

Abstract
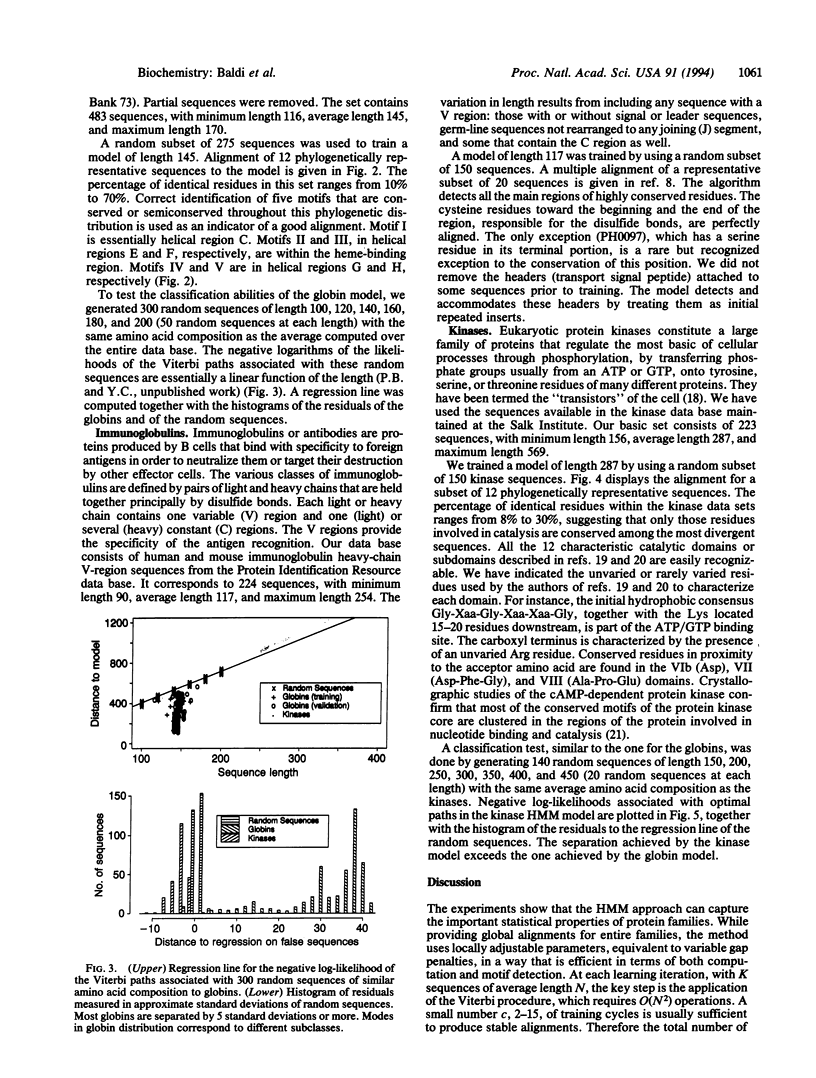
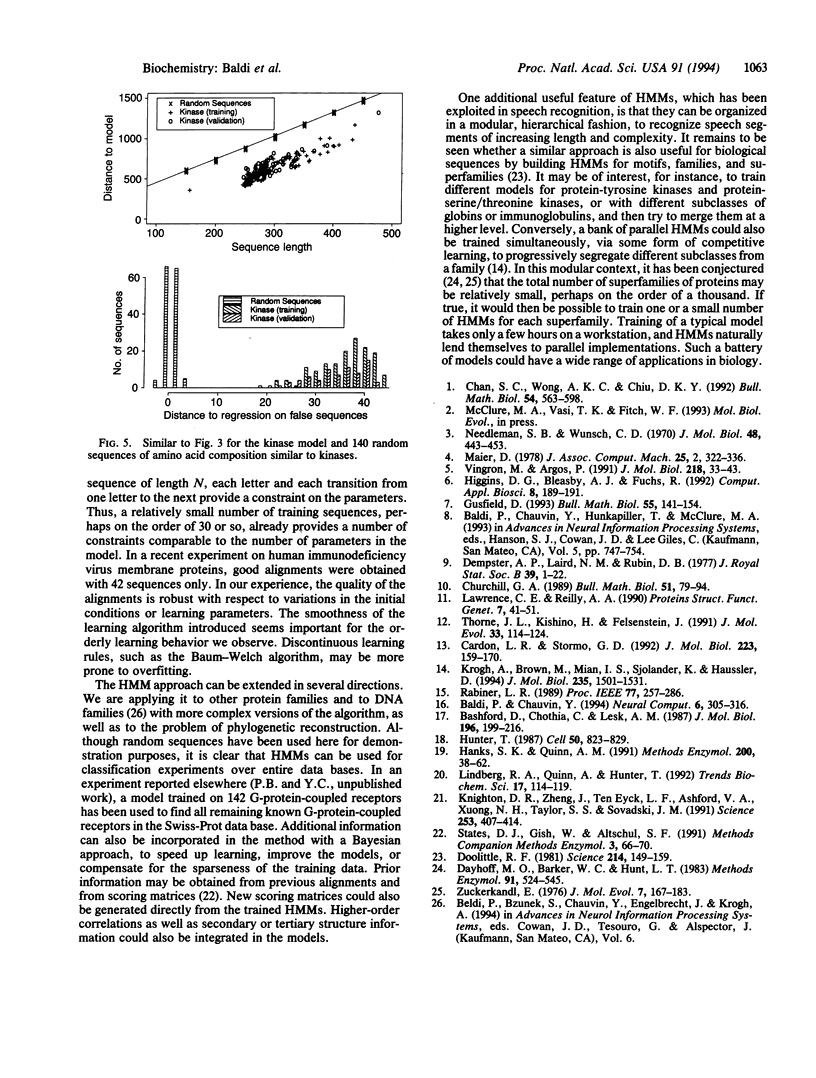
Hidden Markov model (HMM) techniques are used to model families of biological sequences. A smooth and convergent algorithm is introduced to iteratively adapt the transition and emission parameters of the models from the examples in a given family. The HMM approach is applied to three protein families: globins, immunoglobulins, and kinases. In all cases, the models derived capture the important statistical characteristics of the family and can be used for a number of tasks, including multiple alignments, motif detection, and classification. For K sequences of average length N, this approach yields an effective multiple-alignment algorithm which requires O(KN2) operations, linear in the number of sequences.
Full text
PDF



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bashford D., Chothia C., Lesk A. M. Determinants of a protein fold. Unique features of the globin amino acid sequences. J Mol Biol. 1987 Jul 5;196(1):199–216. doi: 10.1016/0022-2836(87)90521-3. [DOI] [PubMed] [Google Scholar]
- Cardon L. R., Stormo G. D. Expectation maximization algorithm for identifying protein-binding sites with variable lengths from unaligned DNA fragments. J Mol Biol. 1992 Jan 5;223(1):159–170. doi: 10.1016/0022-2836(92)90723-w. [DOI] [PubMed] [Google Scholar]
- Chan S. C., Wong A. K., Chiu D. K. A survey of multiple sequence comparison methods. Bull Math Biol. 1992 Jul;54(4):563–598. doi: 10.1007/BF02459635. [DOI] [PubMed] [Google Scholar]
- Churchill G. A. Stochastic models for heterogeneous DNA sequences. Bull Math Biol. 1989;51(1):79–94. doi: 10.1007/BF02458837. [DOI] [PubMed] [Google Scholar]
- Dayhoff M. O., Barker W. C., Hunt L. T. Establishing homologies in protein sequences. Methods Enzymol. 1983;91:524–545. doi: 10.1016/s0076-6879(83)91049-2. [DOI] [PubMed] [Google Scholar]
- Doolittle R. F. Similar amino acid sequences: chance or common ancestry? Science. 1981 Oct 9;214(4517):149–159. doi: 10.1126/science.7280687. [DOI] [PubMed] [Google Scholar]
- Gusfield D. Efficient methods for multiple sequence alignment with guaranteed error bounds. Bull Math Biol. 1993 Jan;55(1):141–154. doi: 10.1007/BF02460299. [DOI] [PubMed] [Google Scholar]
- Hanks S. K., Quinn A. M. Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- Higgins D. G., Bleasby A. J., Fuchs R. CLUSTAL V: improved software for multiple sequence alignment. Comput Appl Biosci. 1992 Apr;8(2):189–191. doi: 10.1093/bioinformatics/8.2.189. [DOI] [PubMed] [Google Scholar]
- Hunter T. A thousand and one protein kinases. Cell. 1987 Sep 11;50(6):823–829. doi: 10.1016/0092-8674(87)90509-5. [DOI] [PubMed] [Google Scholar]
- Knighton D. R., Zheng J. H., Ten Eyck L. F., Ashford V. A., Xuong N. H., Taylor S. S., Sowadski J. M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991 Jul 26;253(5018):407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Krogh A., Brown M., Mian I. S., Sjölander K., Haussler D. Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol. 1994 Feb 4;235(5):1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- Lawrence C. E., Reilly A. A. An expectation maximization (EM) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins. 1990;7(1):41–51. doi: 10.1002/prot.340070105. [DOI] [PubMed] [Google Scholar]
- Lindberg R. A., Quinn A. M., Hunter T. Dual-specificity protein kinases: will any hydroxyl do? Trends Biochem Sci. 1992 Mar;17(3):114–119. doi: 10.1016/0968-0004(92)90248-8. [DOI] [PubMed] [Google Scholar]
- Needleman S. B., Wunsch C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970 Mar;48(3):443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Thorne J. L., Kishino H., Felsenstein J. An evolutionary model for maximum likelihood alignment of DNA sequences. J Mol Evol. 1991 Aug;33(2):114–124. doi: 10.1007/BF02193625. [DOI] [PubMed] [Google Scholar]
- Vingron M., Argos P. Motif recognition and alignment for many sequences by comparison of dot-matrices. J Mol Biol. 1991 Mar 5;218(1):33–43. doi: 10.1016/0022-2836(91)90871-3. [DOI] [PubMed] [Google Scholar]
- Zuckerkandl E. Evolutionary processes and evolutionary noise at the molecular level. I. Functional density in proteins. J Mol Evol. 1976 Apr 9;7(3):167–183. doi: 10.1007/BF01731487. [DOI] [PubMed] [Google Scholar]