

ABSTRACT
It has been shown experimentally that the action of the RSC chromatin remodeler leads to the formation of an irregular, partially remodeled nucleosome, termed a remosome. The remosome contains an extra 30–40 base pairs of DNA compared to a canonical nucleosome. Large-scale molecular simulations have provided information on the probable structure of remosomes and have explained why they remain stable in the absence of RSC. Here we explain how these simulations were carried out and what the resulting remosome models imply in terms of the mechanism of action of RSC. We notably show that local kinks within DNA are key in explaining how extra DNA can be in added to nucleosomes without unduly disturbing DNA-histone binding.
KEYWORDS: chromatin remodeling, DNA, nucleosome, DNA loops, DNA kinks, molecular dynamics, RSC, remosome, SWI/SNF
Understanding the nature of DNA loops formed on nucleosomes is important because of their implication in the process of chromatin remodeling. In the case of the RSC and SWI/SNF remodeler families,1 there is strong evidence that DNA loop formation is an integral part of their functional mechanism.2-4 While loops were typically thought to exist only within the remodeler, groundbreaking work by Shukla et al. showed that nucleosomes containing 20–40 base pairs more DNA than canonical nucleosomes could be isolated after the action of RSC.5 These experiments were performed on a single nucleosome placed within a 255 base pair (bp) DNA fragment and held in position using the so-called 601 sequence.6 The off-center location of the nucleosome binding site showed that the extra DNA drawn into the nucleosome by RSC came more or less equally from both the 5′- and 3′-ends. These modified nucleosomes, termed “remosomes,” were not only stable when isolated from RSC, but also turned out to be substrates for the remodeler, subsequent interaction with RSC leading to nucleosomes displaced to the ends of the DNA substrate. Cryo-electron microscopy showed that remosomes had very irregular structures, clearly contrasting with canonical nucleosome core particles.5 However, these images were not able to provide any detailed structural information. An additional clue to the dramatic impact of the extra DNA within remosomes came from data on DNA accessibility. This was studied using nuclease binding sites inserted at regular intervals within the nucleosome binding sequence. Whereas, within nucleosomes, only sites close to the 5′- and 3′-termini were reactive, within remosomes all sites became accessible to nuclease digestion. Most surprisingly, maximal digestion occurred at the pseudodyad position, normally the site most protected within the nucleosome. The importance of understanding the structure, dynamics and the reasons underlying the stability of nucleosomes has been underlined by recent work from the Dimitrov, Angelov and Bednar groups showing that stable remosomes are also produced by the action of the SWI/SNF remodeler (unpublished data).
Making progress in the analysis of remosomes would clearly benefit from higher resolution structural data. Unfortunately, given the conformational variability observed by Shukla et al. using cryo-electron microscopy, it seems very unlikely that it would be possible to study them crystallographically (even if it were possible to isolate remosomes in sufficient quantities). Their size also precludes NMR spectroscopy as a potential tool. For the present, molecular modeling seems to be the only feasible route to a more detailed understanding of these objects and, in particular, to explaining the origin of their stability.
Given the size of nucleosomes, most modeling to date has been based on simplified representations. Most commonly, long segments of DNA are represented as so-called elastic rods.7-9 These models ignore the atomic structure of the double helix and simply describe its deformability in terms of bending (optionally complemented by twisting and/or stretching), typically using parameters that do not take the base sequence into account. Rod models have been extremely successful in describing the behavior of long segments of isolated DNA and they have the notable advantage of often having fast, analytic solutions. However, when DNA interacts with other species, such as the histone core of the nucleosome, such modeling becomes more difficult and it is necessary to make some assumptions concerning the strength and the distribution of the DNA-protein interactions. In addition, on the relatively short length scale of nucleosomes (or remosomes), it is also clear that local irregularities in DNA structure, not treated by conventional rod models, may become important.
Molecular dynamics offers a route to overcoming these difficulties by representing the system at the atomic scale and taking into account the effects of a realistic water/salt environment.10-13 In this approach, all interactions are treated with a single “force field” that treats factors ranging from atomic bond stretching, valence angle or bond torsion changes, to dispersion/repulsion (Lennard-Jones) interactions and electrostatics. Careful parameterization, and extensive feedback from the user community, enables today's force fields to treat a wide variety of molecules, including both nucleic acids and proteins, in many cases with close to experimental accuracy. Naturally, moving to an atomic-scale representation of macromolecules doesn't come without sacrifices. Exploiting force field models requires computationally expensive numerical solutions, and, most commonly, the generation of dynamic trajectories that involve the numerical integration of Newton's equations of motion.14 Given the rapid movements that occur on the atomic scale, such integration requires evaluating the energy of the system roughly every femtosecond. This makes it difficult to follow movements beyond the microsecond timescale, and also limits the size of the system that can be treated. Lastly, because of the classical (Newtonian) nature of force fields, processes involving chemistry (bond making or breaking, electron transfer, etc.) are necessarily out of reach.
Until very recently systems of the size of remosomes were also out of reach for molecular dynamics. Once we take into account the water molecules and ions that surround remosomes in their natural environment, we have to envisage treating almost half a million atoms. In addition, since we have no detailed information on their structure, we have to simulate their dynamics long enough to allow an initial structural guess to relax into the most stable conformation. Based on studies of other protein-DNA complexes, this is likely to require many hundreds of nanoseconds, much longer than any of the simulations of unmodified nucleosomes carried out to date.15,16 Happily, computer power continues to progress, as do the algorithms and computer codes for performing molecular dynamics simulations. Notably, by running simulations in parallel on hundreds of computational cores (in our case up to 480 cores on the OCCIGEN supercomputer at the CINES center in Montpellier) it has now become feasible to begin to numerically decorticate the remosome.
Our recent publication in Nucleic Acids Research presents results on 2 remosome models that involve either 21 or 42 base pairs (i.e. 2 or 4 helical turns) added to a canonical nucleosome.17 Starting from the high-resolution X-ray structure of the nucleosome,18,19 we made an initial assumption concerning the loop structure. Given the stiffness of the DNA double helix, we chose to detach the central turn of DNA from the nucleosome and then add the extra DNA to form a smooth, circular “epicyclic” loop. The resulting loops were centered on the pseudodyad position. This results in a significant loss of DNA-histone interactions, but maximizes the radius of curvature of the loop DNA (resulting in less bending strain) and also allows smooth transitions between the ends of the loops and the rest of the DNA wound around the core. This choice yields a structure similar to that predicted by a conventional elastic rod model of DNA.
Once we began molecular dynamic simulations of the 2 loops, it became clear that our initial assumption was completely wrong. Within 100–200 nanoseconds, both loops developed sharp kinks that reduced the bending strain in the rest of the loop and also enabled most of the lost DNA-histone contacts to be re-established (see Fig. 1). In fact, the final loop structures resulted in the loss of only 1 or 2 of the 14 key DNA-arginine contacts that exist in canonical nucleosomes.18,19 The resulting loops lengths were subsequently reduced to only 2 or 3 helical turns more than the length of the inserted DNA segment (either 2 or 4 helical turns). After generating trajectories of 1.0 μs for the smaller loop and 0.5 μs for the larger loop, the structures remained stable apart from short-lived detachments of the 5′- or 3′-DNA termini (also observed with canonical nucleosomes20,21) and some local rearrangements of the kinks. In fact, the strain within the remosome loops leads to 2 types of kinks. Crick and Klug originally proposed kinks in 1975 as resulting from the loss of base stacking at a single base pair step.22 Kinks of this type, now termed Type I, have already been seen in molecular simulations of small minicircles, where more extended kinks, termed Type II, and involving the unstacking of 2 steps and breaking the central base pair also occur.23 Here, while the 42 bp insertion leads to a Type II kink, the more strained 21 bp insertion results in a new Type III kink that perturbs 3 steps, and leads to the formation of an unusual staggered pairing following the breakage of 2 successive Watson-Crick base pairs.
Figure 1.
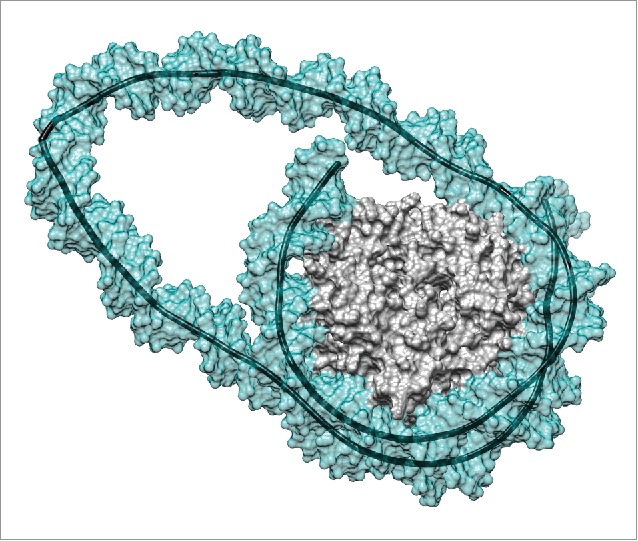
Remosome model resulting from a molecular simulation. This snapshot presents the DNA loop generated by a 42 bp insert at the dyad position.17 The image shows the solvent-excluded surfaces of the histone core (gray) and of the DNA (cyan, partially transparent) as well as the helical axis of the DNA (black). Note the sharp kink within the DNA loop on the left hand side of the image. Molecular graphics were generated with the UCSF Chimera package.27
Implications for RSC remodeling
The first striking result of this study was the apparent stability of the nucleosome loops. The primary reason for this was the recovery of DNA-histone contacts as a result of DNA kinking. Initially, we thought that contacts between DNA and the flexible, cationic histone tails could provide a means of stabilizing the loops. This is still a possibility and indeed it is possible to estimate that roughly 30 positively charged residues within the tails could contact the 42 bp loop placed at the dyad position. However, since the histone tails were excluded from our simulations (due to the difficulty of sampling the conformational states of these unstructured oligopeptides), it is clear that they are not indispensable for maintaining the loops. An alternative source of stabilization is likely to come from the environment. Studies of minicircles currently being carried out with our new analysis techniques,24 show that cations from solution will accumulate on the inside face of strongly curved DNA segments, compensating the increased density of anionic phosphate groups in such regions (unpublished results).
Having established that DNA loops are stable on the nucleosome core we can understand how remosomes become accessible to nuclease attack, as loop migration around the histone core would successively expose all the wrapped DNA. Our initial idea of loop structure involved circular, epicyclic loops that would have been able to change their position with virtually no change in energy. Loop migration would therefore conceivably have occurred at room temperature as a result of thermal agitation. Given the loop structure revealed by the MD simulations, with sharp local kinks, this mechanism seems less likely. In order to displace a loop, it is now necessary to remove the kink before reforming it in another position. Preliminary tests show that causing a loop to move around the histone core by applying external forces is not a simple process. It may therefore be necessary to envisage that loop movement occurs only within RSC and under its ATP-driven action. Our remosome model can indeed be fitted into RSC as shown in Fig. 2, where the 42 bp loop at the dyad position has been placed within the 25 Å electron density map obtained by cryo electron microscopy (kindly provided by Francisco Asturias). In this connection, Chaban et al. remark that their findings are “consistent with the idea that RSC binding leads to separation of DNA from the histones and formation of a bulge around the dyad.”3
Figure 2.
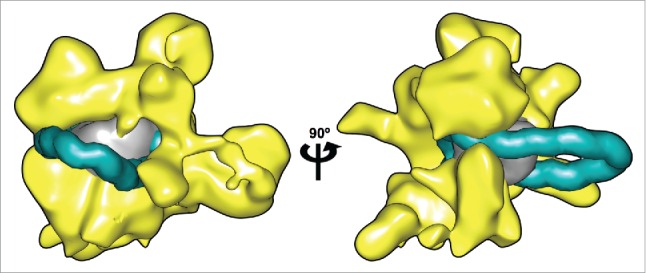
Remosome model placed within the RSC remodeller. Two views of a nucleosome containing a 42 bp insertions at the dyad (DNA shown in cyan, histone core in gray) manually docked inside the 3D reconstruction of the RSC complex calculated from cryo-EM data.3 (shown in yellow). A representative snapshot of the remosome model was taken from a 0.5 μs MD simulation17 converted to a 25 Å-resolution density map to match the RSC data. DNA in the looped nucleosome is shown up to 25 bp from the ends, to account for the possible rearrangements in these zones.3 Molecular graphics were generated with the UCSF Chimera package.27,28
A further point should be made concerning the length and the structure of remosome loops. Concerning their length, it is only possible to build loops without torsional strain if their length is equal to an integer number of helical turns. While a turn contains 10–11 bp on average, the exact number will depend on the base sequence and the length of the looped DNA can thus be expected to vary by several base pairs. Any larger variation would however lead to significant torsion strain that is likely to destroy the loop. In addition to its overall length, there are strong constraints on the position of a kink within a loop. Base sequence may influence the optimal site for loops (although current studies have not yet demonstrated this), but more importantly kinks don't function like ball-and-socket joints. They generally result in DNA bending in a rather constrained direction. This implies that only a small subset of possible kink sites will be compatible with the constraints imposed by the location of the ends of the loop where DNA is bound to the histone core. Within these limits, loops can be expected to vary in position, length and structure, in line with the variability of the shapes of remosome seen experimentally.
Future directions
Simulations are now underway to study loops at alternative locations around the histone core and to analyze the mechanism of loop displacement. Preliminary results on loops near the ends of the nucleosome suggest that even 1 or 2 arginine/minor groove contacts may be sufficient to stabilize a loop. In this connection, it is interesting to note that truncating the C-terminal docking domain of histone H2A weakens interactions with the ends of DNA wrapped around the nucleosome, but also hinders the action of RSC remodeling.25 This apparently counterintuitive finding may be due to the fact that this change could also destabilize loops entering the nucleosome and limit remosome formation.
One feature of loop mobility that remains to be explained is the finding of Liu et al. who created a “peptide bridge” on nucleosomes by linking the H2A histone N- and C-tails.26 This bridge hinders nucleosome mobilization by RSC or SWI/SNF, but does not affect the accessibility of nuclease sites within the DNA of the nucleosomes undergoing remodeling. The authors of this study propose that DNA could thread through the peptide bridge during loop displacement, but this seems difficult to envisage with our remosome model. One possibility is that the bridge might be made bigger by partial denaturation of the termini of H2A, but there is currently no evidence to support this hypothesis.
To better understand DNA-histone interactions, we are currently studying the dynamics of the 14 arginine anchor points around the histone core, and the contribution of other arginine and lysine residues that also interact with the DNA wound around the histone core. We are also analyzing the ion distributions around the remosome models to better understand what role the environment can play in their stability and, lastly, given the findings from molecular simulations, it is important to obtain significantly higher resolution structures for remosomes and hopefully to confirm the importance of DNA kinking.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank GENCI for a generous allocation of supercomputer resources at the CINES center in Montpellier.
Funding
The authors acknowledge funding from the Agence Nationale de la Recherche through project CHROME (ANR-12-BSV5-0017-01).
References
- [1].Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H, Tempst P, Du J, Laurent B, Kornberg RD. RSC, an essential, abundant chromatin-remodeling complex. Cell 1996; 87:1249-60; PMID:8980231; http://dx.doi.org/ 10.1016/S0092-8674(00)81820-6 [DOI] [PubMed] [Google Scholar]
- [2].Zhang Y, Smith CL, Saha A, Grill SW, Mihardja S, Smith SB, Cairns BR, Peterson CL, Bustamante C. DNA translocation and loop formation mechanism of chromatin remodeling by SWI/SNF and RSC. Mol Cell 2006; 24:559-68; PMID:17188033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chaban Y, Ezeokonkwo C, Chung WH, Zhang F, Kornberg RD, Maier-Davis B, Lorch Y, Asturias FJ. Structure of a RSC–nucleosome complex and insights into chromatin remodeling. Nat Struct Mol Biol 2008; 15:1272-1277; http://dx.doi.org/ 10.1038/nsmb.1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lavelle C, Praly E, Bensimon D, Le Cam E, Croquette V. Nucleosome-remodelling machines and other molecular motors observed at the single-molecule level. FEBS J 2011; 278:3596-3607; PMID:21810177 [DOI] [PubMed] [Google Scholar]
- [5].Shukla MS, Syed SH, Montel F, Faivre-Moskalenko C, Bednar J, Travers A, Angelov D, Dimitrov S. Remosomes: RSC generated non-mobilized particles with approximately 180 bp DNA loosely associated with the histone octamer. Proc Natl Acad Sci U S A 2010; 107:1936-41; http://dx.doi.org/ 10.1073/pnas.0904497107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 1998; 276:19-42; http://dx.doi.org/ 10.1006/jmbi.1997.1494 [DOI] [PubMed] [Google Scholar]
- [7].Schiessel H, Widom J, Bruinsma RF, Gelbart WM. Polymer reptation and nucleosome repositioning. Phys Rev Lett 2001; 86:4414-7; PMID:11328188; http://dx.doi.org/ 10.1103/PhysRevLett.86.4414 [DOI] [PubMed] [Google Scholar]
- [8].Kulić IM, Schiessel H. Nucleosome repositioning via loop formation. Biophys J 2003; 84:3197-211; PMID:12719249; http://dx.doi.org/ 10.1016/S0006-3495(03)70044-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Biswas M, Wocjan T, Langowski J, Smith C. DNA bending potentials for loop-mediated nucleosome repositioning. EPL (Europhysics Letters) 2012; 97:38004; http://dx.doi.org/ 10.1209/0295-5075/97/38004 [DOI] [Google Scholar]
- [10].Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Structural Biol 2002; 9:788-788 [DOI] [PubMed] [Google Scholar]
- [11].Mackerell AD, Nilsson L. Molecular dynamics simulations of nucleic acid-protein complexes. Curr Opin Struct Biol 2008; 18:194-9; http://dx.doi.org/ 10.1016/j.sbi.2007.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Van Der Kamp MW, Shaw KE, Woods CJ, Mulholland AJ. Biomolecular simulation and modelling: status, progress and prospects. J Royal Soc Interface 2008; 5:173-190; PMID:18611844; http://dx.doi.org/ 10.1098/rsif.2008.0105.focus [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Karplus M, Lavery R. Significance of Molecular Dynamics Simulations for Life Sciences. Israel J Chem 2014; 54:1042-1051; http://dx.doi.org/ 10.1002/ijch.201400074 [DOI] [Google Scholar]
- [14].Leach A. Molecular modelling principlesand applications. Prentice Hall 2001 [Google Scholar]
- [15].Ettig R, Kepper N, Stehr R, Wedemann G, Rippe K. Dissecting DNA-histone interactions in the nucleosome by molecular dynamics simulations of DNA unwrapping. Biophys J 2011; 101:1999-2008; http://dx.doi.org/ 10.1016/j.bpj.2011.07.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Biswas M, Langowski J, Bishop TC. Atomistic simulations of nucleosomes. WIREs Comput Mol Sci 2013; 3:378-392; http://dx.doi.org/ 10.1002/wcms.1139 [DOI] [Google Scholar]
- [17].Pasi M, Lavery R. Structure and dynamics of DNA loops on nucleosomes studied with atomistic, microsecond-scale molecular dynamics. Nucleic Acids Res 2016; 44:5450-6; PMID:27098037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure ofthe nucleosome core particle at 1.9 a resolution. J Mol Biol 2002; 319:1097-113; PMID:12079350; http://dx.doi.org/ 10.1016/S0022-2836(02)00386-8 [DOI] [PubMed] [Google Scholar]
- [19].Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature 2003; 423:145-50; http://dx.doi.org/ 10.1038/nature01595 [DOI] [PubMed] [Google Scholar]
- [20].Tims HS, Gurunathan K, Levitus M, Widom J. Dynamics of nucleosome invasion by DNA bindingproteins. J Mol Biol 2011; 411:430-448; http://dx.doi.org/ 10.1016/j.jmb.2011.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kenzaki H, Takada S. Partial unwrapping and histone tail dynamics in nucleosome revealed by coarse-grained molecular simulations. PLoS Comput Biol 2015; 11:e1004443; PMID:26262925; http://dx.doi.org/ 10.1371/journal.pcbi.1004443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Crick FH, Klug A. Kinky helix. Nature 1975; 25:5530-533 [DOI] [PubMed] [Google Scholar]
- [23].Lankas F, Lavery R, Maddocks JH. Kinking occurs during moleculars dynamics simulations of small DNA minicircles. Structure 2006; 14:1527-34; http://dx.doi.org/ 10.1016/j.str.2006.08.004 [DOI] [PubMed] [Google Scholar]
- [24].Lavery R, Maddocks JH, Pasi M, Zakrzewska K. Analyzing ion distributions around DNA. Nucleic Acids Res 2014; 42:8138-8149; http://dx.doi.org/ 10.1093/nar/gku504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shukla MS, Syed SH, Goutte-Gattat D, Richard JL, Montel F, Hamiche A, Travers A, Faivre-Moskalenko C, Bednar J, Hayes JJ, et al.. The docking domain of histone H2A is required for H1 binding and RSC-mediated nucleosome remodeling. Nucleic Acids Res 2011; 39:2559-70; http://dx.doi.org/ 10.1093/nar/gkq1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu N, Peterson CL, Hayes JJ. SWI/SNF- and RSC-catalyzed nucleosome mobilization requires internal DNA loop translocation within nucleosomes. Mol Cell Biol 2011; 31:4165-75; PMID:21859889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605-12; http://dx.doi.org/ 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- [28].Goddard TD, Huang CC, Ferrin TE. Visualizing density maps with UCSF Chimera. J Struct Biol 2007; 15:7281-7; PMID:16963278 [DOI] [PubMed] [Google Scholar]