

Abstract
The labor-intensive process of microbial natural product discovery is contingent upon identifying discrete secondary metabolites of interest within complex biological extracts, which contain inventories of all extractable small molecules produced by an organism or consortium. Historically, compound isolation prioritization has been driven by observed biological activity and/or relative metabolite abundance and followed by dereplication via accurate mass analysis. Decades of discovery using variants of these methods has generated the natural pharmacopeia but also contributes to recent high rediscovery rates. However, genomic sequencing reveals substantial untapped potential in previously mined organisms, and can provide useful prescience of potentially new secondary metabolites that ultimately enables isolation. Recently, advances in comparative metabolomics analyses have been coupled to secondary metabolic predictions to accellerate bioactivity and abundance-independent discovery work flows. In this review we will discuss the various analytical and computational techniques that enable MS-based metabolomic applications to natural product discovery and discuss the future prospects for comparative metabolomics in natural product discovery.
Graphical abstract

1 Introduction
Genomic sequencing of both cultivated microorganisms and uncultivated microbiomes has revealed that most of the biosynthetic potential of microorganisms remains inaccessible to date. Even genomes of extensively studied microorganisms contain a large fraction of secondary metabolite biosynthetic gene clusters for which natural products have not been identified.1-3 It is now estimated that the products of greater than 90% of secondary metabolite gene clusters are either not expressed under standard laboratory growth conditions4,5 and/or their products are difficult to identify within extracted metabolomes. Secondary metabolites play crucial roles in the chemical ecology of their producing organisms,6-8 and these roles often correlate translationally into applications in human medicine.9 Therefore, solving the linked problems of secondary metabolite gene expression and the identification of secondary metabolites within metabolomic inventories have become central efforts in the field of natural product discovery.
Natural product biosynthetic potential can be rapidly estimated from genomic sequence data via automated bioinformatics platforms capable of comparing sequenced biosynthetic gene clusters to previously sequenced microorganisms and inferring putative structures of natural products by biosynthetic inference.10-16 Recently, several reviews have described evolving computational tools for biosynthetic gene cluster analysis.17,18 Increasingly sophisticated methodologies have been developed to tackle the biosynthetic gene cluster expression problem, which may be subdivided into heterologous and native approaches. Heterologous strategies endeavour to recapitulate functional secondary metabolic biosynthetic gene clusters in surrogate producers. Gene clusters may be cloned, and/or synthesized and refactored into alternate organisms with the aim of detecting newly produced metabolites in comparison to a clean background.19-24 The success of heterologous expression is dependent upon functional expression within the host organism, which is a function of successful transcription, translation, and precursor availability as discussed in other reviews.19,25,26 Given the phylogenetic diversity of microbial secondary metabolite producers, a number of hurdles must be addressed to successfully identify constructs for functional expression. In addition to optimizing genetic regulatory elements for heterologous expression, differences in protein stability, post-translational modification of biosynthetic enzymes, and precursor availability must be addressed. Alternatively, native expression methods endeavour to activate secondary metabolite production from within the native producer.4,27,28
In addition to refactoring biosynthetic gene clusters via genome editing, many chemical and biological stimuli have been reported over the past few decades that activate secondary metabolite expression. The practice of exposing microorganisms to an array of growth conditions for the purpose of eliciting the production of multiple compounds is not a recent development.29 However, contemporary studies of the impact of stimuli on microbial metabolism, in addition to fulfilling to goals of natural product discovery, now model the chemical ecology and environmental microbiology of microorganisms.4,27,30-32 For example, microbial secondary metabolite producers have shown responses to subinhibitory antibiotic exposure33,34 as well as vertically acquired antibiotic resistance mutations which engender mutations in transcription and translation machinery35-40 have been demonstrated to activate the production of a fraction of previously undiscovered metabolites. Similarly, rare earth metal exposure,41-43 which may affect circulating levels of pleiotropic factors, has been demonstrated to modulate secondary metabolite production. A particularly successful strategy for activating secondary metabolite expression in microbes is via stimulation with competing organisms.44-48 Taken together these phenomenon suggest that secondary metabolites are indeed produced by microorganisms to respond to environmental stimuli45 and this is supported by the apparent biosynthetic gene cluster activation selectivity of various stimulatory methods as well as the intrinsically complex nature of secondary metabolite gene cluster regulation.1,49
Regardless, all categories of genome-prioritized natural product discovery require a means of measuring the modulation of secondary metabolite production within the extracted metabolome of native or heterologous producers, and metabolomics methods have been continually adapted to this task. Metabolomics is often defined as the comprehensive study of small molecules within a biological system and provides a direct measure of detectable secondary metabolite production within an organism of interest. Currently there are two analytical platforms to facilitate metabolome profiling for natural product discovery. Nuclear magnetic resonance (NMR) based metabolomic analyses, reviewed elsewhere,50 are not biased by molecular class and provide enhanced structural information for metabolites but are limited by the inherently low sensitivity of NMR. In contrast, the metabolomic analyses through mass spectrometry (MS), which will be the focus of our review, are exceptionally sensitive but are exclusively biased towards ionisable metabolites. The structural diversity of secondary metabolites, which span a broad range of functionality, molecular weight, and ionization efficiency, renders comprehensive detection of all metabolites through MS a challenging endeavour, and there is no universal approach for bioanalytical detection. For this reason, the development of metabolomics methods with MS strategies necessitates a discussion of contemporary practices and advances in analytical instrumentation.
As the product of the central dogma, the metabolome also contains information regarding a wide variety of cellular processes unrelated, or indirectly related to secondary metabolism. Correspondingly, metabolomics information may encode insights into how secondary metabolite producing organisms respond to chemical and biological stimuli and may also provide a means of investigating the biological mechanisms of newly isolated natural products, antibiotics, and chemotherapeutics, from the metabolomic changes engendered within treated organisms. Secondary metabolites are generally biosynthetic end-products and unlike primary metabolites, they accumulate at higher levels than the fluxes observed in central metabolism.51 Hence, comparatively abundant secondary metabolites are well suited for comparative metabolomics work-flows. Other recent reviews have highlighted some applications of mass spectrometry for the discovery of natural products deriving from plant52 and microbial53 sources. In this review we provide a foundational overview of the analytical techniques that underlie MS-based metabolomic applications to natural product discovery and describe how these various techniques provide differentiating molecular characteristics for detected metabolites. We discuss the computational methods used to process complex metabolomics data and bioinformatics methods that utilize the molecular characteristics of detected metabolites to prioritize and dereplicate leads for natural product discovery. Lastly, we describe how these metabolomic methods are being applied to investigate biological activities for natural products and discuss future prospects for the field.
2 Methods of generating inventories of microbial metabolites
A variety of MS techniques are available to acquire metabolomics data with corresponding advantages and challenges depending on the analytical descriptor(s) that is/are desired. In each method, the end result of the analysis is a set of metabolomic ‘features’, ions with a determined mass-to-charge ratio (m/z) and potentially additional descriptive information. This additional information may include descriptors such as mass accuracy, chromatographic retention time, isotopic envelope, size and shape information, fragmentation data, and topological distribution, among others. A summary of key descriptors and the information they provide in secondary metabolite characterization is provided in Table 1. As the dimensionality of feature characterization has an impact upon subsequent effectiveness of comparative metabolomics analyses, we will briefly discuss in this section commonly utilized methods for MS acquisition and highlight several of these key molecular characteristics that can be obtained with MS.
Table 1.
Overview of analytical descriptors relevant to metabolomics-based natural product discovery
| Analytical Descriptor |
Description | Analytical technique |
|---|---|---|
| Mass accuracy | Deviation of the experimentally determined m/z from the true m/z. Expressed as the mass error (e.g. ppm), with sufficiently small error an exact chemical formula can be determined. |
Mass analyzer
|
|
| ||
| Isotopic modeling |
Comparison of the abundances of specific isotopes in the molecular isotopic envelope. Can provide rapid indication of amount and identity of heteroatoms. |
Chemometrics
|
|
| ||
| Chromatographic retention time |
Time required for fluid-solid phase partitioning across a column. Provides separation on the basis of a differentiating characteristic orthogonal to mass. |
Chromatography
|
|
| ||
| Ion mobility drift time |
Gas-phase electrophoretic separation based on size and shape of the metabolite as ions pass through a gas filled drift tube. |
Ion drift tube
|
|
| ||
| Fragmentation | Tandem MS using ion activation to provide characteristic fragment species. Provides metabolite structural information to prioritize which of multiple isomers are the likely identity for a given elemental formula. |
Ion activation
|
2.1 Mass measurement accuracy
The mass-to-charge ratio (m/z) of detected metabolites is the most useful property used to initiate the process of dereplication. For more than a decade mass analyzers have been able to determine mass accuracy with an error of under 1 ppm.54-57 This level of mass accuracy allows for the determination of elemental composition boundaries for compounds under 600 Da 58,59 when coupled with isotopic mass ratios.60 While advances in Fourier Transform ion cyclotron resonance MS (FTICRMS) can now routinely perform at sub-ppm mass errors, typical instrumentation provides mass errors in the range of 1 to 10 ppm (e.g. time-of-flight MS). Unfortunately, this alone is insufficient to confidently dereplicate features, because of the extensive number of potential isomers for a given elemental composition.61 Early compound dereplication is thereby often dependent on obtaining additional distinguishing characteristics such as those listed in Table 1, or via additional characteristic such as UV/Vis spectrum and biological activity.62,63 It is also noteworthy that MS analysis is predicated on the ability to generate ions of the species of interest. Neutral or poorly ionizing species are transparent to MS, and because of this the number of detectable compounds from a metabolomic extract will vary depending on the analytical methods used during acquisition, in particular the specific ionization source and ionization conditions that are used.
2.2 Isotopic modeling
The isotopic envelope, comprised of both the major and minor isotopic contributions to the elemental formula, provides several opportunities for enhanced characterization information,64 including: (i) the presence of heteroatoms,65 and (ii) isotopic enrichment strategies for relative and absolute quantitation of the abundance of the secondary metabolite.66 The MS analysis of most biological molecules is typically concerning elemental formula comprising C, H, O, and N. The shared characteristic of these elements is that the monoisotopic peak also corresponds to the lowest mass isotope and thus, the lowest mass peak in the envelope is also the highest abundance. However, the vast majority of the periodic table is characterized by isotopic abundances that are somewhat varied from lightest to heaviest mass isotope and their isotopic signatures are oftentimes used in MS-based atomic analyses for identification purposes.67 The presence of heteroatoms, such as chlorine or bromine, are readily discernable in their contribution to the isotopic abundance observed for secondary metabolites and their stoichiometric contribution can be quickly verified through the use of isotopic calculator algorithms.68 Furthermore, these approaches are equally well suited by the addition of non-natural isotopic enrichment or depletion for determining the relative or absolute abundance of the secondary metabolite that is expressed. One such approach termed stable isotope labeling in cell culture (SILAC) has been demonstrated as a facile tool for incorporating enrichment or depletion in experimental protocols.69 Finally, genomic-based structural predictions, implying biosynthetic precursors, can be combined with stable isotope studies to identify targeted metabolites within organisms. 70
2.3 Chromatographic retention time
Liquid chromatography (LC) is one of the most commonly used approaches to separate individual constituents of complex natural product extracts, and various LC methods and their applications have been previously reviewed.71-73 For natural product separations, reversed phase LC, and hydrophobic interaction chromatography are most commonly employed with a water-acetonitrile, or water-methanol gradient.74 This is typically performed on the basis of hydropathy, where reversed phase LC (RPLC) and hydrophobic interaction chromatography (HILIC) are most commonly utilized,75 and column retention will be affected by the ionization of these groups. Mobile phase pH can thereby significantly affect the separation efficiency for natural product extracts. Due to the dependence of compound retention on pH, and to assist ionization, mobile phases are commonly buffered with either acetic acid, trifluoroacetic acid, or formic acid76 to protonate acidic sites and facilitate retention. However, as low pH may suppress detection of negatively charged species in switched scanning modalities, neutral volatile buffers are often preferred.
Liquid chromatography-mass spectrometry (LC-MS) acquisition can take minutes to hours per chromatographic separation, and environmental changes throughout the course of the sample set (column conditioning, instrumental sensitivity and accuracy drift, etc.) can affect the quality of the data. Consequently, for multiple extract samples analysed in a sequential fashion, conditional changes between the start and end of analysis could lead to significant artefactual differences in group metabolomes, which complicate interpretation of subsequent comparative analyses. While challenging, recent reviews have outlined metabolomic experimental design strategies to accommodate these technical problems.77,78
2.4 Size and shape by ion mobility
Additional metabolomic feature information can be obtained by using gas-phase ion mobility-MS (IM-MS), without significantly increasing analysis time over MS-alone.79 The mechanism and utility of IM-MS has been the topic of several recent reviews.79-81 Briefly, in time-dispersive IM-MS, a uniform weak electric field is applied to a post-ionization ion drift tube containing an inert gas, where the ion velocity through the chamber is dependent upon thermal collisions with the background gas and its charge state.82 The number of collisions ions make as they traverse the drift cell are proportional to their collision cross-sectional area, providing distinguishing information regarding an ion’s shape and/or conformation in the gas phase.83 The separations in IM are very low energy in comparison with collisions used for fragmentation analysis, where in IM the ions experience approximately 104 to 106 collisions across a size separation versus 1 or several high energy collisions in collision induced dissociation (CID), respectively. Typical drift tube resolving power of IM-MS is sufficient such that conformationally restricted or extended metabolites, such as cyclic peptides, polycyclic polyketides, and polyenes often possess distinct ion mobility profiles that are obtained over the course of micro to milliseconds. IM-MS is often coupled with time-of-flight (TOF) MS that can rapidly acquire the m/z ratios for ions eluting from the IM-MS cell in a few microseconds. The frequency of data collection allows for sufficient time sampling across chromatographic peaks, which occur over the course of minutes, to be coupled to IM-TOFMS.79,84 When applied to microbial metabolomics the enhanced separation and sensitivity provided by IM-MS has been beneficial for identifying known secondary metabolites, dereplication, and prioritizing features. Our laboratory has previously used IM-MS to help obtain high quality fragmentation data (IM-MS/MS) for all detected ions from crude extracts while comparing the differences between antibiotic resistant and wild-type Nocardiopsis.40 This facilitated the putative identification of several metabolites over-produced in mutant strains. IM-MS has been applied to differentiate halogenated natural products in cyanobacteria85 as well as peptide natural products from cave actinomycetes.86 IM-MS has also been applied to investigate the 3-dimensional structures of lasso peptides, interlocked microbial peptides with a range of bioactivities,87 and this will likely find other useful applications to natural product discovery as the technology becomes more widely available.
2.5 Ion fragmentation for structural information
Both time-dispersive (e.g. TOFMS) and scanning mass spectrometers (e.g. liner quadrupole MS and ion trap MS) can be used to acquire both precursor and fragment ion information (i.e. tandem MS),88 which can provide a wealth of highly specific structural information that can be used to help identify and dereplicate metabolites.89-93 In metabolomics-driven natural product discovery workflows, fragmentation data is commonly collected via an automated data-dependent acquisition method in which the most abundant ions within a scanning cycle are automatically selected for fragmentation. Fragmentation data analysis facilitates natural product dereplication which, as will be discussed in more detail below, is a critical step in the process of natural product discovery.94 There are a variety of methods applied to activate and induce dissociation of target ions, primarily categorized on the basis of how the ion is activated, collisionally, electron attachment, or through photon absorption, where the observed fragment ions will vary based on the method and parameters selected for fragmentation. For small molecules, collision induced dissociation (CID),95 and surface induced dissociation (SID)96 are commonly utilized. The degree of fragmentation observed using these methods depends on the number and degree of scissile bonds within a given molecule as well as the resulting internal ion energies used for analysis. In automated data-dependent tandem mass spectrometric fragmentation analysis, a given single set of dissociation parameters may not be appropriate for every feature of interest within a sample, requiring multiple experiments to determine optimal fragmentation parameters, and to effectively capture fragmentation data for a broad cross section of molecular classes. Ultimately, these methods provide characteristic fragmentation spectra that can be compared to established libraries of secondary metabolite fragmentation data to identify known secondary metabolites within the experimental sample.63 Additionally, tandem mass spectrometric data are useful for elucidating the structures of peptide natural products and have been used in ‘peptidogenomics’ strategies to link ribosomal and non-ribosomal peptide natural products to their cognate biosynthetic gene clusters.97-99
2.6 Leveraging spatiotemporal metabolomics inventories to capture inter-organism interactions
Secondary metabolite producing microorganisms can be cultivated on agar medium 100-102 or in planktonic liquid103-109 culture medium, and several methods have been developed to extract and chromatographically separate resulting metabolomes.110-112 However, microorganisms cultivated as monocultures or mixed cultures on agar may display planar metabolite distributions containing valuable information about chemical pleiotropism, nutrient dependence, and chemical ecology,113-115 and bulk liquid extractions discard the spatial metabolomic feature differentiation that could otherwise be observed.116 Correspondingly, imaging mass spectrometry (IMS) methods have been developed for agar cultivated117 and environmental118-120 microbial samples to provide a second distinguishing ion characteristic, spatial localization. In IMS experiments, the area of a sample is divided into pixels which are individually analysed by the mass spectrometer. Matrix assisted laser desorption ionization (MALDI) is a commonly used ionization technique for IMS.121,122 MALDI requires the application of an ionization matrix to facilitate ionization of cell and agar embedded metabolites. This ablative technique has been applied to visualize spatial temporal distributions of secondary metabolite production in marine cyanobacteria51,120 and to elucidate microbial producers responsible for observed secondary metabolite biosynthesis.119,123 MALDI-IMS has also be used to visualize metabolic exchange between interacting organisms117,124-129 and identify novel antibiotic production in Streptomyces.130 The efficiency of MALDI ionization is matrix dependent, and varies across metabolite classes. Correspondingly, Desorption Electrospray Ionization (DESI) and secondary ion mass spectrometry (SIMS), which do not require the addition of an ionizing matrix, have also been applied to visualize natural product distribution through IMS131-134 among others.135 Determining the spatial distribution of produced secondary metabolites can be useful in natural product research, and as these IMS technologies continue to develop they are expected to become an integral component of metabolomic investigations into microbial secondary metabolites.136 Metabolomic features generated via IMS consist of m/z and its corresponding Cartesian coordinate in agar culture.
3 Preparation of high content mass spectral data for metabolomics studies
3.1 Strategies for formatting data for effective comparative analysis
Typical LC-MS analysis of extracted metabolites results in thousands of detectable features characterized by m/z, and retention time, as well as potentially ion mobility and fragmentation.137 Unbiased manual comparison of features between samples is challenging, especially when analysing a large number of samples. Therefore, it is necessary to automate feature collection from the acquired data in several processing steps that will facilitate data analysis. There are a variety of non-compatible vendor-specific data file formats for mass spectrometric data which originally impeded the development of universal processing software. To address this issue the Protein Standards Initiative (PSI) group138 developed a standardized format, mzData, to facilitate data exchange.139 An additional format, mzXML, was developed by to serve as a standard format for MS and MS/MS data processing.140,141 While both of these formats were popular, the scientific community pushed for a unified standard format to simplify software development. A new format mzML was released to replace both mzXML and mzData formats,142 however, all of these are still commonly used for metabolomics data. Correspondingly, one of the first steps in metabolomics data processing is to convert the data files from a vendor-specific format into one of the appropriate standard formats listed above for the processing software. One common utility for this is ProteoWizard’s MSconvert,143 which also has the ability to pre-filter the data with user defined parameters.
3.2 Methods and considerations for metabolite peak detection and alignment
After data format conversion, metabolite peaks must be identified and extracted from the data and aligned for all samples. A number of reviews have covered and compared the various processing packages and their algorithms.110,144-146 In this section we highlight a few of the common computational methods employed natural product discovery based metabolomics. The initial peak identification can be fairly challenging, as LC-MS ionization methods typically generate high levels of background chemical noise largely from mobile phases and buffers.147 Therefore, the automated processing methods must be able to identify genuine sample features while omitting detected chemical background and instrumental noise, and there have been several algorithms developed to accomplish this task Vectorized peak detection algorithms identify data points above a set intensity threshold in both the m/z and retention time dimensions.148,149 There are also a number of 1-dimensional LC-MS processing algorithms commonly used for peptide analysis which detect peaks by using the isotope patterns in the m/z dimension.150-152 Another of the more common methods involves separating the LC-MS data into extracted ion chromatograms (EIC), each covering a very narrow m/z range. This process is called binning and, while fast and generally effective, this can lead to problems if the bin size is too large or too small. A matched filter153 is commonly applied to EICs to select for m/z peak shapes in the chromatographic time domain, and if features are split between multiple bins due to inappropriate sizing, they can be excluded by the algorithm resulting in false negatives. The traditional XCMS peak detection algorithm, a widely used LC-MS processing software package, sections off 0.1 Dalton wide EICs and then applies a second derivative Gaussian filter that aids in the discrimination of authentic peaks from noise along with a 10:1 signal to noise intensity threshold.154 An alternative to the binning approach for high resolution MS data is the centWave algorithm which identifies ion dense regions of interest in centroid data.155 Peaks are detected along these regions using a continuous wavelet transform, which allows for a much more dynamic range of peak shapes.155,156 The quality and validation of peak detection from increasingly complex datasets remains an area of intense research efforts.157,158
Another consideration in data processing is the tendency for retention times of features to vary between multiple injections due to changes in chromatographic conditions discussed previously. It is therefore necessary to match mass features between samples of an experiment, and align the retention times of matched peaks to generate a discrete feature list. Originally, internal reference standards were used to adjust retention times of each sample.159,160 However, retention time drifts throughout an acquisition are often not linear,154,161 and this also required additional sample preparation steps to incorporate the standards. A variety of algorithms have been developed to align features between sample runs without the use of internal standards. The original XCMS alignment algorithm identified hundreds to thousands of peak groups that are present in a large number of samples. These “well behaved” groups are used as markers to align the remaining detected features. Typically, the number of these markers identified from metabolome extracts is sufficient to cover the chromatographic profile of samples and correctly align the nonlinear retention time drifts. Local regression, LOESS,162 is then used to approximate drifts for regions without sufficient peak markers. Several alignment algorithms have been developed to process LC-MS data,163-169 and in a comparative study of six freely available retention time alignment methods the XCMS algorithm was shown to be the best for processing metabolomics data.170 However, it was noted that the appropriate selection of parameters used for the methods could have a large impact on the data output, such that the apparent success of any particular method is dependent upon the user’s experience. A software package, Isotopologue Parameter Optimization (IPO), was recently released to automatically optimize XCMS parameter settings using natural C13 isotopic peaks.171 This software applies to a variety of different sample types, chromatographic strategies, and instrument methods and aids to simplify and systematize method development while optimizing metabolomics processing for non-experts.
After peak alignment it is common for several mass/retention time features to possess few or even no matches between samples. This may be because some peaks are entirely unique to a subset of experimental samples but can also stem from errors in peak detection due to inappropriate parameter settings, noisy data, etc. Gap-filling is commonly used to ensure these are not false negatives and provide a non-zero value for subsequent statistical analyses. In the absence of a detectable peak, the values obtained through gap filling reflect noise within the region peaks that were detected in other samples. For low abundance features, the integrated noise level over the peak region may be similar to the value determined for the feature, and this can lead to the observation of a metabolite ion that statistically correlates with a single condition while its lower abundance isotopes show no correlations in subsequent statistical analyses.
4 Analysis of metabolomics data in the context of secondary metabolites
The next stage of metabolomics analysis consists of applying one or more methods to compare metabolomics datasets. Depending on the objectives of a given study, several complementary methods may be applied. In the following discussion we review extant methods for comparative metabolomics analysis. To illustrate the application of these methods, we apply them to the analysis of a metabolomics dataset focused on the cytotoxic macrolide producing organism, Nocardiopsis sp FU40, and its exposure to multiple competing organism in mixed culture. In selected mixed culture conditions, this organism increased production of the secondary metabolites called ciromicins, which we highlight throughout data analyses.
4.1 Multivariate statistical analysis and data projections for identification of abundant covarying metabolites
Subsequent to pre-processing, metabolomics data can be analysed through multivariate statistical analyses (MVSA) which simplify and identify significant correlations within the data. Two common methods for metabolomics data analysis are partial least squares (PLS), or projections to latent structures, regression methods and principal component analyses (PCA) reviewed in more detail elsewhere.172,173 Briefly, PLS methods assume that changes within the data are largely driven by a subset of latent variables, which are not themselves measured within the data but are more abstract, such as experimental treatments/stimulants or conditions. With this assumption, a PLS analysis will identify latent vectors within the data that describe the maximal covariance between user defined groups.174,175 Alternatively, PCA makes no assumptions about the data and identifies the sources of the highest variance across the samples to distinguish the samples from one another.176 The fundamental difference between these two analyses is that PLS are supervised with user defined groups while PCA are unsupervised variable reduction methods. Orthogonal signal corrections can be applied to PLS regressions to improve separation between predictive and non-predictive variation.177 The product of these analyses are scoresplots, or projections of samples onto a hyperplane within the data describing sample covariance, from PCA and PLS analyses. Interpretation of scoresplots show the separation of samples based on feature variance to determine which samples are similar (nearby in Cartesian space) and dissimilar (far away) with regards to their most significantly varying features. Replicate analyses of the same sample should cluster within the scoresplot, and in this way scoresplots are a useful means of identifying errors in sample acquisition or data pre-processing. Additionally, a control comprised of pooled samples should locate close to the origin of a PCA plot. Another useful product of the PCA analyses are loadings plots, which show correlations between variables in the data and summarize these variables’ impacts on the scoresplot. Nearby features are positively correlated, while distant features are negatively correlated, and features in the same region as samples in the scoresplot will be more abundantly or uniquely present in those samples.
4.1.1 Strain prioritization via principal component analysis
One approach to the discovery of new natural products has been to prioritize organisms distinguished as metabolically unique through a PCA analysis. There is often a great deal of redundancy in the compounds identified through microbial natural product screening endeavours, and this redundancy can be reduced through the selection of metabolomically diverse microbial strains.178 Under the hypothesis that organisms with similar secondary metabolic potential would cluster in PCA space, Hou et al. analysed 47 microbial strains to demonstrate how MVSA could prioritize strains with diverse secondary metabolic potential.179 Similarly, PCA has been used to prioritize marine microbial symbionts180 as well as phylogenetically similar Streptomyces181 for natural product isolation. Figure 2 demonstrates how metabolically unique organisms are distinguishable along the principal component vectors of the PCA scoresplot. This method may also be useful for identifying new classes of bioactive microbial compounds as has been done for plant extracts.182 However, caveats to this approach include (1) that the correlating features responsible for PCA prioritization of a subset of organisms from a library may not be secondary metabolites, which are generally present in relatively low abundance within crude extracts, and (2) that low abundant secondary metabolites, will not be emphasized by these methods.
Figure 2.
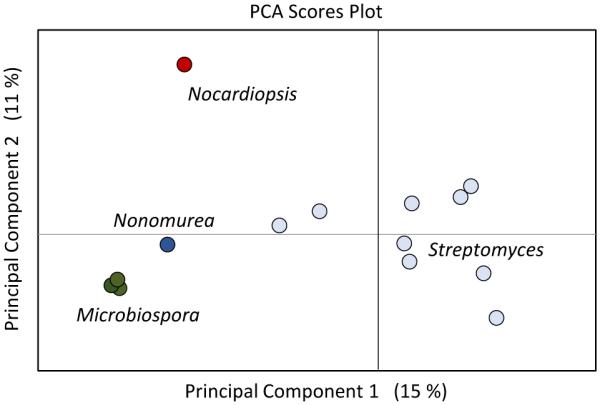
Using a pca scores plot to prioritize microbial producers. a panel of actinomycetes including microbiospora, streptomyces, and nonomurea genera. in this analysis, 14 strains grown under identical conditions were compared and principal component analysis was used to display metabolomic feature variance between the strains. principal component 1 primarily groups streptomyces from other strains, and component 2 further distinguishes nocardiopsis sp. fu40 as metabolomically unique compared to other tested strains. percentages shown in parentheses correspond to the variance between the samples contained within the specific component.
4.1.2 Secondary metabolite prioritization within metabolomics data via principal component and regression analyses
One important application of PCA and PLS metabolomics for natural product discovery is to prioritize induced secondary metabolites in comparative analyses between chemically and/or biologically stimulated and control conditions. Secondary metabolite production can be activated in microorganisms through a variety of chemical and environmental stimulation,29,30,183,184 and PCA and PLS are commonly applied to identify abundantly produced features in these conditions.40,185-187 Binary comparisons using S-plots can be a used to identify group specific features of a PLS model. 188 These graphs separate features by their covariance along the x-axis and their correlation to user defined groups on the y-axis. More simply, more abundant features are farther from the origin on the x-axis, and features with correlations closer to 1 or -1 are likely to be unique or specific to one group or the other. Volcano plots have also recently been used to identify significantly covarying metabolites in binary comparisons of natural product extracts.189 Volcano plots show each features’ statistical significance, p-value, on the y-axis and fold change along the x-axis.190,191 Similarly PCA loadings plots can be used to visualize significant feature differences between sample sets. Figure 3 demonstrates how S-plots, volcano plots, and loadings plots can distinguish induced metabolomic features in our Nocardiopsis case study. The loadings plot tripartite comparison identifies features that correlate with either the Nocardiopsis or Rhodococcus monocultures or a mixed culture where the two compete for nutrients. In this plot the induced cytotoxic macrolactam ciromicin is clearly distinguishable as positively correlated with the mixed culture extract. Similarly, ciromicin was clearly identified through the S-plot and volcano plot comparisons between the Nocardiopsis monoculture and the mixed culture. These methods can be very powerful, and freely available online metabolomics packages, such as XCMS Online192,193 and Metaboanalyst194,195 can perform some routine MVSA data analyses in addition to data pre-processing. An alternative and fairly unique comparative analysis available through XCMS Online is the cloud plot196. These plots convey feature fold changes, m/z, retention time, and statistical distribution in the same Figure, and can perform both binary and multigroup comparisons.193
Figure 3.

mvsa s-, loadings, and volcano plots to identify induced features. (a) the scoresplot reveals group separation between the nocardiopsis monoculture (nf), the rhodococcus wratis competitor monoculture (rw), and the mixed culture (rw&nf). (b) s-plot shows ciromicin significantly correlates (p < 0.1) to the mixed culture group in a binary comparison vs the nocardiopsis monoculture. (c) loadings plot of features shows ciromicin contributes significantly to group differentiation on the pca scoresplot. (d) volcano plot also prioritizes ciromicin ions which have a high correlation (low p-value) on the y-axis and high fold change on the x-axis. shading in panels a and c are used to highlight the data corresponding to the different sample subtypes.
4.2 Discovering molecular inventories of microbial responses via self organizing map analytics
A strength in MVSA analysis of metabolomics datasets is the identification of the most unique and abundant features between small numbers of treatment conditions. However, these methods are limited to displaying data in two or three dimensions and are biased towards the largest differences within the entire dataset. Therefore, the utility of MVSA to represent an experiment diminishes as the number and diversity of samples increases. For instance, it is common to screen a target organism under dozens of stimulus conditions to optimize compound production, or to induce silent biosynthetic gene clusters, and in these cases we have previously demonstrated that an alternative method utilizing Kohonen self-organizing map (SOM) analytics can be more effective at representing multiplexed stimuli data than PCA.184 As discussed above, metabolomic acquisition via LC-MS results in the acquisition of thousands of detectable features. Through SOM analyses these features are organized using an artificial neural network into a 2-dimensional grid based on feature response patterns across all experimental conditions. Features that share similar trend patterns are grouped in nearby nodes of the map as shown in Figure 4. Through multiple iterations, typically several hundred, this organization is improved ultimately resulting in a feature map where features in this case correspond to clusters of similar response trends. Unlike MVSA, SOM analyses improve with increasing amount of data and response conditions (e.g. stimuli), as this leads to more varied response trends which in turn enhances feature organization. A metabolomics workflow – molecular expression dynamics inspector (MEDI), provides an open access methodology for SOM analysis from MS data and is readily applicable to microbial metabolomics.197 In MEDI, each tile, or node, of the grid is coloured based on the centroid intensity of its features to generate heatmaps. Difference maps can be generated by subtractive analysis (e.g. control and stimulus conditions) to readily prioritize abundant and treatment-specific metabolomic features into regions of interest. We applied these SOM analytics to map stimuli-induced metabolomic responses from 23 distinct conditions in Streptomyces coelicolor.184 In this study 16 detected secondary metabolites produced by S. coelicolor were induced in one of the 23 conditions and prioritized through the SOM analysis.184 Application of SOM analyses to investigate metabolomic changes engendered through microbial competition in our Nocardiopsis case study prioritized several metabolites unique to mixed culture conditions including the ciromicins. Additionally, the single SOM analysis recapitulates the results of multiple MVSA analyses. In Figure 5 three PCA loadings plots are compared with three SOM heatmaps from the Nocardiopsis mixed culture example study. When the features held within regions of interest on the SOM maps are sorted by abundance they are highly consistent with the loadings plots from PCA analyses. Indeed, there is correspondence between PCA and the neural networks used for MEDI analysis. As the data and/or conditions become sparser, the SOM heatmap begins to decompose into a similar functional form as PCA.
Figure 4.
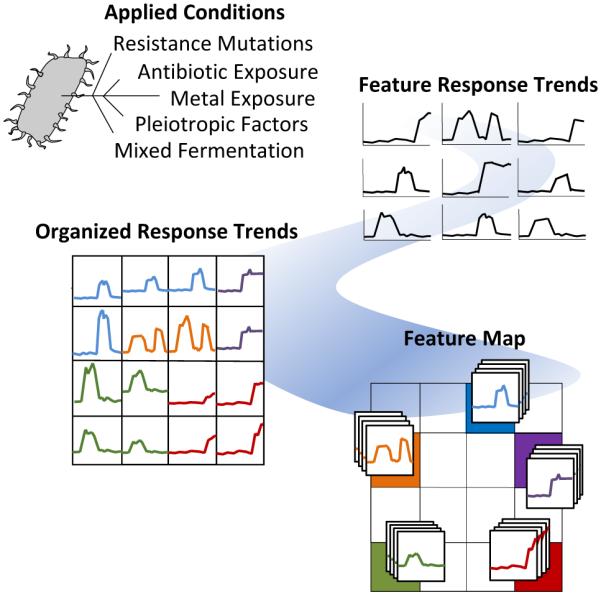
Feature organization within a self-organizing map analysis. feature abundance profiles are illustrated for each feature as a response trend across all experimental conditions shown in the upper right. these trends are organized for similarity as shown on the bottom right. these organized data serve as the basis for visual heatmap representations of the observed metabolomic content of experimental cultures.
Figure 5.

Three example comparisons of prioritized features through principal component and self-organizing map analyses on mixed cultures with nocardiopsis fu40 (nf), rhodococcus wratis (rw), tsukamurella pulmonis (tp), and bacillus subtilis (bs). features prioritized within som regions of interest recapitulate pca tripartite analyses when sorted by abundance, or percentage of the region of interest (% roi). shading in scores and loadings plots used to highlight the data corresponding to the different sample subtypes.
4.3 Molecular networking to reveal structural uniqueness and relatedness in large datasets.
Microorganisms have been extensively mined for natural products throughout much of the past century in the search for new pharmaceuticals, and the rediscovery of known compounds or known families of compounds is quite common. Identifying and removing these rediscovered natural products, a process known as dereplication, is both critical and challenging.59,198 Typically accurate masses or determined molecular formula of extracted compounds are used to search databases of known natural products. However, the large number of isobaric compounds complicate dereplication. UV/Vis absorbance59 spectra and chromatographic retention times199 can be used to further match extracted features to database compounds, and as technologies and databases improve, it is likely that ion mobility will play a role in natural product dereplication as well.85,86,200 Fragmentation spectra acquired through tandem MS is another useful property for dereplication. Metabolite fragmentation patterns observed through MS/MS analysis can be matched to those in databases like PubChem, METLIN201 and MassBank202 to putatively identify MS features. Kernel based machine learning algorithms have recently been applied to dereplicate metabolites using multiple levels of tandem MS,91,203 and while this works well for primary metabolites, public databases for microbial secondary metabolites with fragmentation spectra encompass only a small fraction of known natural products. For example, GNPS: Global Natural Products Social Molecular Networking, the largest natural product public database with MS/MS spectra, contains more than 140,000 natural products,198 and there are an estimated 600,000 published natural compounds.204
Computational methods to generate theoretical fragmentation spectra have been employed to compensate for the lack of experimental data on natural products.205 These in-silico MS/MS spectral databases can further facilitate natural product dereplication when coupled with molecular networking,206 and as both experimental and in-silico database coverage improves, comparisons of fragmentation spectra may become the most useful method of natural product dereplication. In addition to matching fragmentation spectra with database compounds, fragmentation data can be used to cluster related classes of molecules by fragment similarity. Molecular networking analyses cluster families of molecules through vector correlations between fragment ions.207 Yang et al. demonstrated the utility of this approach for natural product discovery by dereplicating 58 natural products from marine and terrestrial microorganisms.208 Molecular networking in this study also identified a number of novel analogs to known compounds, which are more difficult to obtain through other dereplication methods. In Figure 6 we have applied molecular networking to our Nocardiopsis example dataset. Using the network visualizer in the GNPS: Global Natural Products Social Molecular Networking website, fragmentation spectra for each object in the network can be easily viewed and compared to matched reference spectra from the GNPS library. The features in the network can also be coloured by the user-defined group or condition to which they are correlated. In Figure 6 we have highlighted features unique to mixed culture conditions in red. As shown, molecular networking identifies unique features which is unbiased by compound abundance. Several features of this dataset share no significant fragment similarity with the network and are isolated as “self-loops”. In fact, ciromicin A is among these uniquely fragmenting features, and this in itself may be another useful means to prioritize leads, as outlying features may be more structurally unique. Molecular networking analysis can be enhanced by combining additional metabolomics techniques. Klitgaard et al. used a combination of molecular networking and stable isotope labelling to identify novel analogs of nidulanin A and fungisporin in the well-studied fungus Aspergillus nidulans.70 Fragment based clustering in this manner can also be used to identify modified natural products stemming from interactions between organisms. Moree et al. used molecular networking with imaging mass spectrometry to investigate the interkingdom interactions between Pseudomonas and Aspergillus and observed a variety of biotransformed metabolites arising from this microbial competition.209 Similarly, Briand et al. applied molecular networking to identify new compounds and analogs arising from intraspecific interactions between algae.210 The application of molecular networking for the Nocardiopsis mixed culture data shown in Figure 6 links a number features found in the Nocardiopsis monoculture with similarly fragmenting features only detectable in mixed culture. These may represent compounds made by Nocardiopsis that are stimulated or modified in some way by the competitor Tsukamurella.
Figure 6.

Applications of molecular networking to explore data. comparisons of acquired fragmentation spectra to established databases facilitates putative feature identification. connectivity between features shown with blue lines relates structural similarities. reference compounds seeded into the network can identify structural analogs. feature distributions between experimental conditions are indicated by node colouring, red for mixed culture specific, and grey for features detected within the monoculture.
Molecular networking can also prioritize features by linking observed natural products to their cognate biosynthetic gene clusters and gene cluster families99,211 when used in conjunction with genomic sequence analysis. This can be an advantageous means of prioritizing metabolite leads as demonstrated by the work from Kleigrewe et al. where molecular networking was combined with genomic sequence analysis to identify a novel group of acyl amides, termed columbamides, from marine cyanobacteria.212 The Crawford lab has recently employed ‘pathway-targeted’ molecular network analyses to identify metabolites from the colibactin gene cluster, which had been linked to increased virulence in E. coli.213-215 As previously discussed, heterologous hosts are often used for the production of microbial secondary metabolites,216-218 and molecular networking is a useful tool for comparative metabolomics to visualize the output of these heterologous hosts. Schorn et al. used molecular networking to identify novel eponemycin congeners produced through heterologous expression in Streptomyces albus J1046.219 Molecular networking has even been applied to identify virulence factors in pathogenic organisms,220,221 and this method will become more beneficial for natural product discovery as databases and technologies improve.
5. Investigations of secondary metabolite bioactivity
Natural products are intrinsically biologically active, however, the clinical relevance of this activity may not always be discernible. Typically, natural product structure and mode of action are determined fairly late in the natural product discovery pipeline, which contributes to high rediscovery rates. Therefore, prioritizing natural product leads by deep profiling of pharmacologically relevant biological activities would expedite natural product based drug discovery. Natural product extracts are commonly divided into multiple fractions which are then screened to identify the components underlying the desired biological activity.222-225 However, low abundance compounds can often be overlooked in complex extracts, and recently MVSA have been utilized to help link observed fraction bioactivity to detectable features from metabolomic analyses.39,226 Even after correlating metabolites with biological activity, determining the mode of action for active compounds can be difficult and expensive.227-230 One approach has been developed that uses the antibiotic spectrum of activities across different organisms, mode of action profiles (BioMAP), to group similar antibiotics.231 This method was effectively able to cluster antibiotics of the same compound class and led to the identification of a novel naphthoquinone antibiotic, arromycin.231 Gene expression profiling with either the entire transcriptome232,233 or a subset of reporter genes234,235 has also been used to predict modes of action for natural products. However, because these transcriptomic screens are still relatively costly, there is a great interest in applying metabolomics analyses to predict natural product modes of action using either natural product extracts236 or purified compounds.237-242 Vincent et al. have recently shown untargeted metabolomics can effectively identify compound modes of action when specific metabolic pathways are the primary drug target.243 Metabolomic consequences of drug combinations may additionally be able to identify synergism, or antagonism between coadministered drug therapies.241 In a study with M. smegmatis, Halouska et al. observed that antibiotics which share similar biological targets engender similar metabolomic changes and are grouped together through MVSA.239 The group additionally applied their metabolomic methods to investigate antibiotics with unknown biological targets and found them to group with membrane disrupting antibiotics, ampicillin, D‐cycloserine, and vancomycin.239 This methodology could prove very useful to prioritize compounds for isolation. Antimicrobial extracts which separate themselves metabolomically through MVSA or other analyses may exert their activity through a novel biological target or mechanism. In this way pharmaceutically relevant natural products could be prioritized for isolation. These metabolomic analyses have even been applied to investigate the underlying methods by which known antibiotics kill pathogens.244 Another approach, cytological profiling, uses automated image and microscopy analyses to identify phenotypic changes induced from bioactive compounds,245,246 and this method has been used to classify biologically active compounds by their respective modes of action247,248 even within more complex marine derived bacterial extracts.249 A combined approach integrating these phenotypic screens with untargeted metabolomics has recently been developed to predict the modes of action for complex libraries of natural products and prioritize unique bioactive components.250 Applying this method, Compound Activity Mapping, on data from 234 natural product extracts led to the discovery of the quinocinnolinomycins, a new family of natural products implicated to induce endoplasmic reticulum stress based on further cytological profile clustering.250 Ultimately, these multi-omic combinatorial methods may become the preferred means of predicting molecular modes of action. Integrating the phenotypic data from cytological profiling and the transcriptomic functional signature ontologies235 with metabolomics data using one or combinations of the powerful analytical platforms discussed in this review, self-organizing maps, molecular networking, MVSA, etc., could provide new insights into the modes of action of bioactive compounds and greatly facilitate novel drug discovery.
6 Conclusions
Metabolomic analyses are powerful tools for natural product discovery. However, while metabolomics can provide a wealth of information regarding the activity and responses of microorganisms, with current technologies it is practically impossible to analyse the entire metabolome of an organism comprehensively due to variations in ionization efficiency and limitations in detection across wide concentration dynamic range. Instead, only detectable metabolites, which make up a fraction of the total metabolites present, are used to draw conclusions from current studies. While the full transcriptomic and proteomic potential of an organism can be determined through modern genome sequencing, there is no readily discernible limit to the number of metabolites present within organisms, so it is difficult to predict the number of metabolites omitted by current analyses. Due to these limitations, extra care must be taken when drawing conclusions from metabolomics datasets. Nonetheless, metabolomics analyses benefit microbial natural product discovery pipelines in a variety of ways as described in this report. These can be used to prioritize organisms, identify activated compounds from stimuli exposure, prioritize features through bioactivity spectrums or molecular class, and even dereplicate prioritized secondary metabolites. The metabolomics methods described herein may also facilitate investigations into the fundamental purpose behind secondary metabolite production within microbial communities. It is largely unclear how the production of secondary metabolites is regulated in situ as well as which ecological stimuli trigger secondary metabolic production. Such studies would benefit natural product discovery endeavours by facilitating predictions of stimuli to induce secondary metabolite production within the endogenous producer and could additionally provide insight into human health and wellness. Microbial secondary metabolites have a significant impact on human health by means of both isolated pharmaceuticals and compounds produced in situ from within human microbiomes,13,251-255 and metabolomics may be able to offer insights into how these organisms modulate their secondary metabolism in response to diet, medicine, and endogenous host factors.
Comparative metabolomics methods are aiding in unleashing the repressed and/or hidden wealth of microbial secondary metabolism predicted by whole genome sequencing. The combination of complimentary methods (e.g. SOM and molecular networking) has the potential provide new tools to accelerate discovery by comparison. Ultimately, the purpose of these efforts is to identify biological roles for secondary metabolites, be they biochemical, chemical ecological, or translational in human medicine. We feel that the next era in secondary metabolite discovery and application will be facilitated by methods that combine high content biological activity data measurements for metabolites within metabolomes with corresponding multidimensional metabolomic data to illuminate effectors of natural small molecule interactions, and their roles in biological systems.
Figure 1.
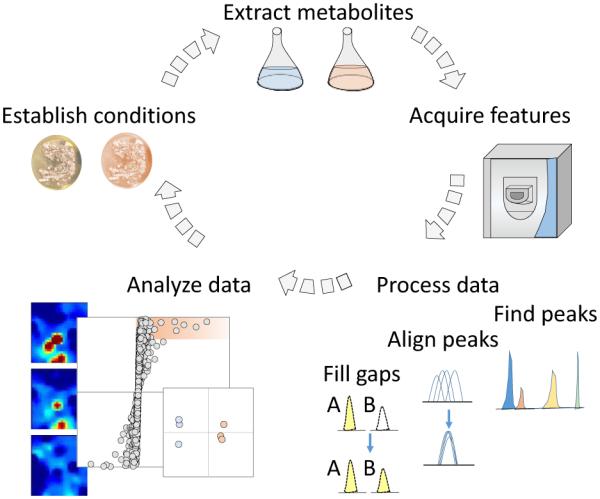
General metabolomics workflow. metabolites are extracted from experimental conditions and detected through ms analysis. ms data is then formatted and processed before undergoing statistical analyses to determine important metabolomic changes between the sample groups. these results may then be used to direct new experiments to optimize secondary metabolite production or test biological hypotheses generated from the initial experiment.
Table 2.
Overview of methods for metabolomic data analysis
| Method | Description | Applications | Disadvantages |
|---|---|---|---|
| Principle component analysis and Projections to latent structures |
MVSA to identify significant covariance within data |
|
|
|
| |||
| Self organizing Maps | Organizes features into a 2- dimensional map based on feature response trends across a variety of experimental conditions |
|
|
|
| |||
| Molecular networking | Organizes features into a connectivity network based on similarities in molecular fragmentation patterns |
|
|
7 Acknowledgements
This work was supported by the National institutes of Health (no. R01GM092218 awarded to B.O.B. and J.A.M., and T32 no. GM0650086 awarded to B.C.C.), the Vanderbilt Institute of Chemical Biology, the Vanderbilt Institute for Integrative Biosystems Research and Education, and the Vanderbilt University College of Arts and Sciences.
Biographies

Brett Covington received a B.S. in Chemistry from Austin Peay State University in 2012. He was awarded with an NIH sponsored Chemistry-Biology Interface training grant in 2013 and is currently a 4th year PhD candidate at Vanderbilt University under the supervision of Brian Bachmann. His research in the natural product discovery section of the Bachmann lab has been primarily focused on the application of untargeted metabolomic methods to identify microbial secondary metabolite responses to environmental stimuli.

Brian Bachmann graduated in 2000 with a PhD in Chemistry from The Johns Hopkins University, studying with Prof Craig Townsend. Following this, he joining Ecopia Biosciences in Montreal, where he was Director of Chemistry, prior to moving to Vanderbilt, where he is currently Professor of Chemistry and Associate Director of the Vanderbilt Institute for Chemical Biology. He has a long standing interest in studying and harnessing the biosynthetic capabilities of living systems in order to radically accelerate the discovery of new bioactive compounds, and revolutionize how molecules are synthesized.

John McLean graduated in 2001 with a PhD in Chemistry from George Washington University. Following postdoctoral research at Forschungszentrum Jülich in Germany and then at Texas A&M University with Prof. David H. Russell, he began at Vanderbilt University in 2006 where he is Stevenson Professor of Chemistry, Director of the Center for Innovative Technology, co-Director of the Automated Biosystems Core, and Deputy Director of the Institute for Integrative Biosystems Research and Education at Vanderbilt University. McLean and colleagues focus on the conceptualization, design, and construction of structural mass spectrometers, specifically targeting complex samples in systems, synthetic, and chemical biology.
8 References
- 1.Bentley SD, Chater KF, Cerdeño-Tárraga AMM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CHH, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream MAA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 2.Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S. Nat. Biotechnol. 2003;21:526–531. doi: 10.1038/nbt820. [DOI] [PubMed] [Google Scholar]
- 3.Zarins-Tutt JS, Barberi TT, Gao H, Mearns-Spragg A, Zhang L, Newman DJ, Goss RJM. Nat. Prod. Rep. 2016;33:54–72. doi: 10.1039/c5np00111k. [DOI] [PubMed] [Google Scholar]
- 4.Scherlach K, Hertweck C. Org. Biomol. Chem. 2009;7:1753–1760. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 5.Walsh CT, Fischbach MA. J. Am. Chem. Soc. 2010;132:2469–2493. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shabuer G, Ishida K, Pidot SJ, Roth M, Dahse H-MM, Hertweck C. Science (New York, N.Y.) 2015;350:670–674. doi: 10.1126/science.aac9990. [DOI] [PubMed] [Google Scholar]
- 7.Findlay BL. ACS Chem. Biol. 2016;11:1502–1510. doi: 10.1021/acschembio.6b00176. [DOI] [PubMed] [Google Scholar]
- 8.Vizcaino MI, Guo X, Crawford JM. J Ind Microbiol Biotechnol. 2014;41:285–299. doi: 10.1007/s10295-013-1356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newman DJ, Cragg GM. J. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 10.Zakrzewski P, Fischbach MA, Weber T. Nucl. Acids Res. 2011;39:339–346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li MHT, Ung PMU, Zajkowski J. BMC Bioinformatics. 2009:10. doi: 10.1186/1471-2105-10-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ju K-SS, Gao J, Doroghazi JR, Wang K-KA, Thibodeaux CJ, Li S, Metzger E, Fudala J, Su J, Zhang JK, Lee J, Cioni JP, Evans BS, Hirota R, Labeda DP, van der Donk WA, Metcalf WW. Proc. Natl. Acad. Sci. U. S. A. 2015;112:12175–12180. doi: 10.1073/pnas.1500873112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA. Cell. 2014;158:1402–1414. doi: 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, Breitling R, Takano E, Medema MH. Nucl. Acids Res. 2015;43:237–243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen PR, Chavarria KL, Fenical W, Moore BS. J. Ind. Microbiol. Biotechnol. 2014;41:203–209. doi: 10.1007/s10295-013-1353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farnet CM, Zazopoulos E. In: Natural Products: Drug Discovery and Therapeutic Medicine. Zhang L, Demain AL, editors. Humana Press; Totowa, NJ: 2005. pp. 95–106. [Google Scholar]
- 17.Ziemert N, Alanjary M, Weber T. Nat. Prod. Rep. 2016 doi: 10.1039/c6np00025h. DOI: 10.1039/C6NP00025H. [DOI] [PubMed] [Google Scholar]
- 18.Medema MH, Fischbach MA. Nat. Chem. Biol. 2015;11:639–648. doi: 10.1038/nchembio.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Juan Pablo G-E, Mervyn JB. J. Ind. Microbiol. Biotechnol. 2013;41:425–431. [Google Scholar]
- 20.Komatsu M, Uchiyama T, mura S, Cane DE, Ikeda H. Proc. Natl. Acad. Sci. U. S. A. 2010;107:2646–2651. doi: 10.1073/pnas.0914833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim E, Moore BS, Yoon YJ. Nat. Chem. Biol. 2015;11:649–659. doi: 10.1038/nchembio.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang X, Li J, Millán-Aguiñaga N, Zhang JJ. ACS Chem. Biol. 2015;10:2841–2849. doi: 10.1021/acschembio.5b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross AC, Gulland LES, Dorrestein PC. ACS Synth. Biol. 2014;4:414–420. doi: 10.1021/sb500280q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donia MS, Ruffner DE, Cao S, Schmidt EW. ChemBioChem. 2011;12:1230–1236. doi: 10.1002/cbic.201000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ongley SE, Bian X, Neilan BA, Müller R. Nat. Prod. Rep. 2013;30:1121–1138. doi: 10.1039/c3np70034h. [DOI] [PubMed] [Google Scholar]
- 26.Luo Y, Enghiad B, Zhao H. Nat. Prod. Rep. 2016;33:174–182. doi: 10.1039/c5np00085h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bibb MJ. Curr. Opin. Microbiol. 2005;8:208–215. doi: 10.1016/j.mib.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 28.Seyedsayamdost MR, Chandler JR, Blodgett JAV. Org. Lett. 2010;12:716–719. doi: 10.1021/ol902751x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bode HB, Bethe B, Höfs R, Zeeck A. ChemBioChem. 2002;3:619–627. doi: 10.1002/1439-7633(20020703)3:7<619::AID-CBIC619>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 30.Rutledge PJ, Challis GL. Nat. Rev. Microbiol. 2015;13:509–523. doi: 10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- 31.Ochi K, Hosaka T. Appl. Microbiol. Biot. 2013;97:87–98. doi: 10.1007/s00253-012-4551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reen FJ, Romano S, Dobson ADW, O'Gara F. Mar. Drugs. 2015;13:4754–4783. doi: 10.3390/md13084754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai Y, Sato S, Tanaka Y, Ochi K, Hosaka T. Appl. Environ. Microbiol. 2015;81:3869–3879. doi: 10.1128/AEM.04214-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W, Ji J, Li X, Wang J, Li S, Pan G, Fan K, Yang K. Proc. Natl. Acad. Sci. U. S. A. 2014;111:5688–5693. doi: 10.1073/pnas.1324253111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tokuyama S, Kaji A, Ikeda H, Ochi K. Appl. Environ. Microbiol. 2009;75:4919–4922. doi: 10.1128/AEM.00681-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsurumi Y, Kodani S, Yoshida M, Fujie A, Ochi K. Nat. Biotechnol. 2009;27:462–464. doi: 10.1038/nbt.1538. [DOI] [PubMed] [Google Scholar]
- 37.Wang G, Hosaka T, Ochi K. Appl. Environ. Microbiol. 2008;74:2834–2840. doi: 10.1128/AEM.02800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ochi K, Okamoto S, Tozawa Y, Inaoka T, Hosaka T, Xu J, Kurosawa K. Adv. Appl. Microbiol. 2003;56:155–184. doi: 10.1016/S0065-2164(04)56005-7. [DOI] [PubMed] [Google Scholar]
- 39.Wu C, Du C, Gubbens J, Choi YH. J. Nat. Prod. 2015;78:2355–2363. doi: 10.1021/acs.jnatprod.5b00276. [DOI] [PubMed] [Google Scholar]
- 40.Derewacz DK, Goodwin CR, McNees RC, McLean JA, Bachmann BO. Proc. Natl. Acad. Sci. U. S. A. 2013;110:2336–2341. doi: 10.1073/pnas.1218524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka Y, Hosaka T, Ochi K. J. Antibiot. (Tokyo) 2010;63:477–481. doi: 10.1038/ja.2010.53. [DOI] [PubMed] [Google Scholar]
- 42.Kawai K, Wang G, Okamoto S, Ochi K. FEMS Microbiol. Lett. 2007;274:311–315. doi: 10.1111/j.1574-6968.2007.00846.x. [DOI] [PubMed] [Google Scholar]
- 43.Haferburg G, Kothe E. J. Basic Microbiol. 2007;47:453–467. doi: 10.1002/jobm.200700275. [DOI] [PubMed] [Google Scholar]
- 44.Abdelmohsen UR, Grkovic T, Balasubramanian S, Kamel MS, Quinn RJ, Hentschel U. Biotechnol. Adv. 2015;33:798–811. doi: 10.1016/j.biotechadv.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Derewacz DK, Covington BC, McLean JA, Bachmann BO. ACS Chem. Biol. 2015;10:1998–2006. doi: 10.1021/acschembio.5b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angell S, Bench BJ, Williams H, Watanabe CMH. Chem. Biol. 2006;13:1349–1359. doi: 10.1016/j.chembiol.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 47.Oh D-C, Kauffman CA, Jensen PR, Fenical W. J. Nat. Prod. 2007;70:515–520. doi: 10.1021/np060381f. [DOI] [PubMed] [Google Scholar]
- 48.Cueto M, Jensen PR, Kauffman C, Fenical W, Lobkovsky E, Clardy J. J. Nat. Prod. 2001;64:1444–1446. doi: 10.1021/np0102713. [DOI] [PubMed] [Google Scholar]
- 49.Brakhage AA. Nat. Rev. Microbiol. 2012;11:21–32. doi: 10.1038/nrmicro2916. [DOI] [PubMed] [Google Scholar]
- 50.Forseth RR, Schroeder FC. Curr. Opin. Chem. Biol. 2011;15:38–47. doi: 10.1016/j.cbpa.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Esquenazi E, Jones AC, Byrum T, Dorrestein PC, Gerwick WH. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5226–5231. doi: 10.1073/pnas.1012813108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jarmusch AK, Cooks GR. Nat. Prod. Rep. 2014;31:730–738. doi: 10.1039/c3np70121b. [DOI] [PubMed] [Google Scholar]
- 53.Krug D, Müller R. Nat. Prod. Rep. 2014;31:768–783. doi: 10.1039/c3np70127a. [DOI] [PubMed] [Google Scholar]
- 54.Bereman MS, Lyndon MM, Dixon RB. Rapid Commun. Mass Spectrom. 2008;22:1563–1566. doi: 10.1002/rcm.3544. [DOI] [PubMed] [Google Scholar]
- 55.Stroh JG, Petucci CJ, Brecker SJ, Huang N. J. Am. Soc. Mass Spectrom. 2007;18:1612–1616. doi: 10.1016/j.jasms.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 56.Sleno L, Volmer DA, Marshall AG. J. Am. Soc. Mass Spectrom. 2005;16:183–198. doi: 10.1016/j.jasms.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Bristow AWT, Webb KS. J. Am. Soc. Mass. Spectrom. 2003;14:1086–1098. doi: 10.1016/S1044-0305(03)00403-3. [DOI] [PubMed] [Google Scholar]
- 58.Kind T, Fiehn O. Bioanal. Rev. 2010;2:23–60. doi: 10.1007/s12566-010-0015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nielsen KF, Månsson M, Rank C, Frisvad JC, Larsen TO. J. Nat. Prod. 2011;74:2338–2348. doi: 10.1021/np200254t. [DOI] [PubMed] [Google Scholar]
- 60.Kind T, Fiehn O. BMC bioinformatics. 2007:8. doi: 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.May JC, McLean JA. Annu. Rev. Anal. Chem. 2016;9:387–409. doi: 10.1146/annurev-anchem-071015-041734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kildgaard S, Mansson M, Dosen I, Klitgaard A. Mar. Drugs. 2014;12:3681–3705. doi: 10.3390/md12063681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.El-Elimat T, Figueroa M, Ehrmann BM, Cech NB, Pearce CJ, Oberlies NH. J. Nat. Prod. 2013;76:1709–1716. doi: 10.1021/np4004307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neumann S, Böcker S. Anal. Bioanal. Chem. 2010;398:2779–2788. doi: 10.1007/s00216-010-4142-5. [DOI] [PubMed] [Google Scholar]
- 65.Hernandes MZ, Moreira DR, de Azevedo Junior WF, Leite AC. Curr. Drug Targets. 2010;11:303–314. doi: 10.2174/138945010790711996. C. S. M. [DOI] [PubMed] [Google Scholar]
- 66.Kim JK, Harada K, Bamba T, Fukusaki E. Biosci. Biotechnol. Biochem. 2005;69:1331–1340. doi: 10.1271/bbb.69.1331. [DOI] [PubMed] [Google Scholar]
- 67.Böcker S, Letzel MC, Lipták Z, Pervukhin A. Bioinformatics. 2009;25:218–224. doi: 10.1093/bioinformatics/btn603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haglund PS, Löfstrand K, Siek K, Asplund L. Mass Spectrom. 2013;2:S0018. doi: 10.5702/massspectrometry.S0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol. Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 70.Klitgaard A, Nielsen JB, Frandsen RJN, Andersen MR, Nielsen KF. Anal. Chem. 2015;87:6520–6526. doi: 10.1021/acs.analchem.5b01934. [DOI] [PubMed] [Google Scholar]
- 71.Bucar F, Wube A, Schmid M. Nat. Prod. Rep. 2013;30:525–545. doi: 10.1039/c3np20106f. [DOI] [PubMed] [Google Scholar]
- 72.Ebada SS, Edrada RA, Lin W, Proksch P. Nat. Protoc. 2008;3:1820–1831. doi: 10.1038/nprot.2008.182. [DOI] [PubMed] [Google Scholar]
- 73.Sticher O. Nat. Prod. Rep. 2008;25:517–554. doi: 10.1039/b700306b. [DOI] [PubMed] [Google Scholar]
- 74.Wolfender JL, Marti G, Thomas A, Bertrand S. J. Chromatogr. A. 2015;1382:136–164. doi: 10.1016/j.chroma.2014.10.091. [DOI] [PubMed] [Google Scholar]
- 75.Månsson M, Phipps RK, Gram L, Munro MH, Larsen TO, Nielsen KF. J. Nat. Prod. 2010;73:1126–1132. doi: 10.1021/np100151y. [DOI] [PubMed] [Google Scholar]
- 76.Nielsen KF, Larsen TO. Front. Microbiol. 2015:6. doi: 10.3389/fmicb.2015.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hounoum BM, Blasco H, Emond P, Mavel S. Trac-Trend. Anal. Chem. 2016;75:118–128. [Google Scholar]
- 78.Beisken S, Eiden M, Salek RM. Expert Rev. Mol. Diagn. 2015;15:97–109. doi: 10.1586/14737159.2015.974562. [DOI] [PubMed] [Google Scholar]
- 79.May JC, McLean JA. Anal. Chem. 2015;87:1422–1436. doi: 10.1021/ac504720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lanucara F, Holman SW, Gray CJ, Eyers CE. Nat. Chem. 2014;6:281–294. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- 81.Cumeras R, Figueras E, Davis CE, Baumbach JI. Analyst. 2015;140:1476–1490. doi: 10.1039/c4an01101e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McLean JA, Ruotolo BT, Gillig KJ, Russell DH. Int. J. Mass Spectrom. 2005;240:301–315. [Google Scholar]
- 83.Creaser CS, Griffiths JR, Bramwell CJ, Noreen S. Analyst. 2004;129:984–994. [Google Scholar]
- 84.Kulchania M, Barnes CAS, Clemmer DE. Int. J. Mass Spectrom. 2001;212:97–109. [Google Scholar]
- 85.Esquenazi E, Daly M, Bahrainwala T. Bioorg. Med. Chem. 2011;19:6639–6644. doi: 10.1016/j.bmc.2011.06.081. [DOI] [PubMed] [Google Scholar]
- 86.Goodwin CR, Fenn LS, Derewacz DK. J. Nat. Prod. 2012;75:48–53. doi: 10.1021/np200457r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jeanne Dit Fouque K, Afonso C, Zirah S, Hegemann JD, Zimmermann M, Marahiel MA, Rebuffat S, Lavanant H. Anal. Chem. 2015;87:1166–1172. doi: 10.1021/ac503772n. [DOI] [PubMed] [Google Scholar]
- 88.Payne AH, Glish GL. Methods Enzymol. 2005;402:109–148. doi: 10.1016/S0076-6879(05)02004-5. [DOI] [PubMed] [Google Scholar]
- 89.Zhu Z-JJ, Schultz AW, Wang J, Johnson CH, Yannone SM, Patti GJ, Siuzdak G. Nat. Protoc. 2013;8:451–460. doi: 10.1038/nprot.2013.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Benton HP, Wong DM, Trauger SA, Siuzdak G. Anal. Chem. 2008;80:6382–6389. doi: 10.1021/ac800795f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dührkop K, Shen H, Meusel M, Rousu J, Böcker S. Proc. Natl. Acad. Sci. U. S. A. 2015;112:12580–12585. doi: 10.1073/pnas.1509788112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hufsky F, Böcker S. Mass spectrom. rev. 2016;9999:1–10. doi: 10.1002/mas.21489. [DOI] [PubMed] [Google Scholar]
- 93.Vaniya A, Fiehn O. Trac-Trend Anal. Chem. 2015;69:52–61. doi: 10.1016/j.trac.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tawfike AF, Viegelmann C, Edrada-Ebel R. Methods Mol. Biol. 2012;1055:227–244. doi: 10.1007/978-1-62703-577-4_17. [DOI] [PubMed] [Google Scholar]
- 95.Kertesz TM, Hall LH, Hill DW, Grant DF. J. Am. Soc. Mass Spectrom. 2009;20:1759–1767. doi: 10.1016/j.jasms.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 96.Wysocki VH, Joyce KE, Jones CM. J. Am. Soc. Mass. Spectrom. 2008;19:190–208. doi: 10.1016/j.jasms.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kersten RD, Yang Y-LL, Xu Y, Cimermancic P, Nam S-JJ, Fenical W, Fischbach MA, Moore BS, Dorrestein PC. Nat. Chem. Biol. 2011;7:794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kersten RD, Ziemert N, Gonzalez DJ, Duggan BM, Nizet V, Dorrestein PC, Moore BS. Proc. Natl. Acad. Sci. U. S. A. 2013;110:4407–4416. doi: 10.1073/pnas.1315492110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nguyen DD, Wu C-HH, Moree WJ, Lamsa A, Medema MH, Zhao X, Gavilan RG, Aparicio M, Atencio L, Jackson C, Ballesteros J, Sanchez J, Watrous JD, Phelan VV, van de Wiel C, Kersten RD, Mehnaz S, De Mot R, Shank EA, Charusanti P, Nagarajan H, Duggan BM, Moore BS, Bandeira N, Palsson BØ, Pogliano K, Gutiérrez M, Dorrestein PC. Proc. Natl. Acad. Sci. U. S. A. 2013;110:2611–2620. doi: 10.1073/pnas.1303471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reeta Rani S, Anil Kumar P, Carlos RS, Ashok P. Biochem. Eng. J. 2009;44:13–18. [Google Scholar]
- 101.Ing-Lung S, Chia-Yu K, Feng-Chia H, Suey-Sheng K, Chienyan H. J. Chin. Inst. Chem. Eng. 2008;39:635–643. [Google Scholar]
- 102.Robinson T, Singh D, Nigam P. Appl. Microbiol. Biotechnol. 2001;55:284–289. doi: 10.1007/s002530000565. [DOI] [PubMed] [Google Scholar]
- 103.Kunze B, Bohlendorf B, Reichenbach H. J. Antibiot. (Tokyo) 2008;61:18–26. doi: 10.1038/ja.2008.104. [DOI] [PubMed] [Google Scholar]
- 104.Abdelfattah MS, Kharel MK, Hitron JA. J. Nat. Prod. 2008;71:1569–1573. doi: 10.1021/np800281f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Surup F, Wagner O, von Frieling J. J. Org. Chem. 2007;72:5085–5090. doi: 10.1021/jo0703303. [DOI] [PubMed] [Google Scholar]
- 106.Rančić A, Soković M, Karioti A, Vukojević J. Environ. Toxicol. Pharmacol. 2006;22:80–84. doi: 10.1016/j.etap.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 107.Yoon TM, Kim JW, Kim JG, Kim WG, Suh JW. J. Antibiot. (Tokyo) 2006;59:640–645. doi: 10.1038/ja.2006.85. [DOI] [PubMed] [Google Scholar]
- 108.Umezawa K, Ikeda Y, Kawase O. J. Chem. Soc. 2001;1:1550–1553. [Google Scholar]
- 109.Murphy B, Anderson K, Borissow C, Caffrey P. Org. Biomol. Chem. 2010;8:3758–3770. doi: 10.1039/b922074g. [DOI] [PubMed] [Google Scholar]
- 110.Zhou B, Xiao J, Tuli L, Ressom HW. Mol. BioSyst. 2012;8:470–481. doi: 10.1039/c1mb05350g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Forcisi S, Moritz F, Kanawati B, Tziotis D. J. Chromatogr. A. 2013;1292:51–65. doi: 10.1016/j.chroma.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 112.Kuehnbaum NL, Britz-McKibbin P. Chem. Rev. 2013;113:2437–2468. doi: 10.1021/cr300484s. [DOI] [PubMed] [Google Scholar]
- 113.Straight PD, Kolter R. Annu. Rev. Microbiol. 2009;63:99–118. doi: 10.1146/annurev.micro.091208.073248. [DOI] [PubMed] [Google Scholar]
- 114.Little AE, Robinson CJ, Peterson SB, Raffa KF, Handelsman J. Annu. Rev. Microbiol. 2008;62:375–401. doi: 10.1146/annurev.micro.030608.101423. [DOI] [PubMed] [Google Scholar]
- 115.Ryan RP, Dow JM. Microbiology. 2008;154:1845–1858. doi: 10.1099/mic.0.2008/017871-0. [DOI] [PubMed] [Google Scholar]
- 116.Watrous JD, Dorrestein PC. Nat. Rev. Microbiol. 2011;9:683–694. doi: 10.1038/nrmicro2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang JY, Phelan VV, Simkovsky R, Watrous JD, Trial RM, Fleming TC, Wenter R, Moore BS, Golden SS, Pogliano K, Dorrestein PC. J. Bacteriol. 2012;194:6023–6028. doi: 10.1128/JB.00823-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lane AL, Nyadong L, Galhena AS. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7314–7319. doi: 10.1073/pnas.0812020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Simmons TL, Coates RC, Clark BR, Engene N, Gonzalez D, Esquenazi E, Dorrestein PC, Gerwick WH. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4587–4594. doi: 10.1073/pnas.0709851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Esquenazi E, Coates C, Simmons L, Gonzalez D, Gerwick WH, Dorrestein PC. Mol. Biosyst. 2008;4:562–570. doi: 10.1039/b720018h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eriksson C, Masaki N, Yao I, Hayasaka T. Mass Spectrom. 2013;2:S0022. doi: 10.5702/massspectrometry.S0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gessel MM, Norris JL, Caprioli RM. J. Proteomics. 2014;107:71–82. doi: 10.1016/j.jprot.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Masatoshi T, Joshawna KN, Niclas E, Eduardo E, Tara B, Pieter CD, William HG. J. Nat. Prod. 2010;73:393–398. [Google Scholar]
- 124.Yu-Liang Y, Yuquan X, Paul S, Pieter CD. Nat. Chem. Biol. 2009;5:885–887. [Google Scholar]
- 125.Bleich R, Watrous JD, Dorrestein PC, Bowers AA, Shank EA. Proc. Natl. Acad. Sci. U. S. A. 2015;112:3086–3091. doi: 10.1073/pnas.1414272112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Johannes K, Martin K, Bernd S, Maria-Gabriele S, Christian H, Ravi Kumar M, Erhard S, Aleš S. Nat. Chem. Biol. 2010;6:261–263. [Google Scholar]
- 127.Gonzalez DJ, Haste NM, Hollands A, Fleming TC, Hamby M, Pogliano K, Nizet V, Dorrestein PC. Microbiology. 2011;157:2485–2492. doi: 10.1099/mic.0.048736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shih C-JJ, Chen P-YY, Liaw C-CC, Lai Y-MM, Yang Y-LL. Nat. Prod. Rep. 2014;31:739–755. doi: 10.1039/c3np70091g. [DOI] [PubMed] [Google Scholar]
- 129.Harn YC, Powers MJ, Shank EA, Jojic V. Bioinformatics. 2015;31:42–50. doi: 10.1093/bioinformatics/btv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wei-Ting L, Roland DK, Yu-Liang Y, Bradley SM, Pieter CD. J. Am. Chem. Soc. 2011;133:18010–18013. [Google Scholar]
- 131.Traxler MF, Watrous JD, Alexandrov T, Dorrestein PC. MBio. 2013;4:e00459–00413. doi: 10.1128/mBio.00459-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vaidyanathan S, Fletcher J, Goodacre R, Lockyer N, Micklefield J, Vickerman J. Anal. Chem. 2008;80:1942–1951. doi: 10.1021/ac701921e. [DOI] [PubMed] [Google Scholar]
- 133.Vaidyanathan S, Fletcher J, Lockyer N, Vickerman J. Appl. Surf. Sci. 2008;255:922–925. doi: 10.1016/j.apsusc.2008.05.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Esquenazi E, Dorrestein PC, Gerwick WH. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7269–7270. doi: 10.1073/pnas.0902840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bhardwaj C, Hanley L. Nat. Prod. Rep. 2014;31:756–767. doi: 10.1039/c3np70094a. [DOI] [PubMed] [Google Scholar]
- 136.Bouslimani A, Sanchez LM, Garg N, Dorrestein PC. Nat. Prod. Rep. 2014;31:718–729. doi: 10.1039/c4np00044g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Patti GJ, Yanes O, Siuzdak G. Nat. Rev. Mol. Cell Biol. 2012;13:263–269. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Orchard S, Hermjakob H, Apweiler R. Proteomics. 2003;3:1374–1376. doi: 10.1002/pmic.200300496. [DOI] [PubMed] [Google Scholar]
- 139.Hermjakob H. Proteomics. 2006;6:34–38. doi: 10.1002/pmic.200600537. [DOI] [PubMed] [Google Scholar]
- 140.Pedrioli PGA, Eng JK, Hubley R, Vogelzang M. Nat. Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 141.Lin SM, Zhu L, Winter AQ, Sasinowski M. Expert Rev. Proteomics. 2005;2:839–845. doi: 10.1586/14789450.2.6.839. [DOI] [PubMed] [Google Scholar]
- 142.Martens L, Chambers M, Sturm M, Kessner D. Mol. Cell Proteomics. 2011:10. doi: 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kessner D, Chambers M, Burke R, Agus D. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hendriks M, Eeuwijk FA, Jellema RH, Westerhuis JA, Reijmers TH, Hoefsloot HCJ, Smilde AK. Trac-Trend. Anal. Chem. 2011;30:1685–1698. [Google Scholar]
- 145.Mahieu NG, Genenbacher JL, Patti GJ. Curr. Opin. Chem. Biol. 2016;30:87–93. doi: 10.1016/j.cbpa.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liland KH. Trac-Trend. Anal. Chem. 2011;30:827–841. [Google Scholar]
- 147.Willem W, Phalp JM, Alan WP. Anal. Chem. 1996;68:3602–3606. [Google Scholar]
- 148.Hastings CA, Norton SM, Roy S. Rapid Commun. Mass Spectrom. 2002;16:462–467. doi: 10.1002/rcm.600. [DOI] [PubMed] [Google Scholar]
- 149.Roy S, Shaler TA, Hill LR, Norton S, Kumar P. Anal. Chem. 2003;75:4818–4826. doi: 10.1021/ac026468x. [DOI] [PubMed] [Google Scholar]
- 150.Monroe ME, Tolić N, Jaitly N, Shaw JL, Adkins JN. Bioinformatics. 2007;23:2021–2023. doi: 10.1093/bioinformatics/btm281. [DOI] [PubMed] [Google Scholar]
- 151.Mueller LN, Rinner O, Schmidt A, Letarte S. Proteomics. 2007;7:3470–3480. doi: 10.1002/pmic.200700057. [DOI] [PubMed] [Google Scholar]
- 152.Sturm M, Bertsch A, Gröpl C, Hildebrandt A. BMC Bioinformatics. 2008:9. doi: 10.1186/1471-2105-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rolf D, Dan B, Karin EM. Anal. Chim. Acta. 2002;454:167–184. [Google Scholar]
- 154.Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. Anal. Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 155.Tautenhahn R, Böttcher C, Neumann S. BMC bioinformatics. 2007:9. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Du P, Kibbe WA, Lin SM. Bioinformatics. 2006;22:2059–2065. doi: 10.1093/bioinformatics/btl355. [DOI] [PubMed] [Google Scholar]
- 157.French WR, Zimmerman LJ, Schilling B, Gibson BW, Miller CA, Townsend RR, Sherrod SD, Goodwin CR, McLean JA, Tabb DL. J. Proteome Res. 2014;14:1299–1307. doi: 10.1021/pr500886y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Aoshima K, Takahashi K, Ikawa M, Kimura T, Fukuda M, Tanaka S, Parry HE, Fujita Y, Yoshizawa AC, Utsunomiya S.-i., Kajihara S, Tanaka K, Oda Y. BMC Bioinformatics. 2014;15:1–14. doi: 10.1186/s12859-014-0376-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dettmer K, Aronov PA, Hammock BD. Mass Spectrom. Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Frenzel T, Miller A, Engel KH. Eur. Food Res. Technol. 2003;216:335–342. [Google Scholar]
- 161.Podwojski K, Fritsch A, Chamrad DC, Paul W. Bioinformatics. 2009;25:758–764. doi: 10.1093/bioinformatics/btp052. [DOI] [PubMed] [Google Scholar]
- 162.Cleveland WS, Grosse E. Stat. Comput. 1991;1:47–62. [Google Scholar]
- 163.Perera V, Zabala MDT, Florance H, Smirnoff N, Grant M. Metabolomics. 2012;8:175–185. [Google Scholar]
- 164.Lange E, Gröpl C, Schulz-Trieglaff O, Leinenbach A, Huber C, Reinert K. Bioinformatics (Oxford, England) 2007;23:273–281. doi: 10.1093/bioinformatics/btm209. [DOI] [PubMed] [Google Scholar]
- 165.Lommen A. Anal. Chem. 2009;81:3079–3086. doi: 10.1021/ac900036d. [DOI] [PubMed] [Google Scholar]
- 166.Bork C, Ng K, Liu Y, Yee A. Biotechnol Prog. 2013;29:394–402. doi: 10.1002/btpr.1680. [DOI] [PubMed] [Google Scholar]
- 167.Voss B, Hanselmann M, Renard BY, Lindner MS, Köthe U, Kirchner M, Hamprecht FA. Bioinformatics (Oxford, England) 2011;27:987–993. doi: 10.1093/bioinformatics/btr051. [DOI] [PubMed] [Google Scholar]
- 168.Katajamaa M, Oresic M. BMC bioinformatics. 2004:6. doi: 10.1186/1471-2105-6-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pluskal T, Castillo S, Villar-Briones A, Oresic M. BMC bioinformatics. 2010:11. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lange E, Tautenhahn R, Neumann S. BMC Bioinformatics. 2008:9. doi: 10.1186/1471-2105-9-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Libiseller G, Dvorzak M, Kleb U, Gander E, Eisenberg T, Madeo F, Neumann S, Trausinger G, Sinner F, Pieber T, Magnes C. BMC bioinformatics. 2014:16. doi: 10.1186/s12859-015-0562-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Worley PR. Curr. Metabolomics. 2013;1:92–107. doi: 10.2174/2213235X11301010092. B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fonville JM, Richards SE, Barton RH. J. Chemometr. 2010;24:636–649. [Google Scholar]
- 174.Wiley Interdiscip. Rev. Comput. Stat. 2010;2:97–106. A. H. [Google Scholar]
- 175.Rosipal R, Krämer N. Lect. Notes Comput. Sc. 2006;3940:34–51. [Google Scholar]
- 176.Bro R, Smilde AK. Anal. Methods. 2014;6:2812–2831. [Google Scholar]
- 177.Bylesjö M, Rantalainen M, Cloarec O. J. Chemometr. 2006;20:341–351. [Google Scholar]
- 178.Genilloud O, González I, Salazar O, Martín J, Tormo JRR, Vicente F. J. Ind. Microbiol. Biotechnol. 2011;38:375–389. doi: 10.1007/s10295-010-0882-7. [DOI] [PubMed] [Google Scholar]
- 179.Hou Y, Braun DR, Michel CR, Klassen JL, Adnani N, Wyche TP, Bugni TS. Anal. Chem. 2012;84:4277–4283. doi: 10.1021/ac202623g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Macintyre L, Zhang T, Viegelmann C, Martinez I, Cheng C, Dowdells C, Abdelmohsen U, Gernert C, Hentschel U, Edrada-Ebel R. Mar. Drugs. 2014;12:3416–3448. doi: 10.3390/md12063416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Forner D, Berrué F, Correa H, Duncan K, Kerr RG. Anal. Chim. Acta. 2013;805:70–79. doi: 10.1016/j.aca.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 182.Norazwana S, Pei Jean T, Khozirah S, Faridah A, Hong Boon L. Anal. Chem. 2014;86:1324–1331. [Google Scholar]
- 183.Grond S, Papastavrou I, Zeeck A. Eur. J. Org. Chem. 2002;2002:3237–3242. [Google Scholar]
- 184.Goodwin CR, Covington BC, Derewacz DK, McNees CR, Wikswo JP, McLean JA, Bachmann BO. Chem. Biol. 2015;22:661–670. doi: 10.1016/j.chembiol.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Krug D, Zurek G, Schneider B, Garcia R, Müller R. Anal. Chim. Acta. 2008;624:97–106. doi: 10.1016/j.aca.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 186.Cortina NS, Krug D, Plaza A, Revermann O, Müller R. Angew. Chem. Int. Ed. Engl. 2012;51:811–816. doi: 10.1002/anie.201106305. [DOI] [PubMed] [Google Scholar]
- 187.Krug D, Zurek G, Revermann O, Vos M, Velicer GJ, Muller R. Appl. Environ. Microbiol. 2008;74:3058–3068. doi: 10.1128/AEM.02863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wiklund S, Johansson E, Sjöström L. Anal. Chem. 2008;80:115–122. doi: 10.1021/ac0713510. [DOI] [PubMed] [Google Scholar]
- 189.González-Menéndez V, Pérez-Bonilla M, Pérez-Victoria I, Martín J, Muñoz F, Reyes F, Tormo J, Genilloud O. Molecules. 2016;21:234. doi: 10.3390/molecules21020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hur M, Campbell AA, Almeida-de-Macedo M, Li L, Ransom N, Jose A, Crispin M, Nikolau BJ, Wurtele ES. Nat. Prod. Rep. 2013;30:565–583. doi: 10.1039/c3np20111b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Albright JC, Henke MT, Soukup AA, McClure RA, Thomson RJ, Keller NP, Kelleher NL. ACS Chem. Biol. 2015;10:1535–1541. doi: 10.1021/acschembio.5b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. Anal. Chem. 2012;84:5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Gowda H, Ivanisevic J, Johnson CH, Kurczy ME, Benton HP, Rinehart D, Nguyen T, Ray J, Kuehl J, Arevalo B, Westenskow PD, Wang J, Arkin AP, Deutschbauer AM, Patti GJ, Siuzdak G. Anal. Chem. 2014;86:6931–6939. doi: 10.1021/ac500734c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Xia J, Wishart DS. Curr. Protoc. Bioinformatics. 2011 doi: 10.1002/0471250953.bi1410s34. Chapter14: Unit14. [DOI] [PubMed] [Google Scholar]
- 195.Xia J, Sinelnikov IV, Han B, Wishart DS. Nucl. Acids Res. 2015;43:W251–257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Patti GJ, Tautenhahn R, Rinehart D, Cho K, Shriver L, Manchester M, Nikolskiy I, Johnson C, Mahieu N, Siuzdak G. Anal. Chem. 2013;85:798–804. doi: 10.1021/ac3029745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Goodwin CR, Sherrod SD, Marasco CC, Bachmann BO, Schramm-Sapyta N, Wikswo JP, McLean JA. Analytical chemistry. 2014;86:6563–6571. doi: 10.1021/ac5010794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gaudêncio SP, Pereira F. Nat. Prod. Rep. 2015;32:779–810. doi: 10.1039/c4np00134f. [DOI] [PubMed] [Google Scholar]
- 199.Eugster PJ, Boccard J, Debrus B, Bréant L, Wolfender J-L, Martel S, Carrupt P-A. Phytochemistry. 2014;108:196–207. doi: 10.1016/j.phytochem.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 200.Stow SM, Goodwin CR, Kliman M. J. Phys. Chem. B. 2014;118:13812–13820. doi: 10.1021/jp509398e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Colin AS, Grace OM, Elizabeth JW, Chuan Q, Sunia AT, Theodore RB, Darlene EC, Ruben A, Gary S. Ther. Drug Monit. 2005;27:747–751. [Google Scholar]
- 202.Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, Oda Y, Kakazu Y, Kusano M, Tohge T, Matsuda F, Sawada Y, Hirai MY, Nakanishi H, Ikeda K, Akimoto N, Maoka T, Takahashi H, Ara T, Sakurai N, Suzuki H, Shibata D, Neumann S, Iida T, Tanaka K, Funatsu K, Matsuura F, Soga T, Taguchi R, Saito K, Nishioka T. J. Mass Spectrom. 2010;45:703–714. doi: 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- 203.Shen H, Dührkop K, Böcker S, Rousu J. Bioinformatics. 2014:30. doi: 10.1093/bioinformatics/btu275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bérdy J. J. Antibiot. (Tokyo) 2012;65:385–395. doi: 10.1038/ja.2012.27. [DOI] [PubMed] [Google Scholar]
- 205.Hufsky F, Scheubert K, Böcker S. Nat. Prod. Rep. 2014;31:807–817. doi: 10.1039/c3np70101h. [DOI] [PubMed] [Google Scholar]
- 206.Allard P-MM, Péresse T, Bisson J, Gindro K, Marcourt L, Pham VC, Roussi F, Litaudon M, Wolfender J-LL. Anal. Chem. 2016;88:3317–3323. doi: 10.1021/acs.analchem.5b04804. [DOI] [PubMed] [Google Scholar]
- 207.Watrous J, Roach P, Alexandrov T, Heath BS, Yang JY, Kersten RD, van der Voort M, Pogliano K, Gross H, Raaijmakers JM, Moore BS, Laskin J, Bandeira N, Dorrestein PC. Proc. Natl. Acad. Sci. U. S. A. 2012;109:1743–1752. doi: 10.1073/pnas.1203689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yang JY, Sanchez LM, Rath CM, Liu X, Boudreau PD, Bruns N, Glukhov E, Wodtke A, de Felicio R, Fenner A, Wong WR, Linington RG, Zhang L, Debonsi HM, Gerwick WH, Dorrestein PC. J. Nat. Prod. 2013;76:1686–1699. doi: 10.1021/np400413s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Moree WJ, Phelan VV, Wu C-H, Bandeira N, Cornett DS, Duggan BM, Dorrestein PC. Proc. Natl. Acad. Sci. U. S. A. 2012;109:13811–13816. doi: 10.1073/pnas.1206855109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Briand E, Bormans M, Gugger M, Dorrestein PC, Gerwick WH. Environ Microbiol. 2015;18:384–400. doi: 10.1111/1462-2920.12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Duncan KR, Crüsemann M, Lechner A, Sarkar A, Li J, Ziemert N, Wang M, Bandeira N, Moore BS, Dorrestein PC, Jensen PR. Chem. Biol. 2015;22:460–471. doi: 10.1016/j.chembiol.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kleigrewe K, Almaliti J, Tian IY, Kinnel RB. J. Nat. Prod. 2015;78:1671–1682. doi: 10.1021/acs.jnatprod.5b00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Vizcaino MI, Crawford JM. Nat. Chem. 2015;7:411–417. doi: 10.1038/nchem.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Trautman CJ. Curr. Top. Med. Chem. 2015;16:1705–1716. doi: 10.2174/1568026616666151012111046. EP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Vizcaino MI, Engel P, Trautman E, Crawford JM. J. Am. Chem. Soc. 2014;136:9244–9247. doi: 10.1021/ja503450q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Luo Y, Li B-Z, Liu D, Zhang L, Chen Y, Jia B, Zeng B-X, Zhao H, Yuan Y-J. Chem. Soc. Rev. 2015;44:5265–5290. doi: 10.1039/c5cs00025d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zhang H, Boghigian BA, Armando J. Nat. Prod. Rep. 2011;28:125–151. doi: 10.1039/c0np00037j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Donia, Ruffner DE, Cao S, Schmidt EW. Chembiochem. 2011;12:1230–1236. doi: 10.1002/cbic.201000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schorn M, Zettler J, Noel JP, Dorrestein PC. ACS Chem. Biol. 2013;9:301–309. doi: 10.1021/cb400699p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gonzalez DJ, Corriden R, Akong-Moore K, Olson J. Chem. Biol. 2014;21:1457–1462. doi: 10.1016/j.chembiol.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gonzalez DJ, Vuong L, Gonzalez IS, Keller N, McGrosso D, Hwang JH, Hung J, Zinkernagel A, Dixon JE, Dorrestein PC. Mol. Cell Proteomics. 2014;13:1262–1272. doi: 10.1074/mcp.M113.031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Macherla VR, Liu J, Sunga M, White DJ. J. Nat. Prod. 2007;70:1454–1457. doi: 10.1021/np0702032. [DOI] [PubMed] [Google Scholar]
- 223.Desjardine K, Pereira A, Wright H. J. Nat. Prod. 2007;70:1850–1853. doi: 10.1021/np070209r. [DOI] [PubMed] [Google Scholar]
- 224.McArthur KA, Mitchell SS, Tsueng G. J. Nat. Prod. 2008;71:1732–1737. doi: 10.1021/np800286d. [DOI] [PubMed] [Google Scholar]
- 225.Teasdale ME, Shearer TL, Engel S. J. Org. Chem. 2012;77:8000–8006. doi: 10.1021/jo301246x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kellogg JJ, Todd DA, Egan JM, Raja HA. J. Nat. Prod. 2016;79:376–386. doi: 10.1021/acs.jnatprod.5b01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Salvador-Reyes LA, Luesch H. Nat. Prod. Rep. 2015;32:478–503. doi: 10.1039/c4np00104d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Johnston CW, Skinnider MA, Dejong CA, Rees PN, Chen GM, Walker CG, French S, Brown ED, Bérdy J, Liu DY, Magarvey NA. Nat. Chem. Biol. 2016 doi: 10.1038/nchembio.2018. DOI: 10.1038/nchembio.2018. [DOI] [PubMed] [Google Scholar]
- 229.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, Soule CK, Alexander B, Li A, Montgomery P, Kotz JD, Hon CS, Munoz B, Liefeld T, Dančík V, Haber DA, Clish CB, Bittker JA, Palmer M, Wagner BK, Clemons PA, Shamji AF, Schreiber SL. Nat. Chem. Biol. 2015;12:109–116. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Farha MA, Brown ED. Nat. Prod. Rep. 2016 doi: 10.1039/c5np00127g. DOI: 10.1039/C5NP00127G. [DOI] [PubMed] [Google Scholar]
- 231.Wong WR, Oliver AG, Linington RG. Chem. Biol. 2012;19:1483–1495. doi: 10.1016/j.chembiol.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 233.Hutter B, Schaab C, Albrecht S, Borgmann M, Brunner NA, Freiberg C, Ziegelbauer K, Rock CO, Ivanov I, Loferer H. Antimicrob. Agents Chemother. 2004;48:2838–2844. doi: 10.1128/AAC.48.8.2838-2844.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hu Y, Potts MB, Colosimo D, Herrera-Herrera ML, Legako AG, Yousufuddin M, White MA, MacMillan JB. J. Am. Chem. Soc. 2013;135:13387–13392. doi: 10.1021/ja403412y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Potts MB, Kim HS, Fisher KW, Hu Y, Carrasco YP. Sci. Signal. 2013:6. doi: 10.1126/scisignal.2004657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yu Y, Yi Z, Liang YZ. FEBS Lett. 2007;581:4179–4183. doi: 10.1016/j.febslet.2007.07.056. [DOI] [PubMed] [Google Scholar]
- 237.Liu Y, Wen J, Wang Y, Li Y, Xu W. Chromatographia. 2010;71:253–528. [Google Scholar]
- 238.Halouska S, Chacon O, Fenton RJ, Zinniel DK, Barletta RG, Powers R. J. Proteome Res. 2007;6:4608–4614. doi: 10.1021/pr0704332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Halouska S, Fenton RJ, Barletta RG, Powers R. ACS Chem. Biol. 2012;7:166–171. doi: 10.1021/cb200348m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Vincent IM, Weidt S, Rivas L, Burgess K, Smith TK, Ouellette M. Int. J. Parasitol. Drugs Drug Resist. 2014;4:20–27. doi: 10.1016/j.ijpddr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Vincent IM, Creek DJ, Burgess K, Woods DJ. PLoS Negl. Trop. Dis. 2012;6:e1618. doi: 10.1371/journal.pntd.0001618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Trochine A, Creek DJ, Faral-Tello P, Barrett MP. PLoS Negl. Trop. Dis. 2014;8:e2844. doi: 10.1371/journal.pntd.0002844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Vincent IM, Ehmann DE, Mills S, Perros M. Antimicrob. Agents Ch. 2016;4:20–27. doi: 10.1128/AAC.02109-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lobritz MA, Belenky P, Porter CB, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, Collins JJ. Proc. Natl. Acad. Sci. U. S. A. 2015;112:8173–8180. doi: 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Perlman ZE, Slack MD, Feng Y, Mitchison TJ. Science. 2004;306:1194–1198. doi: 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- 246.Woehrmann MH, Bray WM, Durbin JK, Nisam SC. Mol. Biosyst. 2013;9:2604–2617. doi: 10.1039/c3mb70245f. [DOI] [PubMed] [Google Scholar]
- 247.Peach KC, Bray WM, Winslow D, Linington PF. Mol. Biosyst. 2013;9:1837–1848. doi: 10.1039/c3mb70027e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Navarro G, Cheng AT, Peach KC. Antimicrob. Agents Chemother. 2014;58:1092–1099. doi: 10.1128/AAC.01781-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schulze CJ, Bray WM, Woerhmann MH, Stuart J. Chem. Biol. 2013;20:285–295. doi: 10.1016/j.chembiol.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kurita KL, Glassey E, Linington RG. Proc. Natl. Acad. Sci. U. S. A. 2015;112:11999–12004. doi: 10.1073/pnas.1507743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Schmidt EW. Nat. Chem. 2015;7:375–376. doi: 10.1038/nchem.2252. [DOI] [PubMed] [Google Scholar]
- 252.Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK. Cell Metab. 2014;20:719–730. doi: 10.1016/j.cmet.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hooper LV, Littman DR, Macpherson AJ. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y. Nat. Med. 2010;16:228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Koppel N, Balskus EP. Cell Chem. Biol. 2016;23:18–30. doi: 10.1016/j.chembiol.2015.12.008. [DOI] [PubMed] [Google Scholar]