HLA-class II DP04 tetramers from Benaroya Research Institute, Seattle, WA, USA. The tetramers were used for staining of TCR transduced cells. The results showed robust staining of cells transduced with TCR C13 (Fig. 5B), whereas TCR D71 did not appear to bind the tetramer. All the T cells binding the tetramer also expressed the suicide/sorter gene RQR8.
TCR expression in primary PBMCs and RQR8-based sorting of transduced T cells
Next, we evaluated TCR expression in primary PBMCs after retroviral transduction. The results showed satisfactory transfection efficiency, with TCR and RQR8 expression in 30–60% of transduced PBMCs (Fig. 6). The two TCRs (C13 and D71) were expressed in comparable percentages. TCRs C13 and D71 were derived from CD4+ Th cells. We therefore noted with interest that both TCRs were expressed also in CD8+ T cells (Fig. 6).
Figure 6.
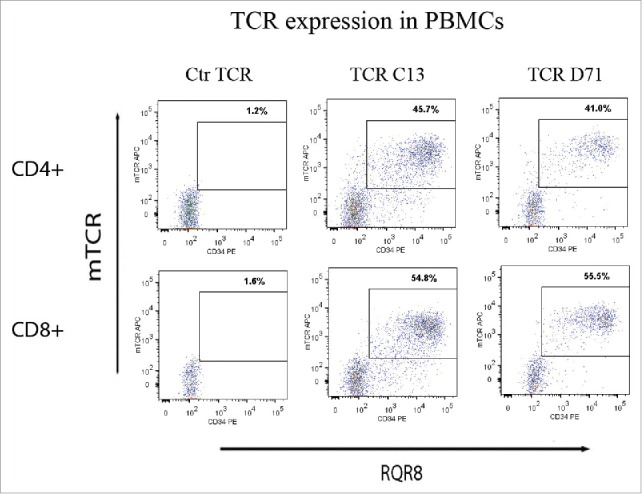
TCR expression in primary T-cells. PBMCs were transduced with TCR-C13_RQR8, TCR-D71_RQR8 or a control TCR and analyzed by flow cytometry. The expression of TCR C13/D71 was measured with a mAb recognizing the murine constant beta region (mCb) incorporated in the TCRs. The expression of marker/suicide gene RQR8 was measured with the mAb QBen10. The figure shows the expression of TCR C13_RQR8 and D71_RQR8 in CD4+ and CD8+ T cells. Left, middle, and right plots show cells transduced with the control TCR (ctr TCR), TCR C13, and TCR D71, respectively. The percentage of cells staining double positive for TCRmCb and RQR8 is given.
We tested the binding capacity of TCR transduced T cells to the CliniMACS QBen10 beads. The sorting process effectively purified the fraction of TCR-C13/RQR8 and TCR-D71/RQR8 expressing T cells (data not shown), as previously reported for other constructs incorporating RQR8.32 It is further desirable to obtain TCR-transduced T cells that can be propagated in vitro and used for functional assays. Such cell lines are not generally available, as T-cell lines like SupT1 are functionally impaired. We took advantage of our biobank of >100 T-cell clones and developed a protocol for transduction of the TCRs into these clones, which grow well in vitro. The results demonstrated that the transduced T-cell clones acquired stable expression of the new TCR, combined with retained ability for propagation in vitro (data not shown).
T-helper TCRs C13 and D71 were functional in both CD4+ and CD8+ T cells
The functionality of TCRs C13 and TCR D71 was tested by transducing primary PBMCs from donors. The transduced T cells were tested for activity against DP4-positive target cells pulsed with GV1001 peptide. The results demonstrated that the TCRs conferred GV1001-specific activity to the recipient T cells. Nearly all (80–95%) transduced T cells displayed GV1001-specific secretion of IFNγ and/or TNFα, as illustrated in Fig. 7. In repeated experiments, the functionality of TCRs C13 and D71 was similar.
Figure 7.
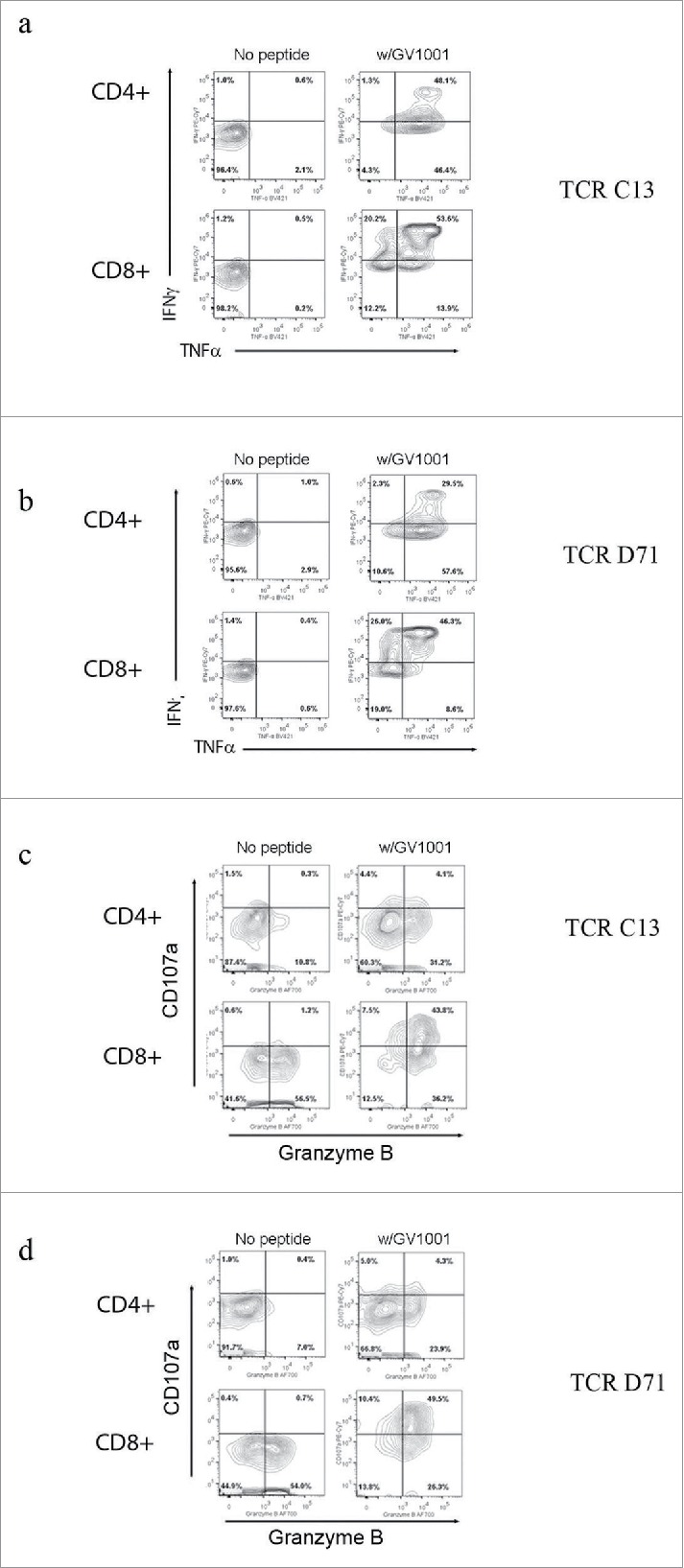
TCR transduced T-cells requires GV1001-specific functionality. Primary T cells were transduced with TCR C13_RQR8 or D71_RQR8. Transduced T cells and non-transduced control T cells were stimulated with irradiated EBV-transformed cells +/− hTERT peptide GV1001. The cultures were incubated overnight and analyzed by flow cytometry. Panels (A) and (B) show INFγ/TNFα staining for C13 and D71, respectively. Percentage of cells in each quartile is given. The transduced TCRs were derived from CD4+ T cells, but conferred GV1001-specific functionality to both CD4+ and CD8+ recipient cells. Panels (C) and (D) show cytotoxic activity of T cells transduced with C13 or D17, measured by use of CD107a and Granzyme B staining.
Interestingly, the Th-derived TCRs C13 and D71 were functional in both CD4+ and CD8+ T cells (Fig. 7A–D). The CD8+ T cells thus acquired HLA class II restricted activity. We detected some differences in functionality between the CD4+ and CD8+ cell population. A larger percentage of CD4+ T cells produced TNFα, whereas the CD8+ T cells produced more INFγ (Fig. 7 A and B). The CD8+ T cells further acquired cytotoxic activity, as demonstrated by DP4/GV1001-retricted secretion of CD107a (Fig. 7 C and D). Most CD4+ T cells did not secrete CD107a. The CD8+ T cells were also more strongly Granzyme B positive (Fig. 7 C and D). Many T cells displayed a polyfunctional profile at the single-cell level, with combined secretion of INFγ, TNFα, and CD107a as well as Granzyme B positivity. This applied both to the TCRs C13 and TCR D71 transduced cells.
Discussion
There is compelling evidence suggesting that effective anticancer responses depend on Th activity,16-18,33-36 but adoptive therapy with Th cells is a largely unexplored strategy. We hypothesize that the employment of millions of activated hTERT-specific Th cells may transform the inflammatory milieu and overcome established tumor tolerance. A few pioneering reports have provided support of this concept.37-39 The Th cells may work through multiple mechanisms, as outlined above, including the induction of epitope spreading. Patient C, that clone C13 was obtained from, developed a complete and durable tumor regression associated with a Th1-weighted and diverse T-cell response, covering multiple epitopes outside of the vaccine peptide. Beyond this anecdotal patient, we have in the GV1001 vaccine studies observed that immune responders have improved survival. This particularly related to those with a durable and broad multi-epitope T-cell response (Refs. [1,2,7,22] and unpublished). The Th-strategy may be a versatile approach for combing with other therapies. Possibly, Th cells may enable CAR T cells to retain their efficacy in solid tumors, synergize with vaccines or immunogenic chemotherapy,40 or induce neo-antigen responses that can be boosted by checkpoint inhibitors.25
Telomerase is universally expressed across cancer forms and important for tumor growth, which probably limits the risk of tumor escape. It is unclear whether hTERT is sufficiently tumor-specific to allow for safe targeting with CTLs.41 However, Th-cell therapy may circumvent key toxicity concerns associated with adoptive CTL therapy. Importantly, Th cells do not directly bind to most host cells, as these are HLA class II negative. The TCRs selected in the present study, C13 and D71, were obtained from T-cell populations that had circulated for years in the respective hosts without causing adverse effects. Further, these two TCRs have not shown any cross-reactivity in tests against panels of EBV cells from different donors. These observations support the safety rationale for the outlined therapy, but there are still reasons for caution. The Th strategy is difficult to test in animal models both with regard to safety and efficacy, as it so heavily depends on the individual patient's ability to mobilize her/his immune system in the context of established tumor tolerance. Although redirected CTLs directly kill their targets, key effector cells in the Th approach may result from epitope spreading and recognize other antigens than telomerase. This is a possible advantage, as it allows the immune system to choose its own targets, similar to what is the case with checkpoint inhibitors. On the other hand, preclinical efficacy and safety data may be of limited value.
It would be highly demanding to establish an informative mouse model. One option may be a humanized mouse model with a full-scale human immune system, in a mouse transgenic for DP4 and human hTERT. Even with such a complex model, the setting is artificial and lack key elements in the Th strategy, including true established tumor tolerance. We therefore rather aim for safe and early clinical testing in patients, by pursing the development of transient redirection of T cells with mRNA encoding the TCR.42 We have established an mRNA platform for CAR T-cell therapy, where the CAR is functional for about a week,43 and develop this approach also for TCRs. The mRNA approach allows for meaningful dose escalation, whereas permanently retargeted T cells can expand and retain their TCR/CAR in vivo. It may be important to perform clinical testing of Th TCRs in different settings and cancer forms, especially as animal models provide limited guidance. In this context, hTERT is an attractive target as it allows for comparing the effect of the same TCR across different cancers. Further, the common HLA restriction (DP4; 76% of Caucasians) of the TCRs studied herein is a key element for feasible clinical development.
To address the safety issue, we have cloned the TCRs together with the suicide/marker gene RQR8, which allows for eliminating the transduced cells with rituximab. The data reported above suggest that the TCRs are expressed well together with RQR8 and that the transduced cells can be effectively purified by use of the clinical grade CliniMACS system. In previous studies, we have shown that RQR8-transduced cells are effectively depleted from mice with rituximab.32 Others have shown that elimination of redirected T cells based on suicide genes may protect against side effects in patients.44
The TCRs studied herein were derived from CD4+ Th cells, but are still functional in CD8+ recipient cells. It will be of interest to study the implications of this finding. We observe that the redirected CD8+ T cells exhibit a larger potential for cytotoxicity, as assessed by CD107a and Granzyme B staining. It is conceivable that these CTL-like, but HLA class II restricted, cells may confer beneficial as well as unwanted effects. Unlike their CD4+ counterparts, CD8+ GV1001-specific cells have not circulated in the vaccinated cancer patients. In the further clinical development of the described TCRs, it is thus important to be aware of potential safety issues. On the other hand, the GV1001-specific CTLs may be able to directly kill tumor cells in HLA class II positive cancers, as well as tumor-infiltrating macrophages and other HLA class II positive stromal cells. The expression of HLA class II has been suggested to represent an immune escape mechanism for melanoma.45 The retargeting of CD8+ CTLs with HLA class II restricted TCRs may counter this mechanism of tumor escape.
TCRs C13 and D71 recognized epitopes naturally processed from a 173 aa hTERT fragment, as described above. We and others have also attempted to synthesize full-length hTERT protein, but this has not been successful, partly due to difficulties in dissolving the protein. This is particularly challenging because of the large size of hTERT and the need to use solvents that are not too toxic for functional T-cell assays. However, the 173 aa hTERT fragment that we synthesized is comparable in size to many full-length proteins, and the recognition of epitopes form this fragment suggests that natural epitopes are recognized. Further, we have observed spontaneous T-cell reactivity toward GV1001 in several cancer patients before vaccination, and not in healthy donors (Ref. [1] and unpublished). The GV1001 vaccine peptide was, moreover, originally validated based on reactivity of CD4+ tumor-infiltrating lymphocytes in a pancreatic cancer patient, spontaneous reactivity among PBMCs from other cancer patients and no reactivity in healthy donors (Patent US7794723 B2; “Antigenic peptides derived from telomerase”). In line with this, others have reported that patients with hTERT positive leukemia have strong T-cell reactivity against GV1001, in contrast to healthy donors and to patients with hTERT negative leukemia.46 In all, there is strong evidence suggesting that the GV1001 epitope is naturally processed and that the TCRs investigated herein recognize naturally processed epitopes. Nevertheless, it is important to investigate whether this applies to all/most patients with hTERT positive tumors and to explore how patients more likely to respond should be selected for therapy. To this aim, we consider developing an ex vivo test system for assessing the ability of each patient´s redirected T cells, produced under full-scale conditions, to recognize APCs loaded with autologous tumor cell fragments.
T-cell clone C13 has a higher avidity than virtually all other T-cell clones in our biobank, including clone D71, and thus receives optimal TCR stimulation at lower antigen concentrations. We found that both clones C13 and D71 produced CD107a, specifically upon stimulation with GV1001, suggesting cytotoxic activity. However, only clone C13 secreted CD107a upon stimulation with the hTERT protein fragment. This observation may be related to the high functional avidity of the C13 clone. In line with this notion, we have in repeated experiments with >30 GV1001-specific T-cell clones observed that all high-avidity clones recognized hTERT protein, whereas some of the low-avidity clones did not. This may reflect that the concentration of epitopes in the hTERT protein experiments does not reach the highest levels in our GV1001 peptide titration experiments.
It is not clear to which extent the avidity is intrinsic to the TCR or reflects other properties of the T cell. Probably, high-affinity TCRs confer higher avidity provided that other factors influencing T-cell responsiveness remain constant. It is thus likely that a high-affinity TCR will confer higher avidity to a particular cell product for adoptive T-cell therapy, produced for a given patient. We observed that the high-avidity clones mounted somewhat lower responses at the highest peptide concentrations. The latter finding is consistent with the notion that high-avidity T cells may be susceptible to exhaustion and activation-induced cell death, as has been reported from studies in chronic infections.47 In the cancer setting, however, the tumor antigen load may not be high enough to trigger this mechanism. In unpublished experiments, we have observed that some high-avidity clones alter their cytokine profile when stimulated with high antigen concentrations. In particular, the IL-10 production appears to increase compared to other cytokines. It is not known whether this would be an issue in the cancer setting. Most investigators consider that high-affinity TCRs are desirable for cancer eradication,47-49 whereas others have raised concerns regarding autoimmune side effects and also suggested that there is an affinity plateau which should not be exceeded.41,50 We aim to use our comprehensive biobank of hTERT-reactive clones to develop a panel of TCRs with different affinities and assess their survival and functionality after infusion into patients. As described above, T-cell clones D71 and C13 had half-maximum proliferation responses at 10−7 and 10−9 M GV1001, respectively.
A Th1 cytokine profile is considered to promote antitumor immune responses and induce cancer senescence,18,19 whereas a Th2 or Tr1-profile may confer tumor tolerance. We have observed that most long-term survivors in the GV1001 trials, including patients C and D, harbored T-cell responses with a Th1-weighted profile, characterized by high levels of INFγ and TNFα and low levels of IL-4 and IL-10 (Refs. [1,2,7,29] and unpublished). A INFγhigh/IL-10low response was associated with low Treg levels and improved survival in the NSCLC patients.7 We did not observe a classic Th1/Th2 dichotomy. Both in T-cell bulk cultures (polyclonal) and in monoclonal cultures, we detected substantial levels of the Th2 cytokines IL5 and IL-13, in spite of high levels of INFγ (Th1) and TNFα (Th1) and low levels of IL-4 (Th2) and IL-10 (Th2/Tr1). As reported above, we observed a polyfunctional and Th1-like profile both in the parent clones C13 and D71 and in donor T cells transduced with C13_RQR8 or BL71_RQR8. Studies of immunity against infection have suggested that polyfunctional T cells are functionally superior51-54, and there is also increasing evidence indicating that polyfunctionality is a hallmark for effective anticancer responses.35,55 Adoptive cell therapy with Th cells with a Th2 or Tr1 profile may cause unwanted augmentation of tumor tolerance. Based on our data, conventional INFγ assays cannot be used as markers of a Th1 profile, as many T cells secrete high levels of INFγ accompanied by even higher levels of IL-10 (Tr1) or Th-2 cytokines. The cytokine profile is likely to vary between cells, even if transduced with the same TCR. It may be useful to include a comprehensive cytokine profiling assay of the T-cell product as part of the quality control up front of adoptive T-cell therapy. The data reported herein indicate that Bioplex analyses of day 2 supernatants are representative. It should though be recalled that the cytokine profiles may also change in vivo.
In summary, we have characterized a series of Th-cell clones targeting the universal tumor antigen hTERT, with regard to properties relevant for use of their TCR for adoptive cell therapy. We have identified and cloned two lead candidate TCRs that are DP4-restricted and thus widely applicable. The TCRs are highly expressed in recipient T cells and confer the desired GV1001-specifcity. The studies demonstrate a polyfunctional cytokine profile in recipient cells and that the TCRs are functional both in CD4+ Th cells and CD8+ CTLs. We hypothesize that therapy with tumor-targeting Th cells may offer a powerful approach for mobilizing the host immune system and overcoming tumor tolerance. The favorable characteristics of the redirected T cells in vitro support the notion that the TCRs may confer clinical benefits, but the strategy is unproven. Both TCRs were derived from long germ survivors without adverse effects, and the incorporation of a suicide/marker gene into the construct further facilitates safe clinical testing. A Th cell strategy may be explored as monotherapy and may also harbor a large potential for synergy with other therapeutic approaches, similar to the physiological role of Th cells as orchestrators of immunity.
Materials and methods
Clinical trials and patients
A series of clinical trials evaluating cancer vaccination with peptide GV1001 were conducted at Oslo University Hospital. The studies were approved by the appropriate regulatory authorities and ethical committees and performed in compliance with the World Medical Association Declaration of Helsinki. Written informed consent was obtained from the patients. Patients C and D had advanced NSCLC and were included in protocol CTN-2000.8,29 Patient E was included in a trial for patients with advanced pancreatic cancer.4,29
Peptides and recombinant hTERT protein fragment
The vaccine peptide GV1001 (hTERT 611-626; EARPALLTSRLRFIPK) was supplied by Pharmexa. Truncated peptides, consisting of sequences contained within GV1001, were supplied by Dr. Ø. Rekdal, University of Tromsø, Norway. The 173 aa recombinant hTERT protein fragment (563-735) was ordered from Genscript or synthesized as previously described.8
PBMC isolation and generation of T-cell clones
PBMCs were obtained prior to start of vaccination and at multiple time points after vaccination. The PBMCs were isolated and frozen as previously described.56 T-cell clones were generated from postvaccination PBMCs as previously described.57 Briefly, limiting dilution seeding was performed (0.3/1/3 T cells/well). The T cells were stimulated with irradiated (30 Gy) allogeneic PBMCs (106 c/mL), PHA (1 µg/mL) and IL-2 (10 U/mL).
Sequences identification and TCR construct design
The TCR sequences were determined as previously described.31 Briefly, mRNA was isolated from pellets of T-cell clones C13 and D71. cDNA was synthesized and 5′-RACE was performed. Sequences were identified (Eurofins) and compared to IMGT database (www.imgt.org) and used for further design. All constructs were generated by in-house gene synthesis using polymerase chain reaction assembly of overlapping oligos unless otherwise specified. Codon optimization used an in-house algorithm (written by M.P. and available upon request). Phusion polymerase, quick Ligase, and NEB5α (New England Biolabs) were used for molecular cloning. Oligonucleotides were purchased from IDTDNA. In addition, the RQR8 tag was cloned upstream of the TCR α and β chains with 2A ribosome skipping sequence. The retroviral vector MP71 was used in all constructs.
TCR expression and T-cell redirection
Generation of RD114-pseudotyped supernatant and transduction of T cells was performed essentially as previously described.31,32 Briefly, PBMC transductions were performed as follows: PBMCs were isolated by Ficoll (GE Healthcare) gradient centrifugation and stimulated with phytohemagglutinin at 5 µg/mL. Interleukin-2 (IL-2) stimulation (100 IU/mL) was added following overnight stimulation. On day 3, PBMCs were harvested, plated on retronectin and retroviral supernatant, and centrifuged at 1000g for 40 min. Retroviral transduction was performed by loading retronectin-coated plates (Takara) with 0.3–0.6 × 106 activated PBMCs suspended in 1 mL retroviral supernatant, centrifuged at 805g for 90 min, and then recovered by overnight incubation at 37 °C. The following day, PBMCs were recovered from the plate and resuspended in complete RPMI supplemented with 50 ng/mL IL-2 (Invitrogen). Purity and viability of transduced cells were assessed by flow cytometry.
T-cell proliferation assays and HLA blocking
T-cell proliferation assays (3H Thymidine) were performed essentially as previously described.56 Irradiated autologous PBMCs or EBV-transformed cells (EBV cells) were used as APCs. T cells were incubated with APCs with or without (w/wo) the relevant antigen (peptide or hTERT protein). T-cell cultures were tested in triplicates. SEM was usually below 10%. T-cell bulk responses were considered antigen-specific when the stimulatory index (SI; response with antigen divided by response without antigen) was above 2. All T-cell clones reported here exhibited SI > 10 .
The following mAbs were used in HLA-blocking experiments, each at 10 μg/mL: W6/32: anti-HLA class I (American Type Culture Collection); anti-HLA-DR (Becton Dickinson); B8/11: anti-HLA-DR (gift from Dr. N. Malissen, Centre d'Immunologie, Marseille, France); SPVL-3: anti-HLA-DQ (gift from Dr. H. Spitz, The Netherlands Cancer Institute, Amsterdam); FN81: anti-HLA-DQ and 22C1: anti-HLA-DP (gifts from Dr. S. Funderud, Radiumhospitalet, Oslo).
Bioplex cytokine assays
T cells were incubated with APCs w/wo the relevant antigen. Bioplex cytokine analyses were performed on supernatants harvested after 1–6 d, according to the manufacturer's protocol (Bio-Rad Laboratories). Supernatants were analyzed in duplicates, each parallel kept separate through T-cell stimulation and Bioplex assays. All T-cell clones reported here exhibited a GV1001-specific response with SI > 50 in cytokine assays.
Flow cytometry
Phycoerythrin (PE)-conjugated tetramers were purchased from Benaroya Research Institute, Seattle, WA, USA. Tetramer staining was performed by incubating the cells with 10 μL tetramer for 30 min at 37 °C, followed by 30 min at RT, addition of surface mAbs and incubation for another 25 min at 4 °C. Finally, the cells were washed with PBS 2% FCS and resuspended in PBS for acquisition. Intracellular staining was performed using the BD Fix/Perm reagent, according to the manufacturer´s protocol (Becton Dickinson). Briefly, the T cells were incubated with antigen (peptide/protein) at a T-cell to target ratio of 1:1.5 for 1 h before addition of BD Golgiplug, BD Golgistop, and CD107a-PE-Cy5 (BD Biosciences). Then, the cells were incubated overnight, before staining with mAbs for flow cytometry. The following reagents were used: QBen10-PE, QBen10-APC (R&D Systems), Fixable Viability Dye eFluor® 780, Granzyme B-PE, aIFNγ-FITC, aINFγ PE-Cy7 (eBioscience), anti-mouse TCR constant beta region (a-mCb) APC, a-mCb V450, aTNFα-BV421, aCD4-BV510, aCD8-FITC, aIL-4 PercP-Cy5.5, aCD4 FITC, aCD4 PE, aCD8 PE (BD Biosciences). Flow cytometry acquisition was performed using Beckman–Coulter Cyan, BD LSRII, or BD Fortessa instruments. The data were analyzed using FlowJo software (Treestar, Inc.).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the doctors and nurses at the Clinical Trial Unit and the staff at the Section for Immunotherapy for their excellent contributions to the vaccine trials that lead to this work.
Funding
This work was supported by the Norwegian Health Region South East, Radiumhospitalets legater and a grant from Oslo University Hospital.
References
- 1.Kyte JA, Gaudernack G, Dueland S, Trachsel S, Julsrud L, Aamdal S. Telomerase peptide vaccination combined with temozolomide: a clinical trial in stage IV melanoma patients. Clin Cancer Res 2011; 17(13):4568-80; PMID:21586625; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0184 [DOI] [PubMed] [Google Scholar]
- 2.Brunsvig PF, Kyte JA, Kersten C, Sundstrom S, Moller M, Nyakas M, Hansen GL, Gaudernack G, Aamdal S. Telomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin Cancer Res 2011; 17(21):6847-57; PMID:21918169; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1385 [DOI] [PubMed] [Google Scholar]
- 3.Hunger RE, Kernland Lang K, Markowski CJ, Trachsel S, Moller M, Eriksen JA, Rasmussen AM, Braathen LR, Gaudernack G. Vaccination of patients with cutaneous melanoma with telomerase-specific peptides. Cancer Immunol Immunother 2011; 60(11):1553-64; PMID:21681371; http://dx.doi.org/ 10.1007/s00262-011-1061-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernhardt SL, Gjertsen MK, Trachsel S, Moller M, Eriksen JA, Meo M, Buanes T, Gaudernack G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer 2006; 95(11):1474-82; PMID:17060934; http://dx.doi.org/ 10.1038/sj.bjc.6603437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266(5193):2011-5; PMID:7605428; http://dx.doi.org/ 10.1126/science.7605428 [DOI] [PubMed] [Google Scholar]
- 6.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer 2008; 8:167-79; PMID:18256617; http://dx.doi.org/ 10.1038/nrc2275 [DOI] [PubMed] [Google Scholar]
- 7.Hansen GL, Gaudernack G, Brunsvig PF, Cvancarova M, Kyte JA. Immunological factors influencing clinical outcome in lung cancer patients after telomerase peptide vaccination. Cancer Immunol Immunother 2015; 64(12):1609-21; PMID:26498005; http://dx.doi.org/ 10.1007/s00262-015-1766-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunsvig PF, Aamdal S, Gjertsen MK, Kvalheim G, Markowski-Grimsrud CJ, Sve I, Dyrhaug M, Trachsel S, Moller M, Eriksen JA et al.. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother 2006; 55(12):1553-64; PMID:16491401; http://dx.doi.org/ 10.1007/s00262-006-0145-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF et al.. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368(16):1509-18; PMID:23527958; http://dx.doi.org/ 10.1056/NEJMoa1215134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M et al.. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6(224):224ra225; PMID:24553386; http://dx.doi.org/26065478 10.1126/scitranslmed.3008226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abken H. Adoptive therapy with CAR redirected T cells: the challenges in targeting solid tumors. Immunotherapy 2015; 7(5):535-44; PMID:26065478; http://dx.doi.org/ 10.2217/imt.15.15 [DOI] [PubMed] [Google Scholar]
- 12.Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, Lewis ID, Brenner MK, Brown MP. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther 2016; 24:1135-49; PMID:27019998; http://dx.doi.org/ 10.1038/mt.2016.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC et al.. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384(9948):1109-17; PMID:25034862; http://dx.doi.org/ 10.1016/S0140-6736(14)60958-2 [DOI] [PubMed] [Google Scholar]
- 14.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P et al.. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015; 373(1):23-34; PMID:26027431; http://dx.doi.org/ 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibney GT, Kudchadkar RR, DeConti RC, Thebeau MS, Czupryn MP, Tetteh L, Eysmans C, Richards A, Schell MJ, Fisher KJ et al.. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res 2015; 21(4):712-20; PMID:25524312; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003; 300(5617):337-9; PMID:12690201; http://dx.doi.org/ 10.1126/science.1082305 [DOI] [PubMed] [Google Scholar]
- 17.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003; 421(6925):852-6; PMID:12594515; http://dx.doi.org/ 10.1038/nature01441 [DOI] [PubMed] [Google Scholar]
- 18.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother 2005; 54(8):721-8; PMID:16010587; http://dx.doi.org/ 10.1007/s00262-004-0653-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C et al.. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013; 494(7437):361-5; PMID:23376950; http://dx.doi.org/ 10.1038/nature11824 [DOI] [PubMed] [Google Scholar]
- 20.Butterfield LH, Ribas A, Dissette VB, Amarnani SN, Vu HT, Oseguera D, Wang HJ, Elashoff RM, McBride WH, Mukherji B et al.. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin Cancer Res 2003; 9(3):998-1008; PMID:12631598 [PubMed] [Google Scholar]
- 21.Lurquin C, Lethe B, De Plaen E, Corbiere V, Theate I, van Baren N, Coulie PG, Boon T. Contrasting frequencies of antitumor and anti-vaccine T cells in metastases of a melanoma patient vaccinated with a MAGE tumor antigen. J Exp Med 2005; 201(2):249-57; PMID:15657294; http://dx.doi.org/ 10.1084/jem.20041378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inderberg-Suso EM, Trachsel S, Lislerud K, Rasmussen AM, Gaudernack G. Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with a single hTERT peptide GV1001. Oncoimmunology 2012; 1(5):670-86; PMID:22934259; http://dx.doi.org/ 10.4161/onci.20426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suso EM, Dueland S, Rasmussen AM, Vetrhus T, Aamdal S, Kvalheim G, Gaudernack G. hTERT mRNA dendritic cell vaccination: complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol Immunother 2011; 60(6):809-18; PMID:21365467; http://dx.doi.org/ 10.1007/s00262-011-0991-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al.. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348(6230):124-8; PMID:25765070; http://dx.doi.org/ 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348(6230):69-74; PMID:25838375; http://dx.doi.org/ 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 26.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT et al.. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 351:1463-9; PMID:26940869; http://dx.doi.org/ 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersen RS, Andersen SR, Hjortso MD, Lyngaa R, Idorn M, Kollgard TM, Met O, Thor Straten P, Hadrup SR. High frequency of T cells specific for cryptic epitopes in melanoma patients. Oncoimmunology 2013; 2(7):e25374; PMID:24073381; http://dx.doi.org/22754759 10.4161/onci.25374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, van Rooij N, Linnemann C, van Buuren MM, Urbanus JH et al.. TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology 2012; 1(4):409-18; PMID:22754759; http://dx.doi.org/ 10.4161/onci.18851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kyte JA, Trachsel S, Risberg B, Thor Straten P, Lislerud K, Gaudernack G. Unconventional cytokine profiles and development of T cell memory in long-term survivors after cancer vaccination. Cancer Immunol Immunother 2009; 58(10):1609-26; PMID:19221745; http://dx.doi.org/ 10.1007/s00262-009-0670-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castelli FA, Buhot C, Sanson A, Zarour H, Pouvelle-Moratille S, Nonn C, Gahery-Segard H, Guillet JG, Menez A, Georges B et al.. HLA-DP4, the most frequent HLA II molecule, defines a new supertype of peptide-binding specificity. J Immunol 2002; 169(12):6928-34; PMID:12471126; http://dx.doi.org/ 10.4049/jimmunol.169.12.6928 [DOI] [PubMed] [Google Scholar]
- 31.Walchli S, Loset GA, Kumari S, Johansen JN, Yang W, Sandlie I, Olweus J. A practical approach to T-cell receptor cloning and expression. PloS one 2011; 6(11):e27930; PMID:22132171; http://dx.doi.org/ 10.1371/journal.pone.0027930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, Marin V, Marafioti T, Chakraverty R, Linch D et al.. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014; 124(8):1277-87; PMID:24970931 [DOI] [PubMed] [Google Scholar]
- 33.Aarntzen EH, De Vries IJ, Lesterhuis WJ, Schuurhuis D, Jacobs JF, Bol K, Schreibelt G, Mus R, De Wilt JH, Haanen JB et al.. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res 2013; 73(1):19-29; PMID:23087058; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1127 [DOI] [PubMed] [Google Scholar]
- 34.Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 2006; 440(7086):890-5; PMID:16612374; http://dx.doi.org/ 10.1038/nature04651 [DOI] [PubMed] [Google Scholar]
- 35.Malandro N, Budhu S, Kuhn NF, Liu C, Murphy JT, Cortez C, Zhong H, Yang X, Rizzuto G, Altan-Bonnet G et al.. Clonal abundance of tumor-specific CD4(+) T cells potentiates efficacy and alters susceptibility to exhaustion. Immunity 2016; 44(1):179-93; PMID:26789923; http://dx.doi.org/ 10.1016/j.immuni.2015.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol 2014; 44(1):69-79; PMID:24114780; http://dx.doi.org/ 10.1002/eji.201343718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 2008; 358(25):2698-703; PMID:18565862; http://dx.doi.org/ 10.1056/NEJMoa0800251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS et al.. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344(6184):641-5; PMID:24812403; http://dx.doi.org/ 10.1126/science.1251102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kayser S, Bobeta C, Feucht J, Witte KE, Scheu A, Bulow HJ, Joachim S, Stevanovic S, Schumm M, Rittig SM et al.. Rapid generation of NY-ESO-1-specific CD4 T1 cells for adoptive T-cell therapy. Oncoimmunology 2015; 4(5):e1002723; PMID:26155389; http://dx.doi.org/23890065 10.1080/2162402X.2014.1002723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 2013; 39(1):74-88; PMID:23890065; http://dx.doi.org/ 10.1016/j.immuni.2013.06.014 [DOI] [PubMed] [Google Scholar]
- 41.Turtle CJ, Hudecek M, Jensen MC, Riddell SR. Engineered T cells for anti-cancer therapy. Curr Opin Immunol 2012; 24(5):633-9; PMID:22818942; http://dx.doi.org/ 10.1016/j.coi.2012.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM et al.. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014; 2(2):112-20; PMID:24579088; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almasbak H, Walseng E, Kristian A, Myhre MR, Suso EM, Munthe LA, Andersen JT, Wang MY, Kvalheim G, Gaudernack G et al.. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther 2015; 22(5):391-403; PMID:25652098; http://dx.doi.org/ 10.1038/gt.2015.4 [DOI] [PubMed] [Google Scholar]
- 44.Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, Durett AG, Dakhova O, Savoldo B, Di Stasi A et al.. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015; 125(26):4103-13; PMID:25977584; http://dx.doi.org/ 10.1182/blood-2015-02-628354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donia M, Andersen R, Kjeldsen JW, Fagone P, Munir S, Nicoletti F, Andersen MH, Thor Straten P, Svane IM. Aberrant expression of MHC class II in melanoma attracts inflammatory tumor-specific CD4+ T-cells, which dampen CD8+ T-cell antitumor reactivity. Cancer Res 2015; 75(18):3747-59; PMID:26183926; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-2956 [DOI] [PubMed] [Google Scholar]
- 46.Kokhaei P, Palma M, Hansson L, Osterborg A, Mellstedt H, Choudhury A.88 Telomerase (hTERT 611-626) serves as a tumor antigen in B-cell chronic lymphocytic leukemia and generates spontaneously antileukemic, cytotoxic T cells. Exp Hematol 2007; 35(2):297-304; PMID:17258078; http://dx.doi.org/ 10.1016/j.exphem.2006.10.006 [DOI] [PubMed] [Google Scholar]
- 47.Vigano S, Utzschneider DT, Perreau M, Pantaleo G, Zehn D, Harari A. Functional avidity: a measure to predict the efficacy of effector T cells? Clin Dev Immunol 2012; 2012:153863; PMID:23227083; http://dx.doi.org/ 10.1155/2012/153863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li LP, Lampert JC, Chen X, Leitao C, Popovic J, Muller W, Blankenstein T. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med 2010; 16(9):1029-34; PMID:20693993; http://dx.doi.org/ 10.1038/nm.2197 [DOI] [PubMed] [Google Scholar]
- 49.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ et al.. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013; 122(6):863-71; PMID:23770775; http://dx.doi.org/ 10.1182/blood-2013-03-490565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, McGary K, Huang K, Boyer J, Corse E et al.. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci USA 2013; 110(17):6973-8; PMID:23576742; http://dx.doi.org/ 10.1073/pnas.1221609110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M et al.. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 2006; 107(12):4781-9; PMID:16467198; http://dx.doi.org/ 10.1182/blood-2005-12-4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL et al.. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med 2007; 13(7):843-50; PMID:17558415; http://dx.doi.org/ 10.1038/nm1592 [DOI] [PubMed] [Google Scholar]
- 53.Duvall MG, Precopio ML, Ambrozak DA, Jaye A, McMichael AJ, Whittle HC, Roederer M, Rowland-Jones SL, Koup RA. Polyfunctional T cell responses are a hallmark of HIV-2 infection. Eur J Immunol 2008; 38(2):350-63; PMID:18200635; http://dx.doi.org/ 10.1002/eji.200737768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kannanganat S, Ibegbu C, Chennareddi L, Robinson HL, Amara RR. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J Virol 2007; 81(16):8468-76; PMID:17553885; http://dx.doi.org/ 10.1128/JVI.00228-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan J, Gnjatic S, Li H, Powel S, Gallardo HF, Ritter E, Ku GY, Jungbluth AA, Segal NH, Rasalan TS et al.. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci USA 2008; 105(51):20410-5; PMID:19074257; http://dx.doi.org/ 10.1073/pnas.0810114105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kyte JA, Kvalheim G, Aamdal S, Saeboe-Larssen S, Gaudernack G. Preclinical full-scale evaluation of dendritic cells transfected with autologous tumor-mRNA for melanoma vaccination. Cancer Gene Ther 2005; 12(6):579-91; PMID:15818380; http://dx.doi.org/ 10.1038/sj.cgt.7700837 [DOI] [PubMed] [Google Scholar]
- 57.Gjertsen MK, Saeterdal I, Saeboe-Larssen S, Gaudernack G. HLA-A3 restricted mutant ras specific cytotoxic T-lymphocytes induced by vaccination with T-helper epitopes. J Mol Med (Berlin, Germany) 2003; 81(1):43-50; PMID:12545248 [DOI] [PubMed] [Google Scholar]